
Abstract
Purpose of Review
This review aims to discuss and summarize the peripheral sensory mechanisms involved in the induction of the early phase of insulin release, known as cephalic phase insulin release (CPIR), triggered by stimuli related to food, particularly sugars.
Recent Findings
At least, two distinct systems on the tongue are responsible for detecting oral sugars. The first system involves the G-protein-coupled receptor Tas1r2/Tas1r3, which can detect not only sugars but also artificial sweeteners and sweet proteins. The second system relies on glucose transporters, specifically recognize and transport monosaccharides. The Tas1r2/Tas1r3 receptor utilizes a signal transduction pathway involving gustducin, phospholipase β2, and transient receptor potential channel M5 to depolarize taste cells. On the other hand, glucose transporters facilitate the transport of monosaccharides into cells, where their degradation produces ATP. This ATP inhibits the metabolic sensor KATP channel, ultimately leading to cell depolarization. Recent studies in mice have demonstrated that glucose transporters and KATP channels, rather than the Tas1r2/Tas1r3 receptor, are essential for the induction of CPIR.
Summary
The detection of sugars in the oral cavity relies on two essential mechanisms: the Tas1r2/Tas1r3 receptor and glucose transporters. Notably, oral glucose transporters are likely to play a significant role in the induction of sugar-induced CPIR. As a result, these two sugar detection systems may have distinct roles in maintaining energy homeostasis within the body.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Insulin plays a crucial role in reducing plasma glucose levels by facilitating the absorption of glucose into muscle, adipose tissue, and liver cells. Its secretion is triggered by blood glucose, resulting in an increase in plasma insulin following food ingestion. However, even before the rise in plasma glucose levels after a meal, the stimulation of sensory systems in the head and oropharynx region initiates an early release of insulin from pancreatic β-cells [1,2,3]. This early phase of insulin release, known as cephalic phase insulin release (CPIR), is elicited by food-related stimuli, particularly sugars such as glucose and sucrose [4,5,6,7]. Sugars are typical tastants for sweet taste, and therefore, other sweet-tasting substances, for example, artificial sweetener saccharin, have also been reported to induce CPIR [1, 5, 8,9,10]. However, conflicting reports exist regarding the ability of artificial sweeteners to elicit CPIR, with some studies showing no such effect [11,12,13,14].
Taste receptors located on the taste cells are responsible for detecting oral sugars. Two types of receptor systems are known to be involved: the Tas1r2/Tas1r3 sweet receptor and glucose transporters. The Tas1r2/Tas1r3 receptor is the primary sensor for sweet tastants, capable of detecting sugars, artificial sweeteners, and even sweet proteins [15]. Studies have demonstrated that mice lacking functional Tas1r2/Tas1r3 receptors exhibit either abolished or significantly reduced gustatory nerve responses to sweeteners [16, 17]. On the other hand, glucose transporters (GLUTs) and sodium-glucose transporters (SGLTs) serve as specific sensors for glucose. In mice, certain gustatory nerve fibers respond to sugars but not artificial sweeteners, and their responses to glucose + NaCl are notably suppressed by phlorizin, an SGLT inhibitor [18••]. Recently, such glucose specific taste response mediated by SGLT was reported to be increased by a multifunctional regulatory peptide adrenomedullin [19•]. This finding provides further support to the notion that glucose transporters serve as sugar sensors in taste receptor cells. These sensory mechanisms likely play significant roles in sugar-induced CPIR.
The sense of taste is a significant component of the sensory system that influences our food intake. However, other sensory cues, such as visual, olfactory, and somatosensory signals, also play crucial roles in determining our food intake. For instance, visual and olfactory cues associated with foods we dislike can inhibit our consumption of those particular foods. Moreover, nociceptive signals in the oral cavity trigger the aperture reflex, which disrupts the ingestion of food bolus. In the central nervous system, these sensory signals are integrated and utilized to elicit appropriate reactions and behaviors. An illustrative example is the experience of many patients who undergo enteral tube feeding, a method that bypasses oral ingestion and swallowing of food. These patients often report experiencing diarrhea [20]. This highlights the fact that not only taste, but also other sensory signals resulting from food intake, can contribute to the initiation of various reactions, including changes in physiology and behavior.
This review aims to provide an overview and analysis of the peripheral sensory mechanisms involved in the sugar-induced CPIR. First, sugar sensing mechanisms on the tongue were summarized. Then contribution of these mechanisms to sugar-induced CPIR is discussed.
