
Abstract
Purpose of Review
This article provides a review of the recent literature related to the FDA-approved drugs that had been repurposed as potential drug candidates against COVID-19. Moreover, we performed a quality pharmacophore study for frequently studied targets, namely, the main protease, RNA-dependent RNA polymerase, and spike protein.
Recent Findings
Ever since the COVID-19 pandemic, the whole spectrum of scientific community is still unable to invent an absolute therapeutic agent for COVID-19. Considering such a fact, drug repurposing strategies seem a truly viable approach to develop novel therapeutic interventions.
Summery
Drug repurposing explores previously approved drugs of known safety and pharmacokinetics profile for possible new effects, reducing the cost, time, and predicting prospective side effects and drug interactions. COVID-19 virulent machinery appeared similar to other viruses, making antiviral agents widely repurposed in pursuit for curative candidates. Our main protease pharmacophoric study revealed multiple features and could be a probable starting point for upcoming research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Towards the end of 2019, a novel beta coronavirus was identified as the causative agent of huge cases of unprecedented pneumonia, reported for the first time in Wuhan city, China. Soon after, the WHO labeled the severe acute respiratory syndrome as COVID-19 and the virus as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1, 2]. Starting on March 2020, the WHO announced that COVID-19 has become a global pandemic [3]. The pandemic affected the whole world, [1, 4], and due to its airborne infectivity, social distancing measures have been imposed by international authorities [5].
As a response to the sudden outbreak of COVID-19, an intensive global research efforts have been devoted to investigate potential therapeutic approaches. Looking at the fast return and the low cost of drug repurposing strategies, it has been implemented as immediate drug discovery pipeline [6•]. Many clinically useful drugs including aspirin and sildenafil which were originally used as inflammation and hypertension have been successfully repurposed for cardiovascular diseases and erectile dysfunction, respectively [7, 8]. Nowadays, molecular docking as an in silico method is very popular in drug discovery researches, because of its ability to illustrate the interactions between the ligand and its biological targets [8, 9]. Pharmacophore modeling — defined as a set of molecular features that enable biologically active ligands to exert a pharmacological effect — practice is widely used in the drug discovery process [10]. Herein, we reviewed and summarized research of FDA-approved drugs repurposed against SARS-CoV-2 virus.
Methods
Workflow
Search Strategy
During the period of COVID-19, the scientific community had published an immense number of studies about repurposing drugs for COVID-19. These studies are scattered on the internet, and no one database will be inclusive enough to all of these studies, aside from Google Scholar. All studies have been collected by the end of 2020/12/30. In order to achieve an efficient search strategy, the following combinations of keywords were applied during the research in the Google Scholar database: (COVID19 OR SARS-CoV-2) AND (Drug repurposing OR Drug repositioning OR Drug re-profiling OR Drug rediscovery) AND (Docking AND Molecular dynamic).
Data Selection and Extraction
Titles and abstracts were first checked for eligibility using specific inclusion and exclusion criteria (Table 1). Selected paper were then completely reviewed as a second stage, while articles that met the exclusion criteria were eliminated.
Primarily, searching revealed a total of 405 articles, after applying the above mentioned criteria above, we ended up with 92 research articles, as presented in the flow diagram (Fig. 1). We further classified them according to SARS-CoV-2 targets: 73 for the main protease, 11 for spike protein, and 8 for replicase complex.

Flow diagram of the screening process of articles included in the study
Pharmacophore Analysis
To perform the pharmacophore study, we had to ensure that all the drugs analyzed bind to the same site on the target. Thus, we classified the drug within the same target according to the binding site. There are various approaches used in literature to define the binding site or the docking site. Nevertheless, we obtained one binding site for each target except the main protease; it gave us two binding sites. Phase from Schrodinger suits was used to generate the pharmacophore hypothesis [11]. As the quality of the pharmacophore was our biggest concern, we used the number of features and the Hyposcore as a measure of quality. The following criteria were implemented: a maximum of 7 features, a minimum of 6 features, and minimum coverage of 50% of the compounds at the question.
Data Description
In silico work on COVID-19, drug discovery started even before the PDB structure of SARS-CoV-2 targets was released; during that time, homology modeling was used to generate the protein structure in question. The generated targets that have been investigated are spike protein, RNA-dependent RNA polymerase (RdRp), main protease, helicase, and papain-like protease. However, from them, we reviewed only the most studied targets (main protease, spike protein, and RdRp) [1, 2].
All papers follow a similar protocol, as shown in Fig. 2a either docking alone or docking and molecular dynamics simulation.

In silico work on COVID-19. a Types and frequencies of the used in silico methods. b Types and frequencies of the used docking software. c Types and frequencies of the used molecular dynamic software
As illustrated in Fig. 2b, the most used software for molecular docking was Autodock vina, followed by Glide and preceded by Autodock 4. The open source feature of Autodock rationalizes its greatest usage frequency. Autodock vina is the most used because it is easier to use compare to Autodock4, and it has have been implemented in many software packages like PyRx. In addition, Autodock vina is faster and more accurate depending on the system and the parameter setting [12].
While docking studies consider the flexibility of the ligand as a rigid structure, molecular dynamic (MD) simulation takes the ligand–protein complex as a dynamic module. It searches the conformational space for the most stable conformation, giving more accurate results. Moreover, due to the cost of running MD simulation, only 48 studies out of 89 confirmed docking results with MD (Fig. 2a). GROMACS and AMBER were the most used free MD software. On the other hand, Desmond was the most used commercial software (Fig. 2c).
