
Abstract
Purpose of Review
To establish a link or causation between periodontitis and Alzheimer’s disease requires studies that first establish an association or correlation between these two diseases, followed by in vitro, animal model, and human studies to identify possible underlying biological mechanisms, and finally assessing the benefits of periodontal therapy in general and targeted therapies against the microbiota and inflammatory responses in periodontitis. This review presents an update on the current correlation and biological mechanisms that link these two diseases, with special emphasis on the keystone periodontal pathogen Porphyromonas gingivalis and its key family of gingipain enzymes.
Recent Findings
Recent evidence for slowing the progression of Alzheimer’s disease through periodontal therapy in general, as well as focused therapies directed against Porphyromonas gingivalis and its gingipains, are presented.
Summary
These intervention studies, together with the recent association and biological mechanism studies, strengthen the evidence for a direct link or causation between these two diseases. In addition, these recent studies support the special role of the dental practitioner in the management of patients with cognitive decline.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: Setting the Stage
The title of this review, “Reality or Yet Another Association” between Alzheimer’s disease and periodontitis, harkens to a common phrase used when determining a link between two diseases or conditions: causation vs. correlation. Over the past several decades, the question of causation vs. correlation, namely whether the chronic and acute microbial dysbiosis and inflammation that are the hallmarks of periodontal disease can have a direct effect on the initiation and/or progression of systemic diseases, has been the subject of extensive basic, translational, and clinical research [1]. For the spectrum of systemic diseases such as cardiovascular diseases, diabetes, pregnancy outcomes, respiratory diseases, renal diseases, rheumatoid arthritis, etc., the establishment of a direct causation has presented a challenge for this field, now commonly called periodontal medicine. To “close the deal” on determining direct causation rather than just correlation involves what we may distill first from association studies, second in discovering underlying biological mechanisms, and third in assessing the direct effects of treating the local dysbiosis and inflammation for periodontal disease on preventing the initiation, slowing the progression, and/or improving the clinical hallmarks of these systemic conditions.
There is a large and compelling body of published scientific evidence demonstrating correlations/associations between inflammatory periodontal disease and these systemic diseases described above, as well as the underlying effects of this disease on critical clinical risk factors and biological markers for these diseases. However, with very few exceptions, well-designed intervention studies to evaluate the benefits of periodontal treatment on the true clinical endpoints of these diseases have been inconclusive. These intervention studies have focused on the reduction or elimination of the entire dysbiotic biofilm and inflammatory response of periodontal disease or targeting groups of periodontopathic flora and/or products of the destructive inflammatory process. This inability to determine causation may be due in part to the inability to achieve a significant reduction in inflammation and microbial load in these patient populations that are more resistant to the resolution of periodontal inflammation [2]. These patients include those with persistent inflammation after treatment, such as those with poor glycemic control and pregnant patients. In addition, these patients include those with cognitive and/or motor impairment who cannot perform adequate daily plaque control. In addition, in some diseases, a large number of subjects and long observation periods required to assess true clinical endpoints of medical outcomes such as stroke and myocardial infarction also present barriers to determining actual causation.
For the patient with Alzheimer’s disease or other cognitive or motor impairments, this third criterion of assessing the potential benefits of periodontal treatment presents a unique challenge. Thus, from a large majority of cross-sectional and longitudinal association studies, the “chicken or egg first” argument can often be made. Namely, is there just a correlation between Alzheimer’s disease and periodontal disease only due to the inability of the Alzheimer’s patient to effectively remove a dysbiotic biofilm with a daily plaque control regimen [3]? Or, does the translocation of the whole pathogenic microbiota, fragments of the microbiota, and or products of the pathogenic microbiota into the brain actually play a key role in the initiation and progression of Alzheimer’s disease? Or is there mutual causation between these two diseases?
While a full discussion of the biological and clinical hallmarks of Alzheimer’s disease would require a more extensive review, understanding possible causation between periodontal disease and Alzheimer’s disease requires an understanding of the classic pathological markers of Alzheimer’s disease, which include the accumulation of amyloid plaque deposits in the brain, a general increase of inflammation in the brain modulated in large part by the microglial cells, and the fragmentation of tau proteins into microfibrillar tangles. And in the past several years, there have been several published expert opinion articles that stress the roles of all three of these phenomena. In addition, factors that have been taken into consideration for the initiation and progression of Alzheimer’s disease include microvascular damage [4], lifestyle, and genetics. All of these events can lead to the more overt clinical signs of loss of neurons, loss of synaptic connections between neurons, reduction in the size of the brain, and loss of cognitive and motor function.
Several investigators in the periodontal disease field as well as the Alzheimer’s disease field, have raised the question of direct causation between the periodontal disease microbiota, as well as changes in the gut microbiota to Alzheimer’s disease [5]. Until very recently, the amyloid-beta hypothesis as the principal driver for Alzheimer’s disease has dominated this research field, while other hypotheses were relegated to secondary areas of research. However, with the need to identify further upstream events that can be targets of therapy and/or prevention, the microbiome/infection hypothesis [6, 7] and the inflammation/host response hypothesis have received more attention as additional events and risk factors for the initiation and progression of Alzheimer’s disease [6,7,8,9] (Fig. 1). And in the past several years, several published expert opinion articles have stressed interactions between all three of these schools of thought. For the patient with periodontal disease, perhaps the most important consideration to a brain linkage is the invasion of various pathogenic flora, including bacteria, viruses, and fungi [8,9,10,11]. Such evidence has raised the critical question as to whether these current approaches are treating the disease at a stage where there is irreversible damage to the structure and function of the brain. Thus, the investigation of events that are earlier or further upstream from the classic markers of amyloid-beta plaque accumulation and tau tangles that appear in clinical Alzheimer’s disease is urgently needed. Restating the central problem of resolving the issue of correlation vs. causation, using a modification of the Bradford-Hill criteria for causation [12], these investigations must include both the correlation/association studies between specific microbial agents and the general microbiota, understanding the biological plausibility of the effects of these classical microbiotas of periodontal disease on markers of Alzheimer’s disease, and most importantly, the effects of targeted therapy on these microbiotas on the incidence and progression of Alzheimer’s disease.