Detection of Sweeteners by Tas1r2/Tas1r3
The discovery of the sweet taste receptor, Tas1r2/Tas1r3, occurred in the early 2000s, as reported by several studies [21,22,23,24,25,26,27]. Tas1r2/Tas1r3 is a G-protein-coupled receptor with a large extracellular domain known as the Venus flytrap domain (VFTD), which is crucial for detecting various sweet compounds. Activation of the Tas1r2/Tas1r3 receptor by sweet compounds in taste cells initiates an intracellular signaling cascade including gustducin [28], phospholipase Cβ2 [29], inositol-1,4,5-triophosphate receptor type 3 [30], and transient receptor potential channel M5 [29, 31]. This cascade leads to taste cell depolarization and the generation of action potentials [32,33,34]. The action potentials trigger the opening of the calcium homeostasis modulator 1/3 (Calhm1/3) channel, which releases ATP from taste cells [35, 36]. The released ATP binds to and activates purinergic P2X2/3 receptors on the gustatory nerve fibers [37], allowing the transmission of sweet signals to the central nervous system.
Tas1r2/Tas1r3 receptors have the capability to detect various substances, including sugars, artificial sweeteners, and sweet proteins. Sugars such as sucrose and glucose, as well as artificial sweeteners like sucralose, have been shown to bind to the VFTD of both Tas1r2 and Tas1r3 [38,39,40]. However, there are differences in sweet sensitivity to aspartame and neotame between humans and rodents. Humans perceive these artificial sweeteners as sweet, while rodents either do not detect them or have a weak preference [41, 42]. This disparity is due to the varying binding affinity of these artificial sweeteners to human or rodent Tas1r2/Tas1r3 receptors [27, 43]. Certain proteins, such as Brazzein, Monelin, and Thaumatin, possess a sweet taste in humans. These sweet proteins can also bind to and activate human Tas1r2/Tas1r3 receptors in heterologous expression assays [44, 45]. Consequently, taste signals mediated by Tas1r2/Tas1r3 receptors are not exclusive to sugars, which are the primary energy sources for our bodies. In mice lacking the Tas1r2 and/or Tas1r3 genes, a preference for sweet compounds, including sugars, is lost [16, 17, 46], suggesting a strong correlation between these signals and innate preference for sweet compounds. However, mice lacking the Tas1r3 gene still exhibit residual responses to sugars, particularly glucose [17], implying the existence of additional receptor(s) involved in sugar detection in the oral cavity.
Oral Sugar Detection by Glucose Transporters
Glucose transporters play a crucial role in facilitating the absorption of dietary carbohydrates in the intestine. Given the similarities between intestinal cells and taste receptor cells, it is plausible that these transporters may also function in detecting sugars in the oral cavity if they are present in taste cells. Interestingly, the sweet receptor Tas1r2/Tas1r3 has been reported to be expressed in intestinal cells [47, 48]. Conversely, several glucose transporters (Glut1, Glut2, Glut4, Glut5, Glut8, and Glut9) and the sodium glucose transporter Sglt1 have been found to be expressed in the taste tissues of rodents [49,50,51]. In the pancreatic islet cells, the ATP-sensitive potassium channel (KATP channel) plays a crucial role in depolarization and insulin secretion, as it is inhibited by ATP generated during glucose metabolism [52, 53]. Histological studies have shown the expression of KATP channel subunits, specifically sulfonylurea receptor 1 (Sur1) and potassium inwardly rectifying channel 6.1 (Kir6.1), in taste cells [33, 51]. Some of these transporters and KATP channel subunits are likely to be expressed in Tas1r3-positive taste cells, suggesting their potential involvement in sweet-sensitive taste cells.
Experimental evidence supporting the role of glucose transporters as sugar sensors was demonstrated through the recording of sugar responses in gustatory nerve fibers [18••]. Gustatory nerve fibers that exhibited the highest response to sucrose were categorized into three groups: phlorizin-sensitive type, phlorizin-insensitive type, and a mixed type. Phlorizin, a competitive inhibitor of Sglt1, was used to determine whether the responses of the phlorizin-sensitive fibers originated from taste cells expressing Sglts. Indeed, certain taste cells displayed apical uptake of a fluorescent glucose analog 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG) via glucose transporters [18••]. Potential mechanisms for glucose detection by taste cells can be outlined as follows: (1) Some taste cells may possess both glucose transporters and KATP channels, (2) Glucose taken up through the apically expressed glucose transporters is metabolized within taste cells, leading to ATP production, (3) Increases in intracellular ATP concentrations result in the closure of KATP channels, depolarizing the taste cells, and (4) Transmitters, possibly ATP, are released to activate gustatory nerve fibers.