Results and Discussion
Main Protease
The fact that no human protease mimics the cleavage activity of the viral main protease; it has been a well-positioned drug target [13••]. The sequence identity shows that 96% of SARS-CoV-2 main protease is identical with other SARS-COV viruses [14]. This high conservation encourages researchers to study inhibitors and other drugs for repurposing them against SARS-COV-2 main protease. Yang’s group was the first group to release the crystal structure of the main protease (PDB ID: 6LU7) [15]. In this paper, we classified the drugs according to the anatomical therapeutic chemical classification system (Table 2).
Anti-infective agents were the most frequent class, with antivirals and tetracycline being the predominant subclasses; thus, they have been considered in the discussion. The following most frequent classes are the antineoplastic and immunomodulating agents (Table 2). However, we did not find any similarity between the original mechanism during our analysis, and they had failed pharmacophore analysis.
Antivirals
Of the 29 antiviral drugs that were reported to bind with the viral main protease, 16 drugs are already protease inhibitors; hence, the hypothesis here is whether they are SARS-COV-2 protease inhibitors too. These drugs are indinavir, ritonavir, lopinavir, nelfinavir, saquinavir, simeprevir, paritaprevir, darunavir, atazanavir, glecaprevir, telaprevir, tipreanavir, brecanavir, grazoprevir, tipranavir, and amprenavir. Furthermore, they work on only two proteases, human immunodeficiency virus type 1 protease and NS3/4A protein. Of these compounds, indinavir was reported eight times with seven different docking software, ritonavir was repeated twelve times with seven different docking software, and finally, lopinavir was repeated eleven times with seven different docking software. All these evidences support their potential as drugs for COVID-19. On the other hand, asunaprevir, ciluprevir, and pleconaril work on “Genome polyprotein,” a complex protein with many functions including protease activity, and they could be considered for further analysis. Lastly, the other ten drugs have diverse targets, and they failed pharmacophore analysis, so we excluded them in pharmacophore analysis.
Tetracyclines
Another class of compounds that peaked our interest is the tetracyclines; we found that five tetracyclines in the literature claim to have inhibitory activities against the main protease. These compounds are oxytetracycline, lymecycline, tetracycline, minocycline, and doxycycline. Tetracyclines are broad-spectrum antibiotics, and they have the general structure of four fused rings. They work by inhibiting protein synthesis; notably, they inhibit the binding of aminoacyl-tRNA to the mRNA translation complex [89].
Pharmacophore Analysis of the Main Protease
Most studies on the main protease have been conducted on the 6LU7 structure, and all of those studies performed their docking analysis on two binding sites. One has cysteine-type endopeptidase activity (CYS145), and the other is a larger protein cavity. It is worth noting that our pharmacophore studies only conducted on the cysteine type, as replicating such analysis has failed on the second one.
The CYS145 was the most studied binding site in 85% of all the studies that used the PDB 6LU7. The number of drugs for this binding site was huge, putting pressure on the phase alignment algorithm. We had to create pharmacophore modules for the predominant classes of drugs, including antivirals, tetracyclines, antineoplastics, immunomodulating, and all combined drugs. As described in the workflow, we used strict parameters, and only tetracyclines and antivirals had survived. For comparison purposes, we included the N3 complexed inhibitor and the binding site hypothesis. We studied quality, type of features, and features alignment for these pharmacophores (Table 3, Fig. 3a).

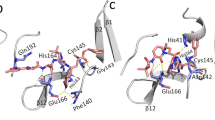
The pharmacophore analysis for targeting SARS-CoV-2 enzymes. a Main protease. b RNA-dependent RNA polymerase
N3 is a co-crystallized inhibitor with 6LU7; its pharmacophore is rich in hydrogen donor features, which is expected because it is a polypeptide, subsequently containing alternating hydrogen bond donor and hydrogen bond acceptor. Tetracycline is also rich in hydrogen donor features, and the antivirals pharmacophore is rich in hydrogen acceptors. The pharmacophore search of antivirals having an anti-protease activity — as its original mechanism of action — was successful with 6 features and a Hyposcore of 1.09 (Table 3). On the other hand, the search for pharmacophores of the other antivirals, where-the original mechanism of action is not anti-protease, had failed due to our strict parameters. These hypotheses contain at least one aromatic ring, and the negative charge feature is only present on the binding site.
Notably, the best scores were obtained with tetracycline pharmacophores; it had seven features and a Hyposcore of 1.35; their structural similarity supported this result (Table 3, Fig. 3a).
Analyzing the table, it seems that tetracycline and N3 inhibitor are the most similar pair of hypotheses, but that is not enough. Pharmacophores have to agree on the location of the features concerning each other, so how do these hypotheses align to each other? They align with at least 3 matches, except N3 inhibitor and tetracycline aligned with 4 matched features. Considering this, we concluded our search for pharmacophores with four hypotheses, and we emphasized the overlapping features of N3 and tetracycline; 2 hydrogen bond donors, 1 hydrogen bond acceptor, and 1 aromatic ring (Fig. 3a).
RNA-Dependent RNA Polymerase
RNA-dependent RNA polymerase (RdRp) is a replicase that operates the synthesis of a complementary RNA strand using RNA [18••, 90]. SARS-CoV-2 RdRp is composed of 932 amino acids forming a framework of SARS-CoV RdRp linked to the Nsp7 and Nsp8 cofactors. Structurally, the RdRp protein is divided into the N-terminal and polymerase domains, which extend from amino acid residues 1 to 397. The polymerase domain is subdivided into three structurally different subunits: the finger, palm, and thumb [18••, 90].