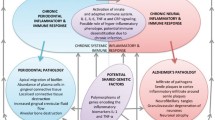
A model for understanding the pathogenesis of Alzheimer’s disease. As shown on the left, histological and pathological features of Alzheimer’s disease within the brain include the accumulation of amyloid plaques, tau fragmentation and tangle formation, and activation of the local inflammatory response through the supporting microglial cells. As shown on the right, an understanding of the pathogenesis of Alzheimer’s disease requires an understanding of upstream events and predisposing factors. Among these is the role of translocation of the microbiome and its products from other diseases and conditions into the brain that can trigger these events within the brain, leading to the overt clinical signs of Alzheimer’s disease (original image created by the author for this review with Biorender.com)
Periodontitis and Alzheimer’s Disease: Moving from Correlation to Biological Plausibility
The first series of questions one may pose regarding direct causation between periodontal diseases and Alzheimer’s disease revolve around correlation/association. Namely, is there a correlation between periodontal disease and Alzheimer’s disease, and is the severity of Alzheimer’s disease correlated with the severity of periodontal disease? Over the past several decades, there has been a large body of both cross-sectional and longitudinal studies that have supported this correlation. In particular, most recent larger-scale cross-sectional or longitudinal studies which examined the correlation of the presence and severity of periodontal diseases have strengthened the evidence for these correlations [13,14,15,16,17,18,19,20,21,22], although some recent studies have not shown definitive correlations between these two diseases [23,24,25]. As an extension of correlation studies between periodontal disease and Alzheimer’s disease, using special imaging techniques to visualize amyloid-beta deposits in the brain, higher levels were observed in patients with clinical attachment loss from periodontal disease, particularly in those patients who also carried the APOE 4 genotype, the most common genetic marker for Alzheimer’s disease risk [26•]. In addition, in patients with early stages of Alzheimer’s disease, changes in serum amyloid-beta levels have been reported to be positively associated with clinical periodontal disease [27].
While a full review of these studies would require a separate manuscript, the reader can refer to a series of excellent and comprehensive reviews regarding this question as well as descriptions of the essential features of these two diseases [9, 28,29,30]. Yet as with all correlation studies between periodontal disease and systemic diseases, the central “chicken and egg” question (which came first) persists until confirmed by an understanding of biological plausibility and effects of periodontal treatment.
Moving from correlation to causation involves moving beyond clinical correlations between periodontitis and Alzheimer’s disease to studies that help establish biological plausibility by examining underlying pathological markers for Alzheimer’s disease. These include studies on correlations between these markers and the presence and levels of periodontopathic microflora intraorally and systemically and the immune response to these bacteria. Pathological markers for the presence and progression of Alzheimer’s disease include levels of amyloid plaque in the brain, the presence of tau tangles, levels of inflammatory markers in the brain, etc. For human studies, these measures can be made by sampling the cerebrospinal fluid, applying newer imaging techniques in the brain, or examining postmortem specimens. These studies can be supplemented by in vitro studies on cultured neurons and in vivo in animal models. Several wild-types and genetically altered mouse models have been studied to determine biological plausibility.
For the patient with periodontal disease, perhaps the most important consideration to a brain linkage is the translocation of various pathogenic flora and associated fragments, including bacteria, viruses, and fungi [8, 10, 31]. Numerous publications have demonstrated that bacteria from the oral cavity but also other sources such as the gastrointestinal tract can translocate to the brain [31], including Borrelia burgdorfei ( associated with Lyme’s disease), Heliobacter pylori [32], and periodontopathic bacteria including Porphyromonas gingivalis (P. gingivalis), Treponema denticola, Aggregatibacter actinomycetemcomitans, Tanerella forsythia, Prevotella intermedia, and Fusobacterium nucleatum [32,33,34,35,36,37]. Dysbiosis of the oral microflora refers to the presence of and/or the elevation of the proportion of these periodontopathic bacteria in the oral cavity. This oral dysbiosis has also been associated with markers of Alzheimer’s disease [33].
Of these bacteria from both oral and nonoral sources, Porphyromonas gingivalis (P. gingivalis) has been the most extensively studied due to its multiple potential roles in both local and systemic inflammation, suppression of the host response, and tissue destruction [34]. When comparing patients who currently have or later develop Alzheimer’s disease, correlations have been reported between levels of circulating antibodies to periodontal pathogens, particularly Porphyromonas gingivalis [ 35,36,37]. Serum antibody titers to P. gingivalis have been reported to be associated with higher levels of both amyloid-beta and T-tau protein in the cerebrospinal fluid [38]. Using markers such as amyloid beta 42/40 ratios and phosphorylated tau protein in cerebrospinal fluid as surrogate markers of Alzheimer’s disease in the brain, higher levels of P. gingivalis DNA in the CSF and in saliva correlate with Alzheimer’s disease [39•, 40, 41]. Correlation studies on post-mortem brain biopsies in animal models and humans have shown that among the periodontal pathogens listed above, there is a preferential invasion of P. gingivalis into areas of the brain responsible for memory, including the hippocampus and cingulate gyrus [42, 43]. This may be partly due to the preferential migration of P. gingivalis to the higher concentrations of iron in the brain, an essential nutrient for P. gingivalis [44].