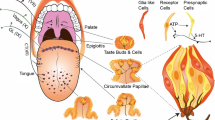
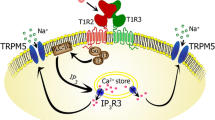
The glucose-transporter dependent system is capable of detecting glucose and monosaccharides, but it can not directly sense di-, oligo-, and polysaccharides such as sucrose and polycose because these molecules can not enter cells through glucose transporters. To enable the detection of di-, oligo-, and polysaccharides by the glucose-transporter dependent system, additional components are necessary to break down these saccharides into monosaccharides. Salivary amylase (Amy1) and pancreatic amylase (Amy2) are enzymes responsible for hydrolyzing starch and producing disaccharides. In the brush border cells of the intestine, various disaccharidases such as maltase-glucoamylase, sucrase-isomaltase, and lactase are expressed. These enzymes further break down the disaccharides into monosaccharides, which are then absorbed into the cells through glucose transporters [54, 55]. Examination of the presence of these amylases and α-glucosidase in taste tissues has revealed that taste cells also express all of these components [56]. Blocking α-glucosidase on the tongue has been shown to decrease gustatory nerve responses specifically to disaccharides (such as sucrose and maltose), but not to monosaccharides (such as glucose and fructose) or noncaloric sweeteners (such as sucralose and SC45647), in both wild-type and Tas1r3-KO mice [56]. This suggests that the α-glucosidase expressed in taste cells plays a crucial role in the detection of disaccharides on the tongue. A summary of possible mechanisms for sugar detection via glucose transporters is summarized in Fig. 1.

Possible mechanism for sugar detection via glucose transporters in taste receptor cells. The glucose transporters present in taste receptor cells have the capability to transfer glucose from the apical side. Glucose undergoes metabolic processes such as glycolysis and oxidative phosphorylation, resulting in the production of ATP. Consequently, intracellular ATP levels are increased. This increase in ATP concentration causes the closure of ATP-sensitive K channels (KATP channels). Subsequently, the taste cell becomes depolarized, leading to the generation of action potentials. Sodium-glucose transporters (Sglts) have the ability to transport both Na+ and glucose. Therefore, the activation of Sglts alone can potentially depolarize taste cells. Disaccharides such as sucrose are enzymatically digested by α-glucosidase expressed on the taste cells. This digestion process produces glucose, which is then transported into the taste cell through glucose transporters.
Sugar Detection and CPIR
Among the five basic tastes (sweet, salty, sour, bitter, and umami), sweet taste has been reported to elicit CPIR in humans [5] and rats [9]. However, one study demonstrated that umami taste also induced CPIR in rats [57]. As mentioned earlier, sweet taste can be evoked by various substances, including sugars, artificial sweeteners, and sweet proteins. Consequently, multiple sweeteners have been tested to determine whether they can induce CPIR. In some reports, the nonnutritive sweetener saccharin was found to induce CPIR in both humans and rats [1, 5, 8,9,10]. However, other studies have shown that oral stimulation with non-nutritive sweeteners did not trigger CPIR [11,12,13,14]. In the case of mice, CPIR was not induced by oral stimulation with various non-nutritive sweeteners, including saccharin, sucralose, acesulfame K, and SC45647. However, sugars such as glucose and sucrose did elicit CPIR in mice [14]. Consistent with these findings, Tas1r3-KO mice still exhibited an increase in plasma insulin 5 min after ingesting a glucose solution, and the changes in plasma insulin were comparable between control B6 mice and Tas1r3-KO mice [14, 58••]. These results collectively suggest that the Tas1r2/Tas1r3 receptor is not necessary for sugar-induced CPIR in mice.
Another potential candidate for sugar detection in the oral cavity, contributing to the induction of CPIR, is the glucose-transporter-mediated system involving glucose transporters (Gluts, Sglts), and the KATP channel. The involvement of the KATP channel in sugar-induced CPIR was investigated using knockout (KO) model mice and pharmacological agents targeting the KATP channel [14]. This study revealed that the increase in plasma insulin 5 min after ingesting a glucose solution was significantly smaller in Sur1-KO mice compared to control mice. Additionally, the mixing of glyburide, a KATP channel closer, or diazoxide, a KATP channel opener, with the glucose solution significantly enhanced or reduced the increase in plasma insulin, respectively. Hence, the KATP channel may play a critical role in sugar-induced CPIR in mice. Regarding the contribution of glucose transporters to sugar-induced CPIR, pharmacological blockers for Gluts and Sglts were applied to the tongues of mice to test whether a blocker mixture could suppress sugar-induced CPIR [58••]. Indeed, treatment with phlorizin and phloretin in the oral cavity of mice significantly inhibited the rapid increase in plasma insulin levels after orally ingesting a glucose solution, although a small increase in plasma insulin remained. However, the non-metabolizable glucose analog methyl-α-D-glucopyranoside (MDG) did not induce CPIR in mice [14, 58••], suggesting that the activation of glucose transporters itself may not contribute to the induction of CPIR in mice. On the other hand, Glendining et al., (2017) demonstrated that plasma insulin dynamics were not significantly different between Sglt1-KO and wild-type control mice [14]. However, in this study, only small changes in plasma insulin occurred after orally ingesting a glucose solution even in wild-type control mice. Collectively, it is possible that oral glucose transporters function in detecting sugars to induce CPIR in mice.