The active site of Nsp12 is situated in the middle of the substrate domain, where the synthesis of RNA takes place when an RNA template is accessed from the template input channel and nucleoside triphosphate (NTP) from NTP input channel [91••]. RdRp has been proposed to be an important target for developing drugs against coronavirus as detailed in Table 4.
Drug Affecting RNA-Dependent RNA Polymerase
In accordance with the inclusion criteria of literature, results showed a handful papers concerning Nsp12. Data reported five antiviral agents targeting the polymerase (remdesivir, tipranavir, tegobuvir, simeprevir, and filibuvir). The second common drugs were binimetinib, palbociclib, lonafarnib, and pegamotecan, which are antineoplastic agents. Other drugs seemed to have miscellaneous pharmacological classes.
Antiviral agents have diverse mechanisms of action and viral targets. Despite this fact, our analysis suggested that antiviral drugs can inhibit the catalytic activity of the Nsp12. Notably, remdesivir is a known nucleoside analog that has been globally well-studied, both in silico and in vitro. Mechanistically, remdesivir is incorporated into the growing viral RNA leading to the termination of the RNA replication. The high affinity of the compound for the active site of RdRp indicates that remdesivir would compete with the natural substrates ribonucleotides and get incorporated into the growing RNA chain. It has been investigated in multiple COVID-19 clinical trials (available at https://clinicaltrials.gov), and granted the FDA Emergency Use Authorization in COVID-19 treatment. While other reported drugs share no similarity in their original clinical use, in silico docking data revealed that definitive interaction with Nsp12 active site is therefore predicted to be a promising inhibitors. Despite the low available data, we performed a pharmacophore analysis based on drugs and the common binding site.
Pharmacophore Analysis of the RNA-Dependent RNA Polymerase
Drug-based analysis was performed on eight FDA-approved drugs of miscellaneous pharmacological classes. The same settings of the main protease pharmacophore were followed, and six-feature pharmacophore was obtained. Among those features, the hydrogen-bond donor was observed only once, while the hydrogen-bond acceptor appeared three times. The remaining two features are reported under the name of the aromatic ring as per Maestro Phase software (Table 3).
The PDB ID 6M71 was the most frequent target observed. The results revealed seven features with a unique feature of negative ionic that was not observed under drug-based hypothesis analysis. Those seven features have 3 hydrogen-bond donors, 1 hydrogen-bond acceptor, and 2 aromatic rings. Comparing the two analyses, they disagree on the hydrogen bond donor and hydrogen bond accepter, but they agree on the aromatic ring, with the unique negative ionic feature reported only for the binding-site based hypothesis study (Table 3, Fig. 3b).
Spike Protein
Spike proteins are a class fusion glycoprotein localized in the surface of SARS-CoV-2. It has a crucial role in viral infection by recognizing the host angiotensin-converting enzyme-2 receptor (ACE2) [98]. It contains two subunits, S1 and S2. The S1 subunit contains a receptor-binding domain (RBD) with a size of 180–200 KD that binds and recognizes the ACE2 receptor, while the S2 subunit mediates viral cell membrane fusion [99••]. The S protein uses the ACE-2 receptor for entry to the host cell. The spike protein is coated with polysaccharide molecules to evade the surveillance of the host immune system during entry; targeting this molecule can potentially decrease the chance of infection owing to the prevention of the viral invasion to the host cells [100]. Much research was conducted computationally to identify the role of repurposed drugs on spike proteins and ACE2 (Table 5).
COVID-19 mechanism of entary is that the spike recognize ACE2 receptor, then the virus can enter the cell; in our review, we classify studies that interrupt this mechanism into three classes: First study that investigate drugs that bind spike protein showed that these drugs such as troxerutin could not be able to recognize ACE2, example of such drug is troxerutin [102••]; second study that investigate drugs that bind ACE2 thus compete with spike protein on ACE2 binding site, example of such a drug is lopinavir [106]; third study that investigate drug binding to the complex of spike and ACE2 (6M0J) and prevent the transition to active state, example of such a drug is polymyxin B [88]. According to the ATC classification system, most of the drugs affecting the spike protein, ACE2, and the complex fall in the anti-infective category (Table 5).
Drug Affecting Spike Protein
As demonstrated in Table 5, thirty-four drugs have been reported to target the spike protein, 6 of them are antivirals (lamivudine, nelfinavir, Dolutegravir, vidarabine, Remdesivir, Daclatasvir), and 3 of them (lamivudine, vidarabine, remdesivir) are nucleoside analogs that originally target the DNA synthesis in viruses with the same exact mechanism of action, and therefore a similar binding mode.
Ivermectin is reported 2 times in the spike docking analysis and is investigated in clinical trials, but its frequency of appearance here does not indicate a potential for being a lead compound, because some in silico trials targeted the ivermectin from the beginning and not as the end result of an exhaustive virtual screening [16].
Drug Affecting the ACE2/Spike Complex
A number of studies investigate the binding of drugs to the ACE2/spike complex after the binding of spike to ACE2 has already been achieved; they seek a drug that can disrupt the interaction of ACE2 with RBD after the binding; 8 drugs were reported targeting ACE2/spike complex, and these drugs are (naltrexone, ivermectin, lopinavir, chloroquine, polymyxin B, colistin, daptomycin, oritavancin). Excluding naltrexone and chloroquine, a common trend in these drugs is that they have a molecular weight above 500 and most of them are above 1000. We run a pharmacophore analysis of these drugs and it had failed due to their complexities and variability.