The relative ease in which P. gingivalis or products of P. gingivalis may invade the brain may be facilitated by the potential of this bacteria to directly invade endothelial cells and secrete a series of proteases called gingipains that can break down the tight junctions of the blood–brain barrier [45] and suppress the local protective host response [46]. In addition, P. Gingivalis can survive in the epithelium [47] and circulating cells of cellular immunity [48], which can then invade the brain as a “Trojan Horse” through the glymphatic system. Translocation of bacteria such as P. gingivalis from the mouth to the brain can lead to local destructive inflammatory reactions as part of the pathogenesis of Alzheimer’s disease [43, 49] and other systemic diseases [50]. Examples include an increase in the secretion of inflammatory mediators from microglial cells, the brain’s principal inflammatory cells [51, 52]. Other autoimmune and destructive inflammatory roles for P. gingivalis include citrullination of host proteins that can set up an autoimmune response [53, 54] and an elevated oxidative stress response that occludes and damage the microvasculature to the brain, leading to microstrokes [4]. In addition, mouse studies have demonstrated that P. gingivialis can impair the clearance rate of amyloid-beta [54] by altering the sleep patterns necessary for the daily clearance of amyloid beta by the glymphatic system [55, 56]. More recent mouse studies on impairment of cognitive function have demonstrated differences in the magnitude of impairment between the four major P. gingivalis serotypes [57•]. In addition, for an older hypothesis for Alzheimer’s disease progression known as the cholinergic hypothesis, P. gingivalis has been implicated in the dysfunction of acetylcholine transmission [58]. A summary of possible effects of mechanisms of P. gingivalis on the pathogenesis of Alzheimer’s disease is summarized in Fig. 2.

Evidence for Porphyromonas gingivalis (P. gingivalis) as a key upstream event in the pathogenesis of Alzheimer’s disease. Evidence from in vitro, animal model, and human studies support the role of P. gingivalis from the periodontal biofilm as an upstream event in the initiation and progression of Alzheimer’s disease: 1. In periodontal inflammation, P. gingivalis, its outer membrane vesicles (OMVs) and key toxins and enzymes can translocate from the plaque biofilm, into the microcirculation of the periodontal tissues, and to the microcirculation in the brain. 2. At the blood–brain barrier in the brain, P. gingivalis, its OMV’s, and in particular its gingipain enzymes, can directly break through this barrier and help promote the formation of amyloid plaques, activate the destructive inflammatory response from microglia, and fragment tau protein and induce tau tangle formation. Gingipains can also cleave apolipoproteins (APOE), which in turn can impair the clearance of amyloid from the brain. 3. P. gingivalis and its gingipains can both directly and indirectly result in cleavage of synapses, death of neurons, and atrophy of areas of the brain. 4. All of these events then lead to the overt clinical signs of Alzheimer’s disease (original image created by the author for this review with Biorender.com)
But whether the entire P. gingivalis bacteria invades the brain may not be as critical as two classes of molecules that have been extensively studied that P. gingivalis employs to evade and neutralize the host response as well as break down healthy brain tissue to access the amino acid nutrients needed for its survival. These are the membrane lipopolysaccharides and the gingipain proteolytic enzymes. For example, the membrane lipopolysaccharides of P. gingivalis per se have been shown to initiate and promote the hallmark pathological and behavioral signs of Alzheimer’s disease in animal models [59, 60].
Recently several investigators have proposed that rather than the translocation of the entire P. gingivalis bacteria in the brain as a driver of Alzheimer’s disease, the smaller outer membrane vesicles (often termed “microbullets”) secreted by P. gingivalis as decoys for the host response and which carry gingipains, lipopolysaccharides, and toxic fimbriae may directly invade the brain tissue [34, 61, 62]. The outer membrane vesicles have been shown in cell culture and rodent studies to increase amyloid-beta plaque levels, reduce clearing amyloid in the brain, and increase microglia activation with the elevated secretion of IL-1 beta [61].
While the role of membrane lipopolysaccharides may be one weapon from P. gingivalis that leads to the initiation and progression of Alzheimer’s disease, a second and more extensively studied area of P. gingivalis pathogenesis are the proteolytic gingipain family of enzymes. These enzymes come in three forms depending on their preferred proteolytic cleavage sites; one lysine gingipain (Kgp) and two arginine gingipains (Rgp1 and 2). Their primary roles are to provide metabolites for the survival of P. gingivalis as well as neutralize and evade the protective host response [62]. As an upstream event in the pathogenesis of Alzheimer’s disease, there is an extensive body of work that demonstrates the biological plausibility of the effects of gingipains on a full range of the “microbial,” “inflammation,” “amyloid beta,” and “tau tangle” schools of thought for the initiation and progression of Alzheimer’s disease and reduction in cognitive function [63].
Starting with evidence for a correlation between gingipains and Alzheimer’s disease in humans, post-mortem studies have shown higher levels of Kgp and Rgp in post-mortem brains with Alzheimer’s disease compared to brains from patients without cognitive decline [39•] and are correlated to the presence of tau tangles in the brain [39•]. Moving on to biological plausibility, there are several published studies using neuron cultures incubated with whole P. gingivalis or gingipains, and in vivo studies from mouse models using oral inoculation of whole P. gingivalis or gingipains in wild-type and genetically susceptible mice [39•, 51, 52, 60, 64, 65]. These studies have reported increases in amyloid beta, tau protein tangles, microglia activation and proliferation, activation of the enzymes that cleave amyloid precursor protein, increases in inflammatory mediators in the brain including IL-1 beta, TNF-alpha, IL-6, degeneration of neurons, neurodegeneration, and loss of synaptic connections [51, 52, 64]. In addition, several of these mice inoculation studies, as well as a study on neuron cultures, have demonstrated the localization and internalization of these gingipains both within neurons and within amyloid beta plaques. In addition, gingipains have been demonstrated to induce the phosphorylation and cleavage of tau proteins, disrupt the neuron cytoskeleton, and promote the loss of synapses [65]. As noted previously, the most well-characterized risk genetic factor for the development of Alzheimer’s disease is the presence of an apolipoprotein 4 genetic variant which has an impaired ability to clear amyloid-beta. Recent evidence has demonstrated that the genetic APOE4 variant is more susceptible to gingipain cleavage, leading to further impairment of transport of amyloid beta (abstract available online at JPAD 6 S24-25: 2019). These observations support a potential interplay between the microbiome and genetic susceptibility to Alzheimer’s disease.