Following the activation of taste cells by sugars, the signals are transmitted to gustatory nerve fibers, the central nervous system, and subsequently, vagus nerve fibers to induce CPIR. The transmission of signals from sweet taste cells to gustatory nerve fibers involves the release of ATP from sweet taste cells via Calhm1/3 and the activation of P2X2/X3 receptors on the gustatory nerve fiber [59•]. However, both Calhm1-KO mice and P2X2/X3 double KO mice still displayed a rapid increase in plasma insulin levels after orally ingesting a glucose solution, similar to their wild-type control mice [14]. This suggests a possibility that Tas1r-independent sugar detection mechanisms might employ a non-purinergic transmission pathway at the synapse between taste cells and gustatory nerve fibers. One potential candidate for this mechanism is peptidergic transmission mediated by glucagon-like peptide-1 (GLP-1). Previous studies have shown that Tas1r3-positive taste cells express GLP-1 [60]. GLP-1 is released from sweet-sensitive taste cells and may function as a neurotransmitter to activate gustatory nerve fibers [61]. This system could operate independently of the channel synapse in sweet-sensitive taste cells and potentially contribute to sensory signaling for the induction of CPIR. Additionally, taste cells are known to release acetylcholine in response to sweet-bitter mixtures [62]. Solitary chemosensory cells (SCCs) in the nasal cavity, which share properties with type II taste bud cells (sweet, bitter, and umami cells), express choline acetyltransferase [63]. Similar types of cells are found in the trachea, auditory tube, urethra, and thymic medulla, all expressing choline acetyltransferase [64,65,66,67]. While the expression of choline acetyltransferase in sweet-sensitive taste cells has not been confirmed, it is plausible that acetylcholine may function as a neurotransmitter from sugar-sensitive taste cells, in addition to purinergic transmission. Further studies are required to elucidate the mechanisms underlying the transmission of signals from sugar-sensitive cells to gustatory nerve fibers.
Other Sensory Cue for CPIR
When animals consume food, not only the sense of taste, but also other sensory systems such as touch and thermal sensors in the oral and pharynx region will be activated. These somatosensory signals elicited by food intake play a significant role in gastrointestinal functions and potentially contribute to the induction of CPIR. In mice, direct infusion of glucose into the gut did not trigger the rapid phase of insulin responses in both Tas1r3-KO and wild-type mice [7, 58••]. Hence, glucose signals from the gut and intestine alone might not be sufficient to induce CPIR. However, when intragastric infusion of glucose was combined with water drinking (without chemical cues from the tongue), a slight but significant increase in plasma insulin levels was observed 5 min after glucose administration [58••]. Consistent with this finding, mice treated with inhibitors for glucose transporters on the tongue still exhibited a small but significant increase in plasma insulin levels 5 min after glucose administration [58••]. Ingestion of MDG with gastric infusion of glucose solution also elicited small but significant rapid increase in plasma insulin level. These results suggest that somatosensory signals from the oropharynx region contribute to the induction of CPIR. However, the rapid phase of insulin response is minimal when chemical cues or signals derived from glucose transporters on the tongue are omitted. Overall, somatosensory signals may have a supportive or synergistic effect on the induction of CPIR in conjunction with oral sugar signals. Somatosensory signals from the oropharynx region propagate through the trigeminal nerve, glossopharyngeal nerve, and vagus nerve. Taste signals, on the other hand, propagate through the facial nerve (chorda tympani nerve and greater petrosal nerve), or glossopharyngeal nerve, and vagus nerve. These signals are likely integrated in nuclei of central nervous system, such as the nucleus of the solitary tract and the dorsal motor nucleus of the vagus nerve. The integration of signals from different modalities would enhance efferent signals to β-cells in the islets, ultimately leading to the induction of CPIR. Further investigation is needed to explore this possibility in future studies.
Conclusion
Although the mechanism for the induction of CPIR is still controversial, taste cells located on the tongue are considered crucial for detecting the chemical signals involved in CPIR. Given that CPIR is prominently elicited by sugars, the sugar detection system on the tongue plays a fundamental role in CPIR. Recent studies have demonstrated that the sugar-specific detection system, comprising glucose transporters and the KATP channel, functions to detect sugars on the tongue and contributes to the induction of CPIR. However, due to the challenges associated with experimental designs, it is important to acknowledge the possibility that other sensory cues may also contribute to the induction of CPIR. Further investigations are necessary to enhance our understanding of the underlying mechanisms involved in CPIR.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Berthoud HR, Trimble ER, Siegel EG, Bereiter DA, Jeanrenaud B. Cephalic-phase insulin secretion in normal and pancreatic islet-transplanted rats. Am J Physiol. 1980. https://doi.org/10.1152/ajpendo.1980.238.4.E336.