Two studies targeting the ACE2 receptor only have been investigated. They seek a compound that can prevent the recognition of the spike to ACE2 [101, 103]. Herein, 10 drugs were reported (lividomycin, burixafor, quisinostat, fluprofylline, pemetrexed, spirofylline, edotecarin, diniprofylline, hydroxychloroquine, chloroquine). Obviously, a drug targeting ACE2 would not be much useful, but those studies were originally part of a bigger investigation and concluded that these compounds bind well to many targets, one of which is ACE2 [103].
Pharmacophore Analysis of the Spike Protein
During pharmacophore analysis, we did not find any consistency in the use of a common PDB structure from the paper that studied the spike protein like what we achieved in the main protease. However, we proceeded with choosing only one PDB (the one with the highest resolution) for binding site pharmacophore with the argument that they all have the similar sequence and thus a similar PDB structure. In the end, the pharmacophore analysis failed due to the diversity of the drugs.
Conclusion
Herein, we reviewed the current research concerning drug repurposing using molecular docking and other in silico drug discovery approaches in the battle against COVID-19. Antiviral drugs were the most studied drugs. The main protease represented a potential target looking at its central role in viral replication and huge availability per our standards. Moreover, studies indicated that the best results of pharmacophore investigation were obtained with tetracyclines and antivirals, which were originally protease inhibitors. We recommend using the models in further research, including virtual screening, which could help the overall efforts in finding effective therapeutic agents against COVID-19.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Macchiagodena M, Pagliai M, Procacci P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem Phys Lett. 2020;750: 137489. https://doi.org/10.1016/j.cplett.2020.137489.
Wang L, Wang Y, Ye D, Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents. 2020;55: 105948. https://doi.org/10.1016/j.ijantimicag.2020.105948.
Hanaei S, Rezaei N. COVID-19: Developing from an outbreak to a pandemic. Arch Med Res. 2020;51:582–4. https://doi.org/10.1016/j.arcmed.2020.04.021.
Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. The Lancet. 2020;395:470–3. https://doi.org/10.1016/s0140-6736(20)30185-9.
McBride O, Murphy J, Shevlin M, Gibson‐Miller J, Hartman TK, Hyland P, et al. Monitoring the psychological, social, and economic impact of the COVID‐19 pandemic in the population: context, design and conduct of the longitudinal COVID‐19 psychological research consortium (C19PRC) study. Int J Methods Psychiatr Re. 2021;30. https://doi.org/10.1002/mpr.1861.
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019;18:41–58. https://doi.org/10.1038/nrd.2018.168
Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5:689–702. https://doi.org/10.1038/nrd2030.
Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–88. https://doi.org/10.1016/0022-2836(82)90153-x.
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discovery. 2004;3:935–49. https://doi.org/10.1038/nrd1549.
Sanders MPA, McGuire R, Roumen L, de Esch IJP, de Vlieg J, Klomp JPG, et al. From the protein’s perspective: the benefits and challenges of protein structure-based pharmacophore modeling. MedChemComm. 2012;3:28–38. https://doi.org/10.1039/c1md00210d.
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput-Aided Mol Des. 2006;20:647–71. https://doi.org/10.1007/s10822-006-9087-6.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. https://doi.org/10.1002/jcc.21334.
Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorgan Med Chem Lett. 2020;30:127377. https://doi.org/10.1016/j.bmcl.2020.127377. This article focuses on the main protease as a target, comparing it with HIV Mpro and SARS‐CoV Mpro. It gives beneficial insights into peptidomimetics and small molecule inhibitors, discussing why repurposing of approved drugs is a strategic option.
Yoshino R, Yasuo N, Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci Rep. 2020;10:12493. https://doi.org/10.1038/s41598-020-69337-9.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93. https://doi.org/10.1038/s41586-020-2223-y.
A Bioinformatics study of structural perturbation of 3CL-protease and the HR2-domain of SARS-CoV-2 induced by synergistic interaction with ivermectins. Biointerface Res Appl Chem. 2020;11:9813–26. https://doi.org/10.33263/briac112.98139826.
Sang P, Tian S-H, Meng Z-H, Yang L-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020;10:15775–83. https://doi.org/10.1039/d0ra01899f.
Yuce M, Cicek E, İnan T, Dağ AB, Kürkçüoğlu Ö, Sungur FA. Repurposing of FDA-approved drugs against active site and potential allosteric drug-binding sites of COVID-19 main protease. Proteins 2021;89:1425–41. https://doi.org/10.1002/prot.26164. This study used guassian and residue model and identified two allosteric binding sites. Flexible docking and molecular dynamics, as well as free energy calculations were performed. Ivermectin, diosmin, and selinexor showed potential results.
Shamsi A, Mohammad T, Anwar S, AlAjmi MF, Hussain A, Rehman MT, et al. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: possible implication in COVID-19 therapy. Biosci Rep 2020;40. 10.1042/BSR20201256.
Francés-Monerris A, Garcia-Iriepa C, Iriepa I, Hognon C, Miclot T, Barone G, et al. Has ivermectin virus-directed effects against SARS-CoV-2? Rationalizing the action of a potential multitarget antiviral agent. ChemRxiv. Cambridge: Cambridge Open Engage; 2020. https://doi.org/10.26434/chemrxiv.12782258.v1
Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol Diversity. 2021;25:383–401. https://doi.org/10.1007/s11030-020-10130-1.
Krishnaprasad B, Maity S, Mehta C, Suresh A, Nayak UY, Nayak Y. In silico drug repurposing of penicillins to target main protease Mpro of SARS-CoV-2. Pharm Sci. 2020;26:S52–62. https://doi.org/10.34172/ps.2020.44.
Eleftheriou P, Amanatidou D, Petrou A, Geronikaki A. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-CoV-2 Virus. Molecules. 2020;25:2529. https://doi.org/10.3390/molecules25112529.