While most research on the role of the periodontal microflora in Alzheimer’s disease has centered on P. gingivalis, several investigators have turned their attention to other bacteria associated with the red complex of periodontal pathogenic flora, in particular for the role of T. denticola in cell culture and animal models. These studies have found similar effects on amyloid-beta production and tau phosphorylation and cleavage [66]. For the family of herpes viruses associated with periodontal disease, several animal models and brain organoid studies have demonstrated the effects of these viruses on pathological hallmarks of Alzheimer’s disease, including activation of microglia, amyloid-beta formation, upregulation of inflammation, and decreases in cognitive function [67,68,69]. In addition, similar effects of viral exposure in newly developed brain organoids [70] have demonstrated increases in beta-amyloid plaque deposition and microglial activation with an expression of destructive inflammatory mediators to neurons and synapses [71]. The potential interplay between herpes viruses and P. gingivalis and its role in Alzheimer’s disease pathology is supported by studies that show the reactivation of herpes viruses, as well as HIV, by exposure of virus-infected cells to P. gingivalis in vitro [72, 73].
Moving from Biological Plausibility to Causation: The effects of General and Targeted Periodontal Therapy on Alzheimer’s Disease
Up to this point, this review has focused on the large body of evidence that supports a correlation/association of periodontal diseases to Alzheimer’s disease. It supports several biological mechanisms for possible causation from cell culture studies, mouse studies, and, where possible, human studies. Nevertheless, to establish actual causation, there is a need to examine the effects of periodontal treatment in general on Alzheimer’s disease initiation or progression or more focussed therapies against specific periodontal pathogens and/or products of the inflammatory and immune host response. General approaches to the treatment of periodontal disease include mechanical debridement by the practitioner, treatment with local and/or systemic antimicrobials where indicated, and plaque control instruction, monitoring, and reinforcement. Several studies have reported the beneficial effects of periodontal treatment in the prevention of dementia [20] or improvements in cognitive function [74, 75]. Such studies support the importance of periodontal treatment as part of a strategy for reducing the risk for Alzheimer’s disease and support a causation role [30, 74].
While these limited number of studies on the benefits of periodontal treatment support a causation role of periodontitis in Alzheimer’s disease, they do not address the central issue of the mutual interactive relationship between reduced cognitive and motor function in Alzheimer’s disease to poorer plaque control and the reemergence of more pathogenic flora. To definitively establish direct causation from periodontitis to Alzheimer’s disease, long or short-term systemic antimicrobial approaches to eliminate pathogens such as P. gingivalis, other periodontal pathogens, viruses, etc., would significantly strengthen this causation hypothesis. For example, the hypothesis that herpes viruses are a key upstream event in the development or progression of Alzheimer’s is indirectly supported by observational studies of patients who frequently take antiherpetic antivirals such as acyclovir and show a lower prevalence and severity of cognitive decline [67, 76].
With the large body of evidence supporting a biological basis for P. gingivalis and its gingipain enzymes and lipopolysaccharides that can translocate from the mouth to the brain, it is understandable that several lines of research have been proposed to test the role of targeted therapies against these two substances. For example, since cathepsin B is essential for P. gingivalis lipopolysaccharide mediated destructive inflammatory pathways, the development of cathepsin B inhibitors may be one approach in treating Alzheimer’s disease, as supported by one animal model study [77].
While the proposed development of cathepsin B inhibitors may be one effective targeted therapeutic approach for inhibiting the potential upstream effects of P. gingivalis in Alzheimer’s disease, the development of specific gingipain inhibitors has received considerable attention. Several strategies and assessments to target P. gingivalis gingipains through either natural molecules, nutrients, or engineered molecules have been published, including one previously developed synthetic inhibitor [78].
Recently, in silico techniques have been used to design a family of small molecule inhibitors for the family of gingipains. These compounds have been shown in neuron cell cultures and in wild-type mice infected orally with gingipains to significantly reduce neural toxicity, the release of destructive inflammatory mediators, and the formation of amyloid plaques and tau tangles [39•]. The development of one candidate lysine gingipain inhibitor (COR388-Atuzuginstat) has been taken through conception, drug design, animal testing, and FDA Phase 1 and Phase 2/3 testing through the multicenter GAIN Trial (ClinicalTrials.gov identifier: NCT03823404). The results from this study were reported in oral presentations at the ClinicalTrials in Alzheimer’s Disease conference in Philadelphia, Pennsylvania in 2021, the Alzheimer’s disease/Parkinson’s disease conference in Barcelona, Spain in 2022, the American Association for Dental Research meeting in Atlanta in 2022 (presentation available at 2022 AADOCR/CADR Annual Meeting Recordings: H. Hasturk: Periodontal Disease and Neurodegenerative Diseases), and the 4th international conference on P. gingivalis in Louisville, Kentucky in 2022 (abstract available at the website for this conference: session V: 2 of 5), with manuscripts in preparation.