Bruce DG, Storlien LH, Furler SM, Chisholm DJ. Cephalic phase metabolic responses in normal weight adults. Metabolism. 1987. https://doi.org/10.1016/0026-0495(87)90106-5.
Teff KL, Mattes RD, Engelman K. Cephalic phase insulin release in normal weight males: verification and reliability. Am J Physiol. 1991. https://doi.org/10.1152/ajpendo.1991.261.4.E430.
Yamazaki M, Sakaguchi T. Effects of D-glucose anomers on sweetness taste and insulin release in man. Brain Res Bull. 1986. https://doi.org/10.1016/0361-9230(86)90126-7.
Just T, Pau HW, Engel U, Hummel T. Cephalic phase insulin release in healthy humans after taste stimulation? Appetit. 2008. https://doi.org/10.1016/j.appet.2008.04.271.
Duskova M, Macourek M, Sramkova M, Hill M, Starka L. The role of taste in cephalic phase of insulin secretion. Prague Med Rep. 2013. https://doi.org/10.14712/23362936.2014.11.
Glendinning JI, Stano S, Holter M, Azenkot T, Goldman O, Margolskee RF, Vasselli JR, Sclafani A. Sugar-induced cephalic-phase insulin release is mediated by a T1r2+T1r3-independent taste transduction pathway in mice. Am J Physiol Regul Integr Comp Physiol. 2015. https://doi.org/10.1152/ajpregu.00056.2015.
Ionescu E, Rohner-Jeanrenaud F, Proietto J, Rivest RW, Jeanrenaud B. Taste-induced changes in plasma insulin and glucose turnover in lean and genetically obese rats. Diabetes. 1988. https://doi.org/10.2337/diab.37.6.773.
Tonosaki K, Hori Y, Shimizu Y, Tonosaki K. Relationships between insulin release and taste. Biomed Res. 2007. https://doi.org/10.2220/biomedres.28.79.
Dhillon J, Lee JY, Mattes RD. The cephalic phase insulin response to nutritive and low-calorie sweeteners in solid and beverage form. Physiol Behav. 2017. https://doi.org/10.1016/j.physbeh.2017.09.009.
Berthoud HR, Powley TL. Identification of vagal preganglionics that mediate cephalic phase insulin response. Am J Physiol. 1990. https://doi.org/10.1152/ajpregu.1990.258.2.R523.
Teff KL, Devine J, Engelman K. Sweet taste: effect on cephalic phase insulin release in men. Physiol Behav. 1995. https://doi.org/10.1016/0031-9384(94)00373-d.
Abdallah L, Chabert M, Louis-Sylvestre J. Cephalic phase responses to sweet taste. Am J Clin Nutr. 1997. https://doi.org/10.1093/ajcn/65.3.737.
Glendinning JI, Frim YG, Hochman A, Lubitz GS, Basile AJ, Sclafani A. Glucose elicits cephalic-phase insulin release in mice by activating KATP channels in taste cells. Am J Physiol Regul Integr Comp Physiol. 2017. https://doi.org/10.1152/ajpregu.00433.2016.
Sanematsu K, Yoshida R, Shigemura N, Ninomiya Y. Structure, function, and signaling of taste G-protein-coupled receptors. Curr Pharm Biotechnol. 2014. https://doi.org/10.2174/1389201015666140922105911.
Zhao GQ, Zhang Y, Hoon MA, Chandrashekar J, Erlenbach I, Ryba NJ, Zuker CS. The receptors for mammalian sweet and umami taste. Cell. 2003. https://doi.org/10.1016/s0092-8674(03)00844-4.
Damak S, Rong M, Yasumatsu K, Kokrashvili Z, Varadarajan V, Zou S, Jiang P, Ninomiya Y, Margolskee RF. Detection of sweet and umami taste in the absence of taste receptor T1r3. Science. 2003. https://doi.org/10.1126/science.1087155.
•• Yasumatsu K, Ohkuri T, Yoshida R, Iwata S, Margolskee RF, Ninomiya Y. Sodium-glucose cotransporter 1 as a sugar taste sensor in mouse tongue. Acta Physiol. 2020. https://doi.org/10.1111/apha.13529. Functional importance of glucose transporters on taste cell in sugar detection was first demonstrated by single fiber recordings of gustatory nerve. In addition, entry of glucose from apical membrane was demonstrated by using fluorescent glucose derivative.
