
Abstract
Carpenter syndrome is a rare autosomal recessive disorder characterized by craniosynostosis, polydactyly and syndactyly. The gene Rab23 was identified in 2007, following which there have been only four reports describing different types of mutations. The authors describe herein a case that was diagnosed solely on the basis of history, antenatal ultrasound findings and fetal photographs taken by a cellular phone. Subsequent molecular studies revealed a novel homozygous mutation in the Rab23 gene. This mutation which converted the stop codon into a coding one, results in the formation of a lengthened 307 amino acids protein in contrast to the wild type protein containing 237 amino acids. The secondary structure prediction by PSIpred revealed that the C-terminal region of the longer mutated Rab23 protein contains several helices/strands resulting in the loss of structural flexibility and consequently, prenylation. This made it possible to offer prenatal testing to this family in its next conception where the fetus was unaffected. This case reports a novel molecular mechanism and the utility of integrating the cellular phone into clinical practice.
Similar content being viewed by others


Introduction
Carpenter syndrome or acrocephalopolysyndactyly type II (OMIM No. 201000)) is an autosomal recessive craniosynostosis disorder characterized by polydactyly, syndactyly, congenital heart disease, umbilical hernia and genital anomalies. Fusion typically affects metopic and sagittal sutures, although severe cases have a kleeblattschadel deformity [1]. Radiographic clues are presence of short to absent middle phalanges and tongue like bony epiphyseal projections in the proximal phalanges [2, 3]. The disorder is commonly caused by mutations in Rab23 gene. Recently, mutations in MEGF8 were shown to cause a Carpenter syndrome subtype frequently associated with defective left–right patterning, probably through perturbation of signaling by hedgehog and nodal family members [4]. The authors describe herein a case that was diagnosed solely on the basis of history, antenatal ultrasound findings and fetal photographs taken by a cellular phone. Subsequent molecular studies revealed a novel homozygous mutation in the Rab23 gene. Prenatal testing was then performed in their next pregnancy which revealed an unaffected fetus.
Report of Case
A nonconsanguineous third gravida lady from Punjab, India, was referred for “abnormal ultrasound findings”. Her antenatal 2D scan at 31 weeks showed a clover leaf like skull deformity in the fetus with polydactyly in both the lower limbs. Other findings included a flat facial profile, long philtrum and short long bones which corresponded to 29 weeks gestation. The thoracic diameter was normal and there was no bowing of long bones.
Their first born was a healthy 8-year-old daughter and the second child, born at term, died within 24 h of life. The father recollected that this neonate had a small, abnormally shaped head and ambiguous genitalia. No investigations had been done and earlier clinical records had been disposed of. The antenatal scans during that conception had reportedly been normal. There were no other affected family members.
Kleeblattschadel deformity or clover leaf skull identified in the ongoing pregnancy is commonly associated with either lethal skeletal dysplasia or a craniosynostosis syndrome. As the thoracic circumference was normal, a lethal dysplasia was excluded and the couple was counseled of the possibility of an autosomal recessive craniosynostosis syndrome. The importance of a definitive diagnosis for postnatal management of the affected infant and for future prenatal diagnosis was discussed with the couple. The family lived at a distance from a tertiary center and planned delivery at a center with limited access to genetic diagnosis. They were advised to obtain photographs, radiographs, and CT with bone window of the neonate after delivery. They were also requested to have blood collected for chromosomal studies, as well for DNA extraction. The lady went into preterm labor at 32 weeks of gestation and the infant died 2 h after birth due to respiratory problems. Autopsy of the neonate was refused.
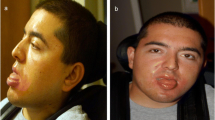
The family returned after 6 months for counseling. There were no clinical notes or anthropometric details of the neonate and radiographs and CT scan had not been obtained. However, the father had taken photographs of the baby using his cell phone which showed a small head with facial dysmorphism—hypertelorism with narrow palpebral fissures, midface hypoplasia and prominent anthelix. Bilateral duplication of the great toe with postaxial polydactyly of one upper limb was present (Fig. 1). It was difficult to assess the same in the opposite limb due to poor visualization. The parents denied any genital ambiguity although there were no pictures to verify the same.