Novak J, Rimac H, Kandagalla S, Pathak P, Grishina M, Potemkin V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J Biomol Struct Dyn. 2021;21:1–14. https://doi.org/10.1080/07391102.2021.1927845.
Jain R, Mujwar S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Struct Chem. 2020;31:2487–99. https://doi.org/10.1007/s11224-020-01605-w.
Hakmi, Hakmi, Morocco. Repurposing of known antivirals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. Bioinformation 2020;16:301. https://doi.org/10.6026/97320630016301.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93. https://doi.org/10.1038/s41586-020-2223-y.
Nayeem SM, Sohail EM, Ridhima G, Reddy MS. Target SARS-CoV-2: computation of binding energies with drugs of dexamethasone/umifenovir by molecular dynamics using OPLS-AA force field. Res Biomed Eng. 2021. https://doi.org/10.1007/s42600-020-00119-y.
Jordaan MA, Ebenezer O, Damoyi N, Shapi M. Virtual screening, molecular docking studies and DFT calculations of FDA approved compounds similar to the non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz. Heliyon. 2020;6: e04642. https://doi.org/10.1016/j.heliyon.2020.e04642.
Mathpal S, Joshi T, Sharma P, Joshi T, Pundir H, Pande V, et al. A dynamic simulation study of FDA drug from zinc database against COVID-19 main protease receptor. J Biomol Struct Dyn. 2020;1–17. https://doi.org/10.1080/07391102.2020.1821785.
Havranek B, Islam SM. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J Biomol Struct Dyn. 2021;39:4304–15. https://doi.org/10.1080/07391102.2020.1776158.
Wafa T, Mohamed K, Mushtaq M, Ul-Haq Z. Wafa TACHOUA, Mohamed KABRINE, Mushtaq M, Ul-Haq Z. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. ChemRxiv. Cambridge: Cambridge Open Engage; 2020. https://doi.org/10.26434/chemrxiv.12318689.v2.
Das S, Sarmah S, Lyndem S, Roy AS. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39:3347–57. https://doi.org/10.1080/07391102.2020.1763201.
Kumar N, Awasthi A, Kumari A, Sood D, Jain P, Singh T, et al. A antitussive noscapine and antiviral drug conjugates as arsenal against COVID-19: a comprehensive chemoinformatics analysis. J Biomol Struct Dyn. 2022;40:101–16. https://doi.org/10.1080/07391102.2020.1808072.
Fintelman-Rodrigues N, Sacramento CQ, Ribeiro Lima C, Souza da Silva F, Ferreira AC, Mattos M, et al. Atazanavir, alone or in combination with ritonavir, inhibits SARS-CoV-2 replication and proinflammatory cytokine production. Antimicrob Agents Chemother 2020;64. https://doi.org/10.1128/AAC.00825-20.
Gupta PSS, Biswal S, Singha D, Rana MK. Binding insight of clinically oriented drug famotidine with the identified potential target of SARS-CoV-2. J Biomol Struct Dyn. 2020;1–7. https://doi.org/10.1080/07391102.2020.1784795.
Ortega JT, Serrano ML, Jastrzebska B. Class A G protein-coupled receptor antagonist famotidine as a therapeutic alternative against SARS-CoV2: an in silico analysis. Biomolecules. 2020;10:954. https://doi.org/10.3390/biom10060954.
Khan A, Ali SS, Khan MT, Saleem S, Ali A, Suleman M, et al. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J Biomol Struct Dyn. 2021;39:4659–70. https://doi.org/10.1080/07391102.2020.1779128.
Khan MA, Mahmud S, Rubayet Ul Alam AS, Rahman ME, Ahmed F, Rahmatullah M. C Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: a molecular docking study. J Biomol Struct Dyn. 2021;39:6317–23. https://doi.org/10.1080/07391102.2020.1796813.
Fadaka AO, Sibuyi NRS, Madiehe AM, Meyer M. Computational insight of dexamethasone against potential targets of SARS-CoV-2. J Biomol Struct Dyn. 2020;1–11. https://doi.org/10.1080/07391102.2020.1819880.
Bharadwaj S, Lee KE, Dwivedi VD, Kang SG. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci. 2020;257: 118080. https://doi.org/10.1016/j.lfs.2020.118080.
Yu R, Chen L, Lan R, Shen R, Li P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents. 2020;56: 106012. https://doi.org/10.1016/j.ijantimicag.2020.106012.
Muralidharan N, Sakthivel R, Velmurugan D, Michael GM. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J Biomol Struct Dyn. 2021;39:2673–8. https://doi.org/10.1080/07391102.2020.1752802.
Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J Biomol Struct Dyn. 2021;39:4647–58. https://doi.org/10.1080/07391102.2020.1779819.
Koulgi S, Jani V, Uppuladinne M, Sonavane U, Nath AK, Darbari H, et al. Drug repurposing studies targeting SARS-nCoV2: an ensemble docking approach on drug target 3C-like protease (3CLpro). (n.d.) https://doi.org/10.26434/chemrxiv.12228831.v1.
Lobo-Galo N, Terrazas-López M, Martínez-Martínez A, Díaz-Sánchez ÁG. FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. J Biomol Struct Dyn. 2020;1–9. https://doi.org/10.1080/07391102.2020.1764393.
Arouche T da S, da Silva Arouche T, Reis AF, Martins AY, Costa JFS, Junior RNC, et al. Interactions between remdesivir, ribavirin, favipiravir, galidesivir, hydroxychloroquine and chloroquine with fragment molecular of the COVID-19 main protease with inhibitor N3 complex (PDB ID:6LU7) using molecular docking. J Nanosci Nanotechnol. 2020;20:7311–23. https://doi.org/10.1166/jnn.2020.18955.
Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn. 2021;39:2607–16. https://doi.org/10.1080/07391102.2020.1751298.
Sharma P, Vijayan V, Pant P, Sharma M, Vikram N, Kaur P, et al. I Identification of potential drug candidates to combat COVID-19: a structural study using the main protease (mpro) of SARS-CoV-2. J Biomol Struct Dyn. 2021;39:6649–59. https://doi.org/10.1080/07391102.2020.1798286.
Chandel V, Raj S, Rathi B, Kumar D. In silico identification of potent COVID-19 main protease inhibitors from FDA approved antiviral compounds and active phytochemicals through molecular docking: a drug repurposing approach. Preprints. 2020. https://doi.org/10.20944/preprints202003.0349.v1.
Gul S, Ozcan O, Asar S, Okyar A, Barıs I, Kavakli IH. In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. J Biomol Struct Dyn. 2021;39:6772–91. https://doi.org/10.1080/07391102.2020.1802346.
Martorana A, Gentile C, Lauria A. In silico insights into the SARS CoV-2 main protease suggest NADH endogenous defences in the control of the pandemic coronavirus infection. Viruses. 2020;12:805. https://doi.org/10.3390/v12080805.
Deshpande RR, Tiwari AP, Nyayanit N, Modak M. In silico molecular docking analysis for repurposing therapeutics against multiple proteins from SARS-CoV-2. Eur J Pharmacol. 2020;886: 173430. https://doi.org/10.1016/j.ejphar.2020.173430.
Sachdeva C, Wadhwa A, Kumari A, Hussain F, Jha P, Kaushik NK. In silico potential of approved antimalarial drugs for repurposing against COVID-19. OMICS: J Integr Biol. 2020;24:568–80. https://doi.org/10.1089/omi.2020.0071.
Kumar Y, Singh H, Patel CN. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health. 2020;13:1210–23. https://doi.org/10.1016/j.jiph.2020.06.016.
Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252: 117652. https://doi.org/10.1016/j.lfs.2020.117652.
Braz HLB, de Moraes Silveira JA, Marinho AD, de Moraes MEA, de Moraes Filho MO, Monteiro HSA, et al. In silico study of azithromycin, chloroquine and hydroxychloroquine and their potential mechanisms of action against SARS-CoV-2 infection. Int J Antimicrob Agents. 2020;56: 106119. https://doi.org/10.1016/j.ijantimicag.2020.106119.
Alabboud M, Javadmanesh A. In silico study of various antiviral drugs, vitamins, and natural substances as potential binding compounds with SARS-CoV-2 main protease. DYSONA - Life Sci. 2020;1:44–63. https://doi.org/10.30493/dls.2020.225404.
Ibrahim MAA, Abdelrahman AHM, Hegazy M-EF. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn. 2021;39:5756–67. https://doi.org/10.1080/07391102.2020.1791958.
Elzupir AO. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J Mol Struct. 2020;1222: 128878. https://doi.org/10.1016/j.molstruc.2020.128878.
Hagar M, Ahmed HA, Aljohani G, Alhaddad OA. Investigation of some antiviral N-heterocycles as COVID 19 drug: molecular docking and DFT calculations. Int J Mol Sci. 2020;21:3922. https://doi.org/10.3390/ijms21113922.
Ferraz WR, Gomes RA, Novaes ALS, Trossini GHG. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med Chem. 2020;12:1815–28. https://doi.org/10.4155/fmc-2020-0165.
Salah M, Belghiti ME, Aitouna AO, Zeroual A, Jorio S, El Alaoui AH, et al. MEDT study of the 1,3-DC reaction of diazomethane with psilostachyin and investigation about the interactions of some pyrazoline derivatives with protease (Mpro) of nCoV-2. J Mol Graph Model. 2021;102: 107763. https://doi.org/10.1016/j.jmgm.2020.107763.
Kumar N, Sood D, van der Spek PJ, Sharma HS, Chandra R. Molecular binding mechanism and pharmacology comparative analysis of noscapine for repurposing against SARS-CoV-2 protease. J Proteome Res. 2020;19:4678–89. https://doi.org/10.1021/acs.jproteome.0c00367.
Lokhande KB, Doiphode S, Vyas R, Venkateswara SK. Molecular docking and simulation studies on SARS-CoV-2 M(pro) reveals mitoxantrone, leucovorin, birinapant, and dynasore as potent drugs against COVID-19. J Biomol Struct Dyn. 2021;39:7294–305. https://doi.org/10.1080/07391102.2020.1805019.
Pant S, Singh M, Ravichandiran V, Murty USN, Srivastava HK. Peptide-like and small-molecule inhibitors against COVID-19. J Biomol Struct Dyn. 2021;39:2904–13. https://doi.org/10.1080/07391102.2020.1757510.
Alamri MA, Qamar MT ul, Mirza MU, Bhadane R, Alqahtani SM, Muneer I, et al. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CL(pro). J Biomol Struct Dyn 2021;39:4936–48. https://doi.org/10.1080/07391102.2020.1782768.
Ancy I, Sivanandam M, Kumaradhas P. Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study. J Biomol Struct Dyn. 2021;39:5368–75. https://doi.org/10.1080/07391102.2020.1786459. This is an in silico study on 2 HIV clinical trials drugs. Molecular docking, molecular dynamics, and free energy calculations revealed that TMB607 is promising against SARS‐COV19.