• Iwata S, Yoshida R, Takai S, Sanematsu K, Shigemura N, Ninomiya Y. Adrenomedullin enhances mouse gustatory nerve responses to sugars via T1R-independent sweet taste pathway. Nutrients. 2023. https://doi.org/10.3390/nu15132941. This study demonstrated that adrenomedullin selectively enhanced sugar responses of gustatory nerves via glucose transporter dependent pathway in taste cells.
Whelan K. Enteral-tube-feeding diarrhoea: Manipulating the colonic microbiota with probiotics and prebiotics. Proc Nutr Soc. 2007. https://doi.org/10.1017/S0029665107005551.
Bachmanov AA, Li X, Reed DR, Ohmen JD, Li S, Tordoff MG, de Jong PJ, Wu C, West DB, Chatterjee A, Ross DA, Beauchamp GK. Positional cloning of the mouse saccharin preference (Sac) locus. Chem Senses. 2001. https://doi.org/10.1093/chemse/26.7.925.
Kitagawa M, Kusakabe Y, Miura H, Ninomiya Y, Hino A. Molecular genetic identification of a candidate receptor gene for sweet taste. Biochem Biophys Res Commun. 2001. https://doi.org/10.1006/bbrc.2001.4760.
Max M, Shanker YG, Huang L, Rong M, Liu Z, Campagne F, Weinstein H, Damak S, Margolskee RF. Tas1r3, encoding a new candidate taste receptor, is allelic to the sweet responsiveness locus Sac. Nat Genet. 2001. https://doi.org/10.1038/ng0501-58.
Montmayeur JP, Liberles SD, Matsunami H, Buck LB. A candidate taste receptor gene near a sweet taste locus. Nat Neurosci. 2001. https://doi.org/10.1038/87440.
Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Mammalian sweet taste receptors. Cell. 2001. https://doi.org/10.1016/s0092-8674(01)00451-2.
Sainz E, Korley JN, Battey JF, Sullivan SL. Identification of a novel member of the T1R family of putative taste receptors. J Neurochem. 2001. https://doi.org/10.1046/j.1471-4159.2001.00292.x.
Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E. Human receptors for sweet and umami taste. Proc Natl Acad Sci USA. 2002. https://doi.org/10.1073/pnas.072090199.
Wong GT, Gannon KS, Margolskee RF. Transduction of bitter and sweet taste by gustducin. Nature. 1996. https://doi.org/10.1038/381796a0.
Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, Zuker CS, Ryba NJ. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003. https://doi.org/10.1016/s0092-8674(03)00071-0.
Hisatsune C, Yasumatsu K, Takahashi-Iwanaga H, Ogawa N, Kuroda Y, Yoshida R, Ninomiya Y, Mikoshiba K. Abnormal taste perception in mice lacking the type 3 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2007. https://doi.org/10.1074/jbc.M705641200.
Damak S, Rong M, Yasumatsu K, Kokrashvili Z, Pérez CA, Shigemura N, Yoshida R, Mosinger B Jr, Glendinning JI, Ninomiya Y, Margolskee RF. Trpm5 null mice respond to bitter, sweet, and umami compounds. Chem Senses. 2006. https://doi.org/10.1093/chemse/bjj027.
Yoshida R, Ohkuri T, Jyotaki M, Yasuo T, Horio N, Yasumatsu K, Sanematsu K, Shigemura N, Yamamoto T, Margolskee RF, Ninomiya Y. Endocannabinoids selectively enhance sweet taste. Proc Natl Acad Sci USA. 2010. https://doi.org/10.1073/pnas.0912048107.
Yoshida R, Noguchi K, Shigemura N, Jyotaki M, Takahashi I, Margolskee RF, Ninomiya Y. Leptin suppresses mouse taste cell responses to sweet compounds. Diabetes. 2015. https://doi.org/10.2337/db14-1462.
Yoshida R, Margolskee RF, Ninomiya Y. Phosphatidylinositol-3 kinase mediates the sweet suppressive effect of leptin in mouse taste cells. J Neurochem. 2021. https://doi.org/10.1111/jnc.15268.
Taruno A, Vingtdeux V, Ohmoto M, Ma Z, Dvoryanchikov G, Li A, Adrien L, Zhao H, Leung S, Abernethy M, Koppel J, Davies P, Civan MM, Chaudhari N, Matsumoto I, Hellekant G, Tordoff MG, Marambaud P, Foskett JK. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature. 2013. https://doi.org/10.1038/nature11906.
Ma Z, Taruno A, Ohmoto M, Jyotaki M, Lim JC, Miyazaki H, Niisato N, Marunaka Y, Lee RJ, Hoff H, Payne R, Demuro A, Parker I, Mitchell CH, Henao-Mejia J, Tanis JE, Matsumoto I, Tordoff MG, Foskett JK. CALHM3 is essential for rapid ion channel-mediated purinergic neurotransmission of GPCR-mediated tastes. Neuron. 2018. https://doi.org/10.1016/j.neuron.2018.03.043.
