
Abstract
Purpose of Review
This review gives a perspective on the current “state of the art” in metabolic drug-drug interaction (DDI) prediction. We highlight areas of successful prediction and illustrate progress in areas where limits in scientific knowledge or technologies prevent us from having full confidence.
Recent Findings
Several examples of success are highlighted. Work done for bitopertin shows how in vitro and clinical data can be integrated to give a model-based understanding of pharmacokinetics and drug interactions. The use of interpolative predictions to derive explicit dosage recommendations for untested DDIs is discussed using the example of ibrutinib, and the use of DDI predictions in lieu of clinical studies in new drug application packages is exemplified with eliglustat and alectinib. Alectinib is also an interesting case where dose adjustment is unnecessary as the activity of a major metabolite compensates sufficiently for changes in parent drug exposure.
Examples where “unusual” cytochrome P450 (CYP) and non-CYP enzymes are responsible for metabolic clearance have shown the importance of continuing to develop our repertoire of in vitro regents and techniques. The time-dependent inhibition assay using human hepatocytes suspended in full plasma allowed improved DDI predictions, illustrating the importance of continued in vitro assay development and refinement.
Summary
During the past 10 years, a highly mechanistic understanding has been developed in the area of CYP-mediated metabolic DDIs enabling the prediction of clinical outcome based on preclinical studies. The combination of good quality in vitro data and physiologically based pharmacokinetic modeling may now be used to evaluate DDI risk prospectively and are increasingly accepted in lieu of dedicated clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Quantification of a drug-drug interaction (DDI) effect in a man is the basis for explicit dose recommendation in drug labels to minimize the risk of adverse events or reduced efficacy, thereby supporting appropriate use of the drug. It is therefore essential that such quantitative DDI assessments are made with confidence. There has been a steady development of in vitro assays and the reagents available for the study of drug metabolism and metabolic enzyme inhibition. This, combined with advances in our capability to extrapolate in vitro data to in vivo, has brought us past a “tipping point” such that applying a model-based synthesis of the available data has become normal in drug-drug interaction assessments [1,2,3,4,5••]. Simple static models, built upon DDI studies reaching back to the 1970s [6•], still find utility in early drug discovery where there are very limited data available for the drug candidate. However, the greatest DDI effects are observed where the metabolism of an orally administered drug is substantially inhibited in the first pass metabolism, potentially in both the intestine and liver. The combination of increased drug reaching the systemic circulation as well as reduced systemic clearance will result in a significantly higher exposure (area under the plasma concentration-time curve [AUC]) than when inhibition of systemic clearance alone is considered. An example of this can be seen when comparing the DDI effect of ketoconazole on alprazolam and midazolam which are low and high clearance cytochrome P450 (CYP) 3A substrates, respectively. In the recent study of Boulenc et al., peak concentration (Cmax) for alprazolam and midazolam were increased by 1.18- and 4.21-fold, whereas AUC was increased by 2.63- and 16.95-fold, respectively, when co-administered with multiple once-daily doses of 400 mg ketoconazole [7]. Mechanistic static models have significantly extended mathematical model usage, by incorporating additional considerations such as intestinal metabolism, enzyme induction, and enzyme inactivation [8, 9]. Nevertheless, these mathematical models cannot capture the full dynamic nature of drug metabolism in vivo since only a fixed concentration of inhibitor is considered. For example, DDI effects on simultaneous co-administration versus staggered dosing situations may be different, especially when the interacting drugs have short half-lives and high first pass metabolism. Details of the different approaches to DDI prediction were recently described in a Pharmaceutical Industry Innovation and Quality working group publication from Bohnert et al. and will not be discussed further in this review [10•].
A more powerful approach to DDI prediction can be taken using physiologically based pharmacokinetic (PBPK) modeling, especially when human pharmacokinetic data are available. Validated PBPK models allow high confidence in prospective DDI predictions [1]. This application of modeling and simulation has been reflected in the regular inclusion of PBPK model information into new drug application (NDA) submissions [4, 5••] and recent use in final drug product labeling text with explicit dosage recommendations (see examples below). Similarly, the simulations may support selection of dose strengths to be developed. In order to generate a well-validated PBPK model for a drug, a large amount of data need to be collected. Such data include pharmacokinetics of drug substance and metabolites, drug solubility and permeability, plasma protein binding, contributions of individual enzymes to hepatic and extrahepatic clearance, enzyme inhibition, inactivation and induction, clearance by non-metabolic routes (e.g., urinary and biliary secretion information), and any existing clinical drug-drug interaction information. Only when a good description of compound pharmacokinetics and metabolism has been established can drug-drug interaction predictions and the consequences for efficacy and safety be adequately addressed.