In brief, this 48-week study enrolled 643 subjects at over 90 centers in the USA and Europe with mild to moderate impairment as the primary inclusion criteria. It allotted subjects 1:1:1 to a daily orally administer 80 mg of COR 388, 40 mg of COR 388, or a placebo pill. In addition to various measures of cognition, several biomarkers and other clinical assessments were performed throughout the study. In addition, and of particular relevance to this review, subgroup analyses were performed for those subjects in the entire study population with detectable levels of P. gingivalis in saliva and subjects with higher antibody titers to P. gingivalis in serum and CSF (while the presence and severity of periodontal disease were not inclusion criteria for this FDA phase 2/3 study, periodontal examinations and supra and subgingival plaque collections were performed on a subgroup of patients in centers with access to appropriate personnel for post hoc analysis).
While the analysis for the total study population of 643 subjects showed no significant trends in the rate of cognitive decline with COR388-Atuzaginstat vs. placebo as measured by the gold standard ADAS-Cog11 test, for the subgroup of patients with detectable P. Gingivalis at baseline, there was a statistically significant slowing of cognitive decline at 48 weeks (unpublished data). In addition, for subjects with higher antibody titers to P. gingivalis in serum or cerebrospinal fluid, there were non-significant trends in the slowing of cognitive decline (unpublished data). In addition, in further support of P. gingivalis as an upstream event in Alzheimer’s disease, for the total study population, there were either statistically significant correlations or trends between reduction in P. gingivalis levels in saliva and slowing of clinical decline using four standard measures of cognitive function (unpublished data).
These results demonstrated proof of concept for the potential role of P. gingivalis in particular, as well as oral dysbiosis in general, as key upstream events in the initiation and progression of Alzheimer’s disease in a large subgroup of this study with mild to moderate cognitive impairment. However, a safety issue emerged for this COR 388-Atuzuginstat compound regarding the percentage of subjects that demonstrated initial transient elevations in their liver enzyme levels. For this reason, at the time of this writing, the COR 388-Atuzuginstat formulation has been put on full clinical hold by the Division of Neurology 1 (DN1) of the FDA. In response to this safety issue, phase 1 human studies for a second candidate lysine gingipain inhibitor (COR 588) have been recently completed (ClinicalTrials.gov identifier: NCT04920903). In these randomized, double-blinded, placebo-controlled studies, the candidate COR 588 molecule demonstrated effective penetration into the central nervous system and inhibition of lysine gingipain. In addition, with regards to the safety profile for this COR 588 gingipain inhibitor, there were no significant adverse events with regard to both clinical and laboratory measures (unpublished data).
Lessons Learned and Future Directions
For the question posed in the title of this review, it is evident that pursuing the role of translocation of P. gingivalis and its toxic products to the brain may be one of the key upstream events in a significant proportion of the Alzheimer’s susceptible population that have detectable levels of this bacteria in saliva and/or an elevated immune response to this bacterium. Focusing on this pathogen is justified when considering that most patients with moderate to severe periodontal disease carry P. gingivalis in their subgingival plaque. In addition, P. gingivalis may be a keystone pathogen in developing a more general dysbiosis of the plaque biofilm [79]. With these concepts in mind, the dental practitioner may have a pivotal role in identifying periodontal disease and removing as much of the biofilm through mechanical means and local and systemic antimicrobials. However, there is the probability of reestablishment of a P. Gingivalis has driven dysbiotic biofilm between dental maintenance visits where there is inaccessibility of P. gingivalis to mechanical debridement by the dental practitioner and due to the reduced motor and cognitive skills of the patient with Alzheimer’s disease to remove all accessible biofilms on a daily basis. Therefore, directed therapies such as systemic administration of gingipain inhibitors may be one of several approaches to address the upstream events and risks of Alzheimer’s disease before the clinical appearance of declines in cognitive and motor function. Such targeted long-term approaches to P. gingivalis and periodontal treatment, in general, can be considered part of a personalized medicine/personalized dentistry approach that would combine targeted approaches to bacteria with drugs to reduce amyloid, reduce the inflammatory response in the mouth and the brain, lifestyle modifications, and assessment and modification of other risk factors.
The story continues.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Tuganbaev T, Yoshida K, Honda K. The effects of oral microbiota on health. Science. 2022;376(6596):934–6. https://doi.org/10.1126/science.abn1890.
Armitage GC. Effect of periodontal therapy on general health–is there a missing component in the design of these clinical trials? J Clin Periodontol. 2008;35(12):1011–2. https://doi.org/10.1111/j.1600-051X.2008.01327.x.
Ma KS, Hasturk H, Carreras I, Dedeoglu A, Veeravalli JJ, Huang JY, et al. Dementia and the risk of periodontitis: a population-based cohort study. J Dent Res. 2022;101(3):270–7. https://doi.org/10.1177/00220345211037220.
Rokad F, Moseley R, Hardy RS, Chukkapalli S, Crean S, Kesavalu L, et al. Cerebral oxidative stress and microvasculature defects in TNF-alpha expressing transgenic and porphyromonas gingivalis-infected ApoE-/- mice. J Alzheimers Dis. 2017;60(2):359–69. https://doi.org/10.3233/JAD-170304.
Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18(6):794–9. https://doi.org/10.1038/nn.4017.
Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):979–84. https://doi.org/10.3233/JAD-160152.
Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol. 2020;16(4):193–7. https://doi.org/10.1038/s41582-020-0323-9.
Pisa D, Alonso R, Rabano A, Rodal I, Carrasco L. Different brain regions are infected with fungi in Alzheimer’s disease. Sci Rep. 2015;5:15015. https://doi.org/10.1038/srep15015.