Finger TE, Danilova V, Barrows J, Bartel DL, Vigers AJ, Stone L, Hellekant G, Kinnamon SC. ATP signaling is crucial for communication from taste buds to gustatory nerves. Science. 2005. https://doi.org/10.1126/science.1118435.
Nie Y, Vigues S, Hobbs JR, Conn GL, Munger SD. Distinct contributions of T1R2 and T1R3 taste receptor subunits to the detection of sweet stimuli. Curr Biol. 2005. https://doi.org/10.1016/j.cub.2005.09.037.
Nie Y, Hobbs JR, Vigues S, Olson WJ, Conn GL, Munger SD. Expression and purification of functional ligand-binding domains of T1R3 taste receptors. Chem Senses. 2006. https://doi.org/10.1093/chemse/bjj053.
Maîtrepierre E, Sigoillot M, Le Pessot L, Briand L. Recombinant expression, in vitro refolding, and biophysical characterization of the N-terminal domain of T1R3 taste receptor. Protein Expr Purif. 2012. https://doi.org/10.1016/j.pep.2012.03.006.
Sclafani A, Abrams M. Rats show only a weak preference for the artificial sweetener aspartame. Physiol Behav. 1986. https://doi.org/10.1016/0031-9384(86)90228-3.
Bachmanov AA, Tordoff MG, Beauchamp GK. Sweetener preference of C57BL/6ByJ and 129P3/J mice. Chem Sens. 2001. https://doi.org/10.1093/chemse/26.7.905.
Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X. Different functional roles of T1R subunits in the heteromeric taste receptors. Proc Natl Acad Sci USA. 2004. https://doi.org/10.1073/pnas.0404384101.
Jiang P, Ji Q, Liu Z, Snyder LA, Benard LM, Margolskee RF, Max M. The cysteine-rich region of T1R3 determines responses to intensely sweet proteins. J Biol Chem. 2004. https://doi.org/10.1074/jbc.M406779200.
Ohta K, Masuda T, Tani F, Kitabatake N. Introduction of a negative charge at Arg82 in thaumatin abolished responses to human T1R2-T1R3 sweet receptors. Biochem Biophys Res Commun. 2011. https://doi.org/10.1016/j.bbrc.2011.08.033.
Yamase Y, Huang H, Mitoh Y, Egusa M, Miyawaki T, Yoshida R. Taste responses and ingestive behaviors to ingredients of fermented milk in mice. Foods. 2023. https://doi.org/10.3390/foods12061150.
Mace OJ, Affleck J, Patel N, Kellett GL. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J Physiol. 2007. https://doi.org/10.1113/jphysiol.2007.130906.
Margolskee RF, Dyer J, Kokrashvili Z, Salmon KS, Ilegems E, Daly K, Maillet EL, Ninomiya Y, Mosinger B, Shirazi-Beechey SP. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci USA. 2007. https://doi.org/10.1073/pnas.0706678104.
Merigo F, Benati D, Cristofoletti M, Osculati F, Sbarbati A. Glucose transporters are expressed in taste receptor cells. J Anat. 2011. https://doi.org/10.3390/nu15132941.
Toyono T, Seta Y, Kataoka S, Oda M, Toyoshima K. Differential expression of the glucose transporters in mouse gustatory papillae. Cell Tissue Res. 2011. https://doi.org/10.1007/s00441-011-1210-x.
Yee KK, Sukumaran SK, Kotha R, Gilbertson TA, Margolskee RF. Glucose transporters and ATP-gated K+ (KATP) metabolic sensors are present in type 1 taste receptor 3 (T1r3)-expressing taste cells. Proc Natl Acad Sci USA. 2011. https://doi.org/10.1073/pnas.1100495108.
Aizawa T, Komatsu M, Asanuma N, Sato Y, Sharp GWG. Glucose action ‘beyond ionic events’ in the pancreatic beta cell. Trends Pharmacol Sci. 1998. https://doi.org/10.1016/s0165-6147(98)01273-5.
Ashcroft FM, Rorsman P. KATP channels and islet hormone secretion: new insights and controversies. Nat Rev Endocrinol. 2013. https://doi.org/10.1038/nrendo.2013.166.
Hauri HP, Sterchi EE, Bienz D, Fransen JA, Marxer A. Expression and intracellular transport of microvillus membrane hydrolases in human intestinal epithelial cells. J Cell Biol. 1985. https://doi.org/10.1083/jcb.101.3.838.
Robayo-Torres CC, Quezada-Calvillo R, Nichols BL. Disaccharide digestion: clinical and molecular aspects. Clin Gastroenterol Hepatol. 2006. https://doi.org/10.1016/j.cgh.2005.12.023.