Improvements in in vitro technologies and the buildup of system knowledge (enzyme abundance, physiological parameters, effect of disease, age, sex, and polymorphism status) have allowed increasingly realistic computational models of the human body to be developed [11]. Confidence in competitive CYP inhibition measurement and consequent DDI prediction is typically high. In contrast, although availability, consistency, and sensitivity of time-dependent inhibition (TDI) measurement have improved considerably [12], challenges still exist in the quantitative extrapolation of TDI data. This is especially true in complex situations, for example, where time-dependent inhibition is combined with active uptake or enzyme induction. The human immunodeficiency virus (HIV) drug ritonavir, used to boost the bioavailability of antiviral agents such as saquinavir by inhibition of CYP3A4, is an example of a complex case. As well as being a CYP3A4 substrate, ritonavir inhibits, inactivates, and induces CYP3A4 [13,14,15]. It also inhibits and induces other drug-metabolizing enzymes [16].
The other facet of DDI assessment, that of victim DDIs, can be made with confidence for drugs principally metabolized by well-characterized metabolic enzymes (e.g., CYPs 1A2, 2C8, 2C9, 2C19, 2D6, 3A4). However, model validation is more difficult and prediction confidence is lower for enzymes such as aldehyde oxidase (AO), flavin monooxygenases (FMOs) and UGP-glucuronosyltransferases (UGTs) where human pharmacokinetic data for selective substrates and for in vivo interactions with inhibitors are lacking.
This review draws on recent Roche experiences combined with key literature examples to provide an overview of the current state of the art in DDI prediction and ongoing developments in the field. The structures of the drugs featured in this review, together with information relevant to their metabolic DDIs, can be found in Table 1.
The Recent Past: Mechanistic Understanding of DDIs Through Retrospective Modeling
Bitopertin Case Study—Drug-Drug Interaction with CYP3A4 Inhibitors
Bitopertin inhibits the glycine transporter type 1 (GlyT1), which is expressed in the central nervous system and in peripheral tissues, mainly in erythroid cells [17, 18]. Bitopertin is cleared slowly and almost exclusively by oxidative metabolism, primarily via CYP3A4 (fm(CYP3A enzymes) > 90% in vitro) with less than 0.1% of the administered dose excreted in the urine as unchanged drug [19•]. The half-life is approximately 2 days.
The pharmacokinetics of bitopertin was predicted prior to clinical studies using a PBPK model developed on the basis of non-clinical data [20]. After entry into the clinic, the model-predicted pharmacokinetics were found to be in close agreement with observations and the model was refined [21] and then applied to simulate the potential for drug-drug interactions. The clinical effect of CYP3A4 inhibition on bitopertin exposure was assessed in two studies in healthy volunteers with open-label, two-period, fixed-sequence designs [19•].
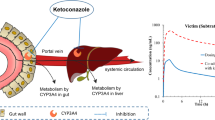
Ketoconazole, a strong CYP3A4 inhibitor, increased the bitopertin AUC from 0 to 312 h (AUC0–312 h) 4.2-fold (90% confidence interval [CI] 3.5–5.0) while erythromycin, a moderate CYP3A4 inhibitor, increased the AUC from time zero to infinity (AUC0–inf) 2.1-fold (90% CI 1.9–2.3). The AUC0–inf ratios predicted by PBPK modeling for these interactions were in good agreement at 7.7 and 1.9, respectively (note that the AUC0–312 h ratio underestimated the full DDI to some extent). The effect on Cmax was minor, <25% for both inhibitors. This was consistent with a high absolute bioavailability as simulated by PBPK for bitopertin with very limited first pass extraction in both the intestine and the liver. After discontinuation of ketoconazole, the bitopertin elimination half-life decreased, becoming similar to that observed in the absence of ketoconazole indicating the reversibility of the CYP3A4/5 inhibition (Fig. 1). For bitopertin, therefore, an excellent picture of the pharmacokinetics and a model describing the CYP3A-mediated drug-drug interactions could be developed and retrospectively validated using emerging clinical data. Details of the PBPK model can be found in Supplementary Table 1. DDI study and simulation data are also available in the Supplementary Materials.

Effect of ketoconazole on exposure of bitopertin. Symbols are mean (±standard deviation) plasma concentration-time profiles after administration of 400 mg/day ketoconazole (filled black circles), bitopertin 10 mg alone (empty blue squares), or concurrently with ketoconazole (filled red triangles). The lines are the plasma concentrations simulated with a PBPK model in GastroPlus. Single dose of bitopertin alone (dashed blue line), bitopertin with ketoconazole (dotted red line), ketoconazole 17 days (solid black line)
Current State of the Art: Interpolation and Limited Prospective DDI Prediction Gain Regulatory Acceptance
PBPK models have initially found use in incorporating the results of DDI studies into an overall description of the pharmacokinetics, then in interpolating results from DDI studies with strong probe inhibitors/inducers for the enzyme of interest (“mechanistic DDI study”) to DDIs with moderate and mild inhibitors/inducers. In addition, PBPK models have been applied to extrapolation of DDI results to subpopulations such as organ failure, geriatrics, or certain phenotypes of the involved metabolic enzymes where it is often ethically and/or practically challenging to investigate DDI [22, 23]. Such simulations have been used for guiding dose adjustment in drug labels in lieu of actual clinical study results since 2009 [1]. Ibrutinib and eliglustat are two examples selected to illustrate how a PBPK model was developed for drugs mainly metabolized by CYP3A and CYP2D6, respectively, and applied to DDI assessment which were accepted in final product labels.