Han Y, Wang Z, Ren J, Wei Z, Li J. Potential inhibitors for the novel coronavirus (SARS-CoV-2). Brief Bioinform. 2021;22:1225–31. https://doi.org/10.1093/bib/bbaa209.
Gimeno A, Mestres-Truyol J, Ojeda-Montes MJ, Macip G, Saldivar-Espinoza B, Cereto-Massagué A, et al. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int J Mol Sci. 2020;21:3793. https://doi.org/10.3390/ijms21113793.
Meyer-Almes F-J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and structure based drug design. Comput Biol Chem. 2020;88: 107351. https://doi.org/10.1016/j.compbiolchem.2020.107351.
Al-Zahrani AA. Rutin as a promising inhibitor of main protease and other protein targets of COVID-19: in silico study. Nat Prod Commun. 2020;15:1934578X20953951. https://doi.org/10.1177/1934578X20953951.
Liang J, Karagiannis C, Pitsillou E, Darmawan KK, Ng K, Hung A, et al. Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface. Comput Biol Chem. 2020;89: 107372. https://doi.org/10.1016/j.compbiolchem.2020.107372.
Al-Zaqri N, Pooventhiran T, Alsalme A, Warad I, John AM, Thomas R. Structural and physico-chemical evaluation of melatonin and its solution-state excited properties, with emphasis on its binding with novel coronavirus proteins. J Mol Liq. 2020;318: 114082. https://doi.org/10.1016/j.molliq.2020.114082.
Mukherjee S, Dasgupta S, Adhikary T, Adhikari U, Panja SS. Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J Biomol Struct Dyn. 2021;39:7322–34. https://doi.org/10.1080/07391102.2020.1804458.
Gahlawat A, Kumar N, Kumar R, Sandhu H, Singh IP, Singh S, et al. Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J Chem Inf Model. 2020;60:5781–93. https://doi.org/10.1021/acs.jcim.0c00546.
Nayeem SM, Sohail EM, Ridhima G, Srinivasa RM. Target SARS-CoV-2: computation of binding energies with drugs of dexamethasone/umifenovir by molecular dynamics using OPLS-AA force field. Res Biomed Eng. 2021. https://doi.org/10.1007/s42600-020-00119-y.
Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res. 2021;52:38–47. https://doi.org/10.1016/j.arcmed.2020.09.013.
Achilonu I, Iwuchukwu EA, Achilonu OJ, Fernandes MA. Targeting the SARS-CoV-2 main protease using FDA-approved isavuconazonium, a P2–P3 α-ketoamide derivative and pentagastrin: an in-silico drug discovery approach. J Mol Graph Model. 2020;101:2. https://doi.org/10.1016/j.jmgm.2020.107730.
Al-Khafaji K, AL-Duhaidahawi D, Tok TT. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J Biomol Struct Dyn. 2020;1–9. https://doi.org/10.1080/07391102.2020.1764392.
Hage-Melim LI da S, da Silva Hage-Melim LI, Federico LB, de Oliveira NKS, Francisco VCC, Correia LC, et al. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020;256:117963. https://doi.org/10.1016/j.lfs.2020.117963.
Rahman MM, Saha T, Islam KJ, Suman RH, Biswas S, Rahat EU, et al. Virtual screening, molecular dynamics and structure-activity relationship studies to identify potent approved drugs for COVID-19 treatment. J Biomol Struct Dyn. 2021;39:6231–41. https://doi.org/10.1080/07391102.2020.1794974. In this study, more than 1600 FDA‐approved drugs were subjected to molecular docking and molecular dynamics, as well as free energy calculations. A quantitative structure‐activity relationship was performed, and the accuracy was 82% in agreement with molecular docking results.
Fiorucci D, Milletti E, Orofino F, Brizzi A, Mugnaini C, Corelli F. Computational drug repurposing for the identification of SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn. 2021;39:6242–8. https://doi.org/10.1080/07391102.2020.1796805.
Mitra K, Ghanta P, Acharya S, Chakrapani G, Ramaiah B, Doble M. Dual inhibitors of SARS-CoV-2 proteases: pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J Biomol Struct Dyn. 2021;39:6324–37. https://doi.org/10.1080/07391102.2020.1796802.
Mittal L, Kumari A, Srivastava M, Singh M, Asthana S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J Biomol Struct Dyn. 2021;39:3662–80. https://doi.org/10.1080/07391102.2020.1768151.
Zhu W, Xu M, Chen CZ, Guo H, Shen M, Hu X, et al. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol Transl Sci. 2020;3:1008–16. https://doi.org/10.1021/acsptsci.0c00108.
Chatterjee S, Maity A, Chowdhury S, et al. In silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J Biomol Struct Dyn. 2021;39:5290–303. https://doi.org/10.1080/07391102.2020.1787228.
Maffucci I, Contini A. In silico drug repurposing for SARS-CoV-2 main proteinase and spike proteins. J Proteome Res. 2020;19:4637–48. https://doi.org/10.1021/acs.jproteome.0c00383.
Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother. 2003;47:3675. https://doi.org/10.1128/AAC.47.12.3675-3681.2003.
Battisti V, Wieder O, Garon A, Seidel T, Urban E, Langer T. A computational approach to identify potential novel inhibitors against the coronavirus SARS-CoV-2. Mol Inf. 2020;39:2000090. https://doi.org/10.1002/minf.202000090.
Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: an in silico drug repurposing for COVID-19. F1000Research. 2020;9:1166. https://doi.org/10.12688/f1000research.26359.1. This study performed molecular docking of FDA‐approved drugs against viral RNA department RNA polymerase as a potential target. Top‐ranked hits were subjected to extra precision docking and molecular dynamics. It was recommended that pitavastatin, ridogrel, and rosoxacin could be further optimized in preclinical and clinical research.