Sukumaran SK, Yee KK, Iwata S, Kotha R, Quezada-Calvillo R, Nichols BL, Mohan S, Pinto BM, Shigemura N, Ninomiya Y, Margolskee RF. Taste cell-expressed α-glucosidase enzymes contribute to gustatory responses to disaccharides. Proc Natl Acad Sci USA. 2017. https://doi.org/10.1073/pnas.1520843113.
Niijima A, Togiyama T, Adachi A. Cephalic-phase insulin release induced by taste stimulus of monosodium glutamate (umami taste). Physiol Behav. 1990. https://doi.org/10.1016/0031-9384(90)90247-2.
•• Takamori M, Mitoh Y, Horie K, Egusa M, Miyawaki T, Yoshida R. Sugar signals from oral glucose transporters elicit cephalic-phase insulin release in mice. J Physiol Sci. 2023. https://doi.org/10.1186/s12576-023-00875-3. This study showed that sugar induced CPIR is inhibited by oral treatment of glucose transporter inhibitors, suggesting that oral sugar detection mediated by glucose transporter is crucial for sugar-induced CPIR.
• Taruno A, Nomura K, Kusakizako T, Ma Z, Nureki O, Foskett JK. Taste transduction and channel synapses in taste buds. Pflugers Arch. 2020. https://doi.org/10.1007/s00424-020-02464-4. A comprehensive review on taste transduction and signal transmission from taste cells to gustatory nerve fibers.
Shin YK, Martin B, Golden E, Dotson CD, Maudsley S, Kim W, Jang HJ, Mattson MP, Drucker DJ, Egan JM, Munger SD. Modulation of taste sensitivity by GLP-1 signaling. J Neurochem. 2009. https://doi.org/10.1111/j.1471-4159.2008.05397.x.
Takai S, Yasumatsu K, Inoue M, Iwata S, Yoshida R, Shigemura N, Yanagawa Y, Drucker DJ, Margolskee RF, Ninomiya Y. Glucagon-like peptide-1 is specifically involved in sweet taste transmission. FASEB J. 2015. https://doi.org/10.1096/fj.14-265355.
Dando R, Roper SD. Acetylcholine is released from taste cells, enhancing taste signalling. J Physiol. 2012. https://doi.org/10.1113/jphysiol.2012.232009.
Saunders CJ, Christensen M, Finger TE, Tizzano M. Cholinergic neurotransmission links solitary chemosensory cells to nasal inflammation. Proc Natl Acad Sci USA. 2014. https://doi.org/10.1073/pnas.1402251111.
Krasteva G, Canning BJ, Hartmann P, Veres TZ, Papadakis T, Mühlfeld C, Schliecker K, Tallini YN, Braun A, Hackstein H, Baal N, Weihe E, Schütz B, Kotlikoff M, Ibanez-Tallon I, Kummer W. Cholinergic chemosensory cells in the trachea regulate breathing. Proc Natl Acad Sci USA. 2011. https://doi.org/10.1073/pnas.1019418108.
Krasteva G, Hartmann P, Papadakis T, Bodenbenner M, Wessels L, Weihe E, Schütz B, Langheinrich AC, Chubanov V, Gudermann T, Ibanez-Tallon I, Kummer W. Cholinergic chemosensory cells in the auditory tube. Histochem Cell Biol. 2012. https://doi.org/10.1007/s00418-012-0911-x.
Deckmann K, Filipski K, Krasteva-Christ G, Fronius M, Althaus M, Rafiq A, Papadakis T, Renno L, Jurastow I, Wessels L, Wolff M, Schütz B, Weihe E, Chubanov V, Gudermann T, Klein J, Bschleipfer T, Kummer W. Bitter triggers acetylcholine release from polymodal urethral chemosensory cells and bladder reflexes. Proc Natl Acad Sci USA. 2014. https://doi.org/10.1073/pnas.1402436111.
Panneck AR, Rafiq A, Schütz B, Soultanova A, Deckmann K, Chubanov V, Gudermann T, Weihe E, Krasteva-Christ G, Grau V, del Rey A, Kummer W. Cholinergic epithelial cell with chemosensory traits in murine thymic medulla. Cell Tissue Res. 2014. https://doi.org/10.1007/s00441-014-2002-x.
Funding
Open access funding provided by Okayama University. This study is funded by the JSPS KAKENHI grants JP21K19601 (R.Y.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
No.
Competing Interests
The author declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoshida, R. Sensory Systems for Sugar-Induced Cephalic Phase Insulin Release. Curr Oral Health Rep 10, 117–123 (2023). https://doi.org/10.1007/s40496-023-00347-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40496-023-00347-y