Facial dysmorphism showing hypertelorism with narrow palpebral fissures, midface hypoplasia and prominent anthelix with inset showing preaxial polydactaly of foot
A MEDLINE database search for autosomal recessive craniosynostosis syndromes revealed 102 listings. However on refining the search criteria by addition of polydactyly the list was narrowed to two disorders—Carpenter syndrome and Baller–Gerold syndrome. Cardinal features of Baller–Gerold are radial ray hypoplasia/aplasia and craniosynostosis [5]. There were no apparent radial ray defects in the baby. Additionally, hypogenitalism has not been reported in Baller–Gerold thus favoring a clinical diagnosis of Carpenter syndrome in the baby. To confirm the clinical suspicion, Rab23 gene was analyzed.
Molecular Analysis
After seeking consent from the parents, the stored DNA of the affected child was retrieved and analyzed. The six coding exons of Rab23 were amplified with primers designed using Primer3 software (primer sequences available on request). PCR was performed using 10 pmol of each primer, 1U of Taq DNA polymerase, 1.5 mM MgCl2 and 0.1 mM dNTPs in the recommended buffer with the initial denaturation at 94 °C for 5 min, 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s and extension at 72 °C for 45 s. Sequencing was carried out on ABI 3500 analyzer covering all the six coding exons encompassing nearly 80 bps of the exon intron boundaries.
Results
The authors identified a novel stop codon mutation in homozygous state at position c.1093T > G, (p.Stop238E) in the Rab23 gene. This was considered to be the likely cause of Carpenter syndrome in the present case. The stop codon mutation converts the stop codon TAA into GAA (glutamic acid) thereby resulting in the formation of a lengthened protein (Fig. 2). The Stop238E in the present case extends the open reading frame of the coding sequence in the exon 7 of the Rab23 gene up to nucleotide 918 followed by a subsequent stop codon sequence. This results in an additional 69 amino acid residue sequence and a longer mutated Rab23 protein containing 306 amino acid residues in contrast to the wild type (WT) protein with 237 amino acid residues. The parents were heterozygous for the point mutation. Since Carpenter syndrome is an autosomal recessive disorder, the couple were counseled that there would be a 25 % risk of recurrence and since prenatal testing could be performed as the molecular basis was now delineated. During their fourth conception, the couple desired fetal testing and the chorionic villous sampling at 11 weeks revealed an unaffected fetus.

Secondary structure prediction of Rab23 protein sequence by PSIpred. The wild type Rab23 protein contains 1–237 aa only. The residues shaded in red (238–306 aa) are the extra amino acids in the mutated Rab23 protein. The disordered residues (1–3 aa and 172–234 aa) predicted by DISOPRED have been shaded in green
Discussion
After experiencing child loss, couples contemplate future pregnancy with a great deal of trepidation. They often consult a geneticist for preconceptional counseling or in early pregnancy in order to avoid a recurrence of the problem. However, very often in India the investigations in the propositus are incomplete or records of the same have been disposed of by the parents as an attempt to forget the past. The present case highlights the fact that in spite of obtaining advice pertaining to the necessary investigations, complete work up was not done for various reasons. This case is unique as the diagnosis of Carpenter syndrome was established based on the photographs taken with a cellular phone camera.
Carpenter syndrome is a rare autosomal recessive condition with a varied phenotypic representation. A number of cases of Carpenter syndrome have been reported based on clinical examination findings and investigations, the recent one being from Bangladesh [6]. However, the conformation by genetic analysis of the Rab23 gene is limited to a few case reports. Jenkins et al. [1] first showed that Carpenter syndrome results from mutations in Rab23 gene. He identified a single homozygous mutation (p.L145X) in ten of the 16 mutation positive families, describing a considerable founder effect in affected individuals of northern European descent. Alessandri et al. [7] identified c.86dupA mutation, resulting in a premature stop codon (p.Y29X), in four members with Carpenter syndrome with variable severity of craniosynostosis in a large family from Comoros Islands. In 2013, a novel eight nucleotide deletion mutation was reported in a consanguineous Emirati family extending the spectrum of Rab23 mutation [8].