Kamer AR, Craig RG, Niederman R, Fortea J, de Leon MJ. 2020 Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol. 2000;83(1):242–71. https://doi.org/10.1111/prd.12327.
Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis. 2008;13(4):437–49. https://doi.org/10.3233/jad-2008-13408.
Watts JC, Prusiner SB. Beta-amyloid prions and the pathobiology of Alzheimer's disease. Cold Spring Harb Perspect Med. 2018;8(5). https://doi.org/10.1101/cshperspect.a023507.
Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295–300.
Kaye EK, Valencia A, Baba N, Spiro A 3rd, Dietrich T, Garcia RI. Tooth loss and periodontal disease predict poor cognitive function in older men. J Am Geriatr Soc. 2010;58(4):713–8. https://doi.org/10.1111/j.1532-5415.2010.02788.x.
Stewart R, Weyant RJ, Garcia ME, Harris T, Launer LJ, Satterfield S, et al. Adverse oral health and cognitive decline: the health, aging and body composition study. J Am Geriatr Soc. 2013;61(2):177–84. https://doi.org/10.1111/jgs.12094.
Iwasaki M, Kimura Y, Ogawa H, Yamaga T, Ansai T, Wada T, et al. Periodontitis, periodontal inflammation, and mild cognitive impairment: a 5-year cohort study. J Periodontal Res. 2019;54(3):233–40. https://doi.org/10.1111/jre.12623.
Iwasaki M, Yoshihara A, Kimura Y, Sato M, Wada T, Sakamoto R, et al. Longitudinal relationship of severe periodontitis with cognitive decline in older Japanese. J Periodontal Res. 2016;51(5):681–8. https://doi.org/10.1111/jre.12348.
Shin HS, Shin MS, Ahn YB, Choi BY, Nam JH, Kim HD. Periodontitis is associated with cognitive impairment in elderly Koreans: results from the Yangpyeong cohort study. J Am Geriatr Soc. 2016;64(1):162–7. https://doi.org/10.1111/jgs.13781.
Chen CK, Wu YT, Chang YC. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimers Res Ther. 2017;9(1):56. https://doi.org/10.1186/s13195-017-0282-6.
Tzeng NS, Chung CH, Yeh CB, Huang RY, Yuh DY, Huang SY, et al. Are chronic periodontitis and gingivitis associated with dementia? A nationwide, retrospective, matched-cohort study in Taiwan. Neuroepidemiology. 2016;47(2):82–93. https://doi.org/10.1159/000449166.
Lee YT, Lee HC, Hu CJ, Huang LK, Chao SP, Lin CP, et al. Periodontitis as a modifiable risk factor for dementia: a nationwide population-based cohort study. J Am Geriatr Soc. 2017;65(2):301–5. https://doi.org/10.1111/jgs.14449.
Ide M, Harris M, Stevens A, Sussams R, Hopkins V, Culliford D, et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS ONE. 2016;11(3):e0151081. https://doi.org/10.1371/journal.pone.0151081.
Choi S, Kim K, Chang J, Kim SM, Kim SJ, Cho HJ, et al. Association of chronic periodontitis on Alzheimer’s disease or vascular dementia. J Am Geriatr Soc. 2019;67(6):1234–9. https://doi.org/10.1111/jgs.15828.
Arrive E, Letenneur L, Matharan F, Laporte C, Helmer C, Barberger-Gateau P, et al. Oral health condition of French elderly and risk of dementia: a longitudinal cohort study. Community Dent Oral Epidemiol. 2012;40(3):230–8. https://doi.org/10.1111/j.1600-0528.2011.00650.x.
Okamoto N, Morikawa M, Tomioka K, Yanagi M, Amano N, Kurumatani N. Association between tooth loss and the development of mild memory impairment in the elderly: the Fujiwara-kyo study. J Alzheimers Dis. 2015;44(3):777–86. https://doi.org/10.3233/JAD-141665.
Naorungroj S, Schoenbach VJ, Wruck L, Mosley TH, Gottesman RF, Alonso A, et al. Tooth loss, periodontal disease, and cognitive decline in the atherosclerosis risk in communities (ARIC) study. Community Dent Oral Epidemiol. 2015;43(1):47–57. https://doi.org/10.1111/cdoe.12128.
• Kamer AR, Pirraglia E, Tsui W, Rusinek H, Vallabhajosula S, Mosconi L, et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging. 2015;36(2):627–33. https://doi.org/10.1016/j.neurobiolaging.2014.10.038. Using live imaging techniques on human subjects for amyoid beta plaque deposition in the brain, this is a key study that demonstrates the additive effects of periodontal disease and genetic susceptibility with the APOE4 variant to amyloid beta deposition.
Gil-Montoya JA, Barrios R, Santana S, Sanchez-Lara I, Pardo CC, Fornieles-Rubio F, et al. Association between periodontitis and amyloid beta peptide in elderly people with and without cognitive impairment. J Periodontol. 2017;88(10):1051–8. https://doi.org/10.1902/jop.2017.170071.
Thomson WM, Barak Y. Tooth Loss and Dementia: A Critical Examination. J Dent Res. 2020:22034520957233. https://doi.org/10.1177/0022034520957233.
Jungbauer G, Stahli A, Zhu X, Auber Alberi L, Sculean A, Eick S. 2022 Periodontal microorganisms and Alzheimer disease - a causative relationship? Periodontol. 2000;89(1):59–82. https://doi.org/10.1111/prd.12429.
Harding A, Kanagasingam S, Welbury R, Singhrao SK. Periodontitis as a risk factor for Alzheimer’s disease: the experimental journey so far, with hope of therapy. Adv Exp Med Biol. 2022;1373:241–60. https://doi.org/10.1007/978-3-030-96881-6_13.
Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol. 2015;7:29143. https://doi.org/10.3402/jom.v7.29143.
Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17(2):113–8. https://doi.org/10.1046/j.0902-0055.2001.00100.x.
Kamer AR, Pushalkar S, Gulivindala D, Butler T, Li Y, Annam KRC, et al. Periodontal dysbiosis associates with reduced CSF Abeta42 in cognitively normal elderly. Alzheimers Dement (Amst). 2021;13(1):e12172. https://doi.org/10.1002/dad2.12172.
Singhrao SK, Olsen I. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol. 2019;11(1):1563405. https://doi.org/10.1080/20002297.2018.1563405.
Kamer AR, Craig RG, Pirraglia E, Dasanayake AP, Norman RG, Boylan RJ, et al. TNF-alpha and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Neuroimmunol. 2009;216(1–2):92–7. https://doi.org/10.1016/j.jneuroim.2009.08.013.
Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 2012;8(3):196–203. https://doi.org/10.1016/j.jalz.2011.04.006.
Noble JM, Scarmeas N, Celenti RS, Elkind MS, Wright CB, Schupf N, et al. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS ONE. 2014;9(12):e114959. https://doi.org/10.1371/journal.pone.0114959.
Laugisch O, Johnen A, Maldonado A, Ehmke B, Burgin W, Olsen I, et al. Periodontal pathogens and associated intrathecal antibodies in early stages of Alzheimer’s disease. J Alzheimers Dis. 2018;66(1):105–14. https://doi.org/10.3233/JAD-180620.
• Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. https://doi.org/10.1126/sciadv.aau3333. To date, the most comprehensive examination of the effects of P. gingivalis and its gingipain enzymes as key upstream events in the pathogeneis of Alzheimers Disease, and the potential therapeutic benefits of small molecule gingipain inhibitors.
Noble JM, Borrell LN, Papapanou PN, Elkind MS, Scarmeas N, Wright CB. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. J Neurol Neurosurg Psychiatry. 2009;80(11):1206–11. https://doi.org/10.1136/jnnp.2009.174029.
Merchant AT, Yi F, Vidanapathirana NP, Lohman M, Zhang J, Newman-Norlund RD et al. Antibodies against periodontal microorganisms and cognition in older adults. JDR Clin Trans Res. 2022:23800844211072784. https://doi.org/10.1177/23800844211072784.
Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–77. https://doi.org/10.3233/JAD-121918.
Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing. 2018;15:6. https://doi.org/10.1186/s12979-017-0110-7.
Olsen I. Porphyromonas gingivalis may seek the Alzheimer’s disease brain to acquire iron from its surplus. J Alzheimers Dis Rep. 2021;5(1):79–86. https://doi.org/10.3233/ADR-200272.
Nonaka S, Kadowaki T, Nakanishi H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem Int. 2022;154:105282. https://doi.org/10.1016/j.neuint.2022.105282.
Osorio C, Kanukuntla T, Diaz E, Jafri N, Cummings M, Sfera A. The post-amyloid era in Alzheimer’s disease: trust your gut feeling. Front Aging Neurosci. 2019;11:143. https://doi.org/10.3389/fnagi.2019.00143.
Lee JS, Spooner R, Chowdhury N, Pandey V, Wellslager B, Atanasova KR, et al. In situ intraepithelial localizations of opportunistic pathogens, Porphyromonas gingivalis and Filifactor alocis, in Human Gingiva. Curr Res Microb Sci. 2020;1:7–17. https://doi.org/10.1016/j.crmicr.2020.05.001.
Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas gingivalis infection induces amyloid-beta accumulation in monocytes/macrophages. J Alzheimers Dis. 2019;72(2):479–94. https://doi.org/10.3233/JAD-190298.
Olsen I. Porphyromonas gingivalis-Induced neuroinflammation in Alzheimer’s disease. Front Neurosci. 2021;15:691016. https://doi.org/10.3389/fnins.2021.691016.
Silvestre FJ, Lauritano D, Carinci F, Silvestre-Rangil J, Martinez-Herrera M, Del Olmo A. Neuroinflammation, Alzheimers disease and periodontal disease: is there an association between the two processes? J Biol Regul Homeost Agents. 2017;31(2 Suppl 1):189–96.
Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE. 2018;13(10):e0204941. https://doi.org/10.1371/journal.pone.0204941.
Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis. 2017;3:15. https://doi.org/10.1038/s41514-017-0015-x.
Bartold PM, Lopez-Oliva I. 2020 Periodontitis and rheumatoid arthritis: an update 2012–2017. Periodontol. 2000;83(1):189–212. https://doi.org/10.1111/prd.12300.
Olsen I, Taubman MA, Singhrao SK. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J Oral Microbiol. 2016;8:33029. https://doi.org/10.3402/jom.v8.33029.
Takayama F, Hayashi Y, Wu Z, Liu Y, Nakanishi H. Diurnal dynamic behavior of microglia in response to infected bacteria through the UDP-P2Y6 receptor system. Sci Rep. 2016;6:30006. https://doi.org/10.1038/srep30006.
Nedergaard M, Goldman SA. Glymphatic failure as a final common pathway to dementia. Science. 2020;370(6512):50–6. https://doi.org/10.1126/science.abb8739.
• Diaz-Zuniga J, More J, Melgar-Rodriguez S, Jimenez-Union M, Villalobos-Orchard F, Munoz-Manriquez C, et al. Alzheimer’s disease-like pathology triggered by Porphyromonas gingivalis in wild type rats is serotype dependent. Front Immunol. 2020;11:588036. https://doi.org/10.3389/fimmu.2020.588036. A study that adds to the understanding of the potential key upstream role of P. gingivalis in Alzheimers disease by demonstrating differences in the pathogenic potential between the four major serotypes of P. gingivalis.