Ribaudo G, Ongaro A, Oselladore E, Zagotto G, Memo M, Gianoncelli A. A computational approach to drug repurposing against SARS-CoV-2 RNA dependent RNA polymerase (RdRp). J Biomol Struct Dyn. 2020;4:1101–8. https://doi.org/10.1080/07391102.2020.1822209.
Choudhury S, Moulick D, Saikia P, Mazumder MK. Evaluating the potential of different inhibitors on RNA-dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2: a molecular modeling approach. Armed Forces Med J India. 2021;77:S373–8. https://doi.org/10.1016/j.mjafi.2020.05.005.
Copertino DC, Duarte RRR, Powell TR, Rougvie MM, Nixon DF. Montelukast drug activity and potential against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J Med Virol. 2021;93:187–9. https://doi.org/10.1002/jmv.26299.
Ruan Z, Liu C, Guo Y, He Z, Huang X, Jia X, et al. SARS-CoV-2 and SARS-CoV: virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J Med Virol. 2021;93:389–400. https://doi.org/10.1002/jmv.26222.
Ahmad J, Ikram S, Ahmad F, Rehman IU, Mushtaq M. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) - a drug repurposing study. Heliyon. 2020;6: e04502. https://doi.org/10.1016/j.heliyon.2020.e04502.
Mutlu O, Ugurel OM, Sariyer E, Ata O, Inci TG, Ugurel E, et al. Targeting SARS-CoV-2 Nsp12/Nsp8 interaction interface with approved and investigational drugs: an in silico structure-based approach. J Biomol Struct Dyn. 2020;1–13. https://doi.org/10.1080/07391102.2020.1819882.
Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–9. https://doi.org/10.1038/s41564-020-0688-y.
Huang Y, Yang C, Xu X-F, Xu W, Liu S-W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 2020;41:1141–9. https://doi.org/10.1038/s41401-020-0485-4. A very important article provides an in‐depth knowledge of the structure, function, and antivirals development strategies. Despite its early publishing date, it discussed the importance of the conserved regions when developing neutralizing antibodies. The study suggested that EK1C4, an antibody against conserved regions in S2 subunit, could be promising against both SARS‐COV and SARS‐COV19.
Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003;77:8801–11. https://doi.org/10.1128/jvi.77.16.8801-8811.2003.
Celı KI, Onay-Besı Kcı A, Ayhan-Kilcigı LG. Approach to the mechanism of action of hydroxychloroquine on SARS-CoV-2: a molecular docking study. J Biomol Struct Dyn. 2021;39:5792–8. https://doi.org/10.1080/07391102.2020.1792993.
Somadi G, Sivan SK. Identification of therapeutic target in S2 domain of SARS nCov-2 Spike glycoprotein: key to design and discover drug candidates for inhibition of viral entry into host cell. J Theor Comput Chem. 2020;19:2050028. https://doi.org/10.1142/s0219633620500285. This article revealed the importance of S2 subunit in spike protein and indicated its role during viral entry into host cells. HR1 and CH region in S2 domain were identified as suggested binding sites. The FDA approved virtual screening against this site showed promising candidates like Troxerutin, Thymopentin, and Daclatasvir.
Panda PK, Arul MN, Patel P, Verma SK, Luo W, Rubahn H-G, et al. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci Adv. 2020;6:eabb8097. https://doi.org/10.1126/sciadv.abb8097.
Prajapat M, Shekhar N, Sarma P, Avti P, Singh S, Kaur H, et al. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J Mol Graph Model. 2020;101:107716. https://doi.org/10.1016/j.jmgm.2020.107716. In this study 2456, FDA‐approved drugs were screened against RBD in S1 subunit and subjected to molecular dynamics and free energy calculations. Riboflavin, fenoterol, cangrelor, and vidarabine emerged out as molecules with the best stability.
Musarrat F, Chouljenko V, Dahal A, Nabi R, Chouljenko T, Jois SD, et al. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J Med Virol. 2020;92:2087–95. https://doi.org/10.1002/jmv.25985.
Adeoye AO, Oso BJ, Olaoye IF, Tijjani H, Adebayo AI. Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. J Biomol Struct Dyn. 2021;39:3469–79. https://doi.org/10.1080/07391102.2020.1765876.
Cavasotto CN, Di Filippo JI. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol Inf. 2021;40:2000115. https://doi.org/10.1002/minf.202000115.
Shekhar N, Sarma P, Prajapat M, Avti P, Kaur H, Raja A, et al. In silico structure-based repositioning of approved drugs for spike glycoprotein S2 domain fusion peptide of SARS-CoV-2: rationale from molecular dynamics and binding free energy calculations. mSystems. 2020;5. https://doi.org/10.1128/msystems.00382-20. In this study, 2,625 FDA‐approved small molecules were subjected to molecular docking and molecular dynamics and free energy calculations against the conserved S2 subunit. Chloramphenicol succinate, imipenem, and imidurea emerged as the molecules with the best binding at the fusion peptide hydrophobic pocket.
Author information
Authors and Affiliations
Contributions
All authors listed have significantly contributed to the development and the writing of this article.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Nanodrugs
Rights and permissions
About this article

Cite this article
Fadlalla, M., Ahmed, M., Ali, M. et al. Molecular Docking as a Potential Approach in Repurposing Drugs Against COVID-19: a Systematic Review and Novel Pharmacophore Models. Curr Pharmacol Rep 8, 212–226 (2022). https://doi.org/10.1007/s40495-022-00285-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-022-00285-w