The present study demonstrates a novel molecular basis for the Carpenter syndrome due to a homozygous point mutation in the stop codon of the Rab23 gene. There are two possibilities for the molecular pathophysiology of the present case. Firstly, all Rab proteins have several regions that come together in the three dimensional structure of the protein to form a GTP/GDP binding pocket. They also have a common C-terminal prenylation motif, consisting of the last four amino acids. After translation, lipid modification occurs at this motif following geranylgeranylation, which is essential for targeting of Rabs to specific membranes, and thus for their subsequent function. Due to the mutation in the stop codon, the C-terminal prenylation is impaired effecting the targeting of Rab23 to the specific function site. In support, secondary structure prediction by PSIpred revealed that the C-terminal region of the wild type Rab23 protein sequence (178–237 aa) is almost completely present as random coil. The flexibility of this coil/loop structure is possibly important for easy access to the prenylation motif [9]. However, the C-terminal region of the longer mutated Rab23 protein (238–306 aa) contains several helices/strands (Fig. 2) resulting in the loss of structural flexibility and, consequently, prenylation. Furthermore, the presence and absence of flexibility in the C-terminal region of the wild type and mutated Rab23 protein, respectively, is substantiated by the prediction of dynamically disordered regions by DISOPRED (Fig. 2) [10].
Secondly, the longer mutated Rab23 protein might be unstable due to which there would be presence of detectable mRNA but decreased enzyme activity in the cells. In corroboration, the analysis of WT and mutated Rab23 proteins by ProtParam (www.expasy.org) showed that instability index (II) of the mutated protein (II: 35.73) was slightly higher than the WT protein (II: 33.95). In addition, a similar finding has been reported in the adenine phosphoryl transferase gene and also in non-classic and classic 3β-HSD deficiency congenital adrenal hyperplasia due to a mutation in the stop codon resulting in an extended open reading frame of the gene [11]. Thus, reduced flexibility (which causes inaccessibility of prenylation motif) and higher instability of the mutated Rab23 protein probably acted in conjunction, leading to Carpenter syndrome in the present case.
Conclusion
This case demonstrates the utility of integrating the mobile phone into clinical practice. It also reports a novel mutation with an interesting molecular mechanism as a cause of Carpenter syndrome which made prenatal testing possible in this family.
References
Jenkins D, Seelow D, Jehee FS, et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007;80:1162–70.
Robinson LK, James HE, Mubarak SJ, et al. Carpenter syndrome: natural history and clinical spectrum. Am J Med Genet. 1985;20:461–9.
Cohen DM, Green JG, Miller J, et al. Acrocephalopolysyndactyly type II—Carpenter syndrome: clinical spectrum and an attempt at unification with Goodman and Summit syndromes. Am J Med Genet. 1987;28:311–24.
Twigg SRF, Lloyd D, Jenkins D, et al. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am J Hum Genet. 2012;91(5):897–905.
Van Maldergem L, Siitonen HA, Jalkh N, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–52.
Begum S, Khatun N, Rayhan SM, et al. Carpenter syndrome: a case report. Mymensingh Med J. 2012;21(3):547–9.
Alessandri JL, Dagoneau N, Laville JM, et al. RAB23 mutation in a large family from Comoros Islands with Carpenter syndrome. Am J Med Genet A. 2010;152A:982–6.
Ben-Salem S, Begum MA, Ali BR, et al. A novel aberrant splice site mutation in RAB23 leads to an eight nucleotide deletion in the mRNA and is responsible for Carpenter Syndrome in a consanguineous Emirati family. Mol Syndromol. 2013;3(6):255–61.
McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–5.
Ward JJ, McGuffin LJ, Bryson K, et al. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–9.
Pang S, Wang W, Rich B, et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3β-hydroxysteroid dehydrogenase (3β-HSD) gene causing, respectively, nonclassic and classic 3β-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87(6):2556–63.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Movva, S., Kotecha, U.H., Sharma, D. et al. Prenatal Diagnosis and Elucidation of a Novel Molecular Mechanism in Carpenter Syndrome. J. Fetal Med. 1, 89–93 (2014). https://doi.org/10.1007/s40556-014-0017-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40556-014-0017-8