Patel S, Howard D, Chowdhury N, Derieux C, Wellslager B, Yilmaz O, et al. Characterization of human genes modulated by Porphyromonas gingivalis highlights the ribosome, hypothalamus, and cholinergic neurons. Front Immunol. 2021;12:646259. https://doi.org/10.3389/fimmu.2021.646259.
Leira Y, Iglesias-Rey R, Gomez-Lado N, Aguiar P, Campos F, D’Aiuto F, et al. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch Oral Biol. 2019;99:120–5. https://doi.org/10.1016/j.archoralbio.2019.01.008.
Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation. 2018;15(1):37. https://doi.org/10.1186/s12974-017-1052-x.
Liu Y, Wu Z, Nakanishi Y, Ni J, Hayashi Y, Takayama F, et al. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep. 2017;7(1):11759. https://doi.org/10.1038/s41598-017-12173-1.
Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett. 2012;333(1):1–9. https://doi.org/10.1111/j.1574-6968.2012.02579.x.
Dioguardi M, Crincoli V, Laino L, Alovisi M, Sovereto D, Mastrangelo F et al. The role of periodontitis and periodontal bacteria in the onset and progression of Alzheimer’s disease: a systematic review. J Clin Med. 2020;9(2). https://doi.org/10.3390/jcm9020495.
Ilievski V, Bhat UG, Suleiman-Ata S, Bauer BA, Toth PT, Olson ST, et al. Oral application of a periodontal pathogen impacts SerpinE1 expression and pancreatic islet architecture in prediabetes. J Periodontal Res. 2017;52(6):1032–41. https://doi.org/10.1111/jre.12474.
Haditsch U, Roth T, Rodriguez L, Hancock S, Cecere T, Nguyen M, et al. Alzheimer’s disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimers Dis. 2020. https://doi.org/10.3233/JAD-200393.
Tang Z, Cheng X, Su X, Wu L, Cai Q, Wu H. Treponema denticola Induces Alzheimer-like tau hyperphosphorylation by activating hippocampal neuroinflammation in mice. J Dent Res. 2022:220345221076772. https://doi.org/10.1177/00220345221076772.
Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: possible mechanisms and signposts. FASEB J. 2017;31(8):3216–26. https://doi.org/10.1096/fj.201700360.
De Chiara G, Piacentini R, Fabiani M, Mastrodonato A, Marcocci ME, Limongi D, et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019;15(3):e1007617. https://doi.org/10.1371/journal.ppat.1007617.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 2018;99(1):56-63 e3. https://doi.org/10.1016/j.neuron.2018.06.030.
Cairns DM, Rouleau N, Parker RN, Walsh KG, Gehrke L, Kaplan DL. A 3D human brain-like tissue model of herpes-induced Alzheimer’s disease. Sci Adv. 2020;6(19):eaay8828. https://doi.org/10.1126/sciadv.aay8828.
Hao X, Li Z, Li W, Katz J, Michalek SM, Barnum SR, et al. Periodontal infection aggravates C1q-mediated microglial activation and synapse pruning in Alzheimer’s mice. Front Immunol. 2022;13:816640. https://doi.org/10.3389/fimmu.2022.816640.
Imai K, Victoriano AF, Ochiai K, Okamoto T. Microbial interaction of periodontopathic bacterium Porphyromonas gingivalis and HIV-possible causal link of periodontal diseases to AIDS progression. Curr HIV Res. 2012;10(3):238–44. https://doi.org/10.2174/157016212800618183.
Chen C, Feng P, Slots J. 2020 Herpesvirus-bacteria synergistic interaction in periodontitis. Periodontol. 2000;82(1):42–64. https://doi.org/10.1111/prd.12311.
Rolim Tde S, Fabri GM, Nitrini R, Anghinah R, Teixeira MJ, Siqueira JT, et al. Evaluation of patients with Alzheimer’s disease before and after dental treatment. Arq Neuropsiquiatr. 2014;72(12):919–24. https://doi.org/10.1590/0004-282X20140140.
Schwahn C, Frenzel S, Holtfreter B, Van der Auwera S, Pink C, Bulow R, et al. Effect of periodontal treatment on preclinical Alzheimer’s disease-results of a trial emulation approach. Alzheimers Dement. 2022;18(1):127–41. https://doi.org/10.1002/alz.12378.
Harris SA, Harris EA. Molecular mechanisms for herpes simplex virus type 1 pathogenesis in Alzheimer’s disease. Front Aging Neurosci. 2018;10:48. https://doi.org/10.3389/fnagi.2018.00048.
Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017;65:350–61. https://doi.org/10.1016/j.bbi.2017.06.002.
Olsen I, Potempa J. Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J Oral Microbiol. 2014;6. https://doi.org/10.3402/jom.v6.24800.
Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–25. https://doi.org/10.1038/nrmicro2873.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The multi-center GAIN Trial protocol and the COR 588 Phase 1 Trial protocol described with results in this review were approved by an institutional review board at each site and were conducted in accordance with Good Clinical Practice guidelines. All of the participants or their legal representatives provided written informed consent. The sponsor (Cortexyme) designed and funded the trial, supplied treatment and placebo, and oversaw the contracted research organization.
Competing Interests
Mark I. Ryder, the author of this review, previously served as a member of the Clinical Advisory Board, holds stock, and receives consultant fees from Cortexyme Inc./Quince Therapeutics.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Clinical Periodontics
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ryder, M.I. The Link Between Periodontitis and Alzheimer’s Disease: Reality or Yet Another Association. Curr Oral Health Rep 9, 157–166 (2022). https://doi.org/10.1007/s40496-022-00319-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40496-022-00319-8