Ibrutinib Case Study—Model-Based Interpolation of CYP3A4 Inhibition DDIs
Ibrutinib is a Bruton’s tyrosine kinase inhibitor developed for treatment of leukemia. Ibrutinib is completely absorbed after oral administration and extensively metabolized in the intestine and liver mostly by CYP3A4 and lesser extent by CYP2D6 [24]. The absolute bioavailability of ibrutinib 560 mg (approved dose) was 3.9 and 8.4% in the fasted and fed states, respectively [25]. The intestinal and hepatic bioavailability (Fg and Fh) evaluated after oral (140 mg) and intravenous (100 μg, 13C6 labeled) administration in the fed state were determined as 47.0 and 15.9%, respectively, in an IV microdose study with grapefruit juice pretreatment [25]. A PBPK model was developed by integrating available physicochemical properties, in vitro experiments, and clinical pharmacokinetic (PK) data [26]. The intrinsic clearance of ibrutinib in human liver microsomes was inhibited 95.8% in the presence of 1 μM of a strong CYP3A inhibitor, ketoconazole [27], and this information was incorporated into the PBPK model for DDI simulations with various CYP3A4 modulators. Capability of the PBPK model to predict CYP3A4 DDI for ibrutinib as substrate was examined by predicting fold increase in Cmax and AUC of ibrutinib in the presence of ketoconazole and compared to the observations in the clinical study [28] (predicted vs. observed: 19- and 29- fold for Cmax and 28- and 24-fold for AUC). Subsequently, the PBPK model was verified by showing consistency between prospectively simulated fold decrease in Cmax and AUC of ibrutinib in the presence of a CYP3A inducer, rifampicin, and the observations [28] (predicted vs. observed: 11- and 13-fold for Cmax and 10- and 10-fold for AUC). The verified PBPK model was then used to simulate unstudied clinical DDIs with mild (fluvoxamine, azithromycin), moderate (diltiazem and erythromycin), and strong (voriconazole, clarithromycin, itraconazole) CYP3A inhibitors to guide dose reduction from 560 to 140 mg in concurrent administrations with moderate CYP3A4 inhibitors.
The PBPK model simulations of DDI with moderate (efavirenz) and strong (carbamazepine) CYP3A inducers supported ibrutinib dose of 560 mg in co-administrations with moderate CYP3A inducers since predicted exposure was within defined therapeutic exposure range [27]. The DDI risk assessment and dose modification guidance for ibrutinib based on the PBPK modeling and simulations were submitted in new drug applications and approved in the USA [27, 29], Canada [30], European Union [31], and Japan [32] and used in drug labels.
Eliglustat Case Study—Model-Based Extrapolation to Polymorphic CYP2D6 Phenotype Individuals
Eliglustat is an oral glucosylceramide synthase inhibitor and indicated to treat symptoms of Gaucher disease type 1 [33]. This drug is extensively metabolized by CYP2D6 (fmCYP2D6 = 86%) and to a lesser extent by CYP3A4 (fmCYP3A4 = 14%). Clinical DDI investigations in CYP2D6 intermediate metabolizers (IMs) and extensive metabolizers (EMs) showed increase in AUC of eliglustat by approximately 5-fold (IMs) to 10-fold (EMs) when co-administered with paroxetine (strong CYP2D6 and weak CYP3A4 inhibitor) and by 4-fold (in both IMs and EMs) when co-administered with ketoconazole. Eliglustat is a time-dependent inhibitor of CYP2D6 and multiple dose PK exhibited dose- and time-dependent behavior. Multiple doses of eliglustat increased AUC of metoprolol (CYP2D6 substrate) by approximately 2-fold in EMs and IMs. A PBPK model of eliglustat was developed and its ability to describe PK in different CYP2D6 phenotypes including poor metabolizers (PMs) and to predict clinical DDIs was confirmed. The PBPK model was then used for predicting DDIs with moderate inhibitors of CYP2D6 (terbinafine) and CYP3A4 inhibitors (fluconazole) in EMs and IMs. Moreover, DDIs with moderate to strong CYP3A4 inhibitors (fluconazole and ketoconazole) in PMs were predicted using the PBPK model since CYP3A4 inhibition effect on eliglustat has not been clinically investigated in PMs. Predicted fold increase in AUC0–24 h of eliglustat in concomitant administration with ketoconazole in PMs was 6.2 [34], higher than that in EMs and IMs, due to higher dependency on elimination through CYP3A4 metabolism, and concomitant use with strong CYP3A inhibitors is contraindicated in this population. The PBPK model enabled not only interpolations from DDI with strong enzyme inhibitors to the moderate inhibitors but also extrapolations of DDIs to other CYP2D6 phenotypes which complemented DDI risk assessments of eliglustat as a dual CYP2D6 and CYP3A4 substrate across CYP2D6 phenotypes. Dosage adjustment guidance in the drug label approved by FDA [34] based on these clinical studies and PBPK model predictions are summarized in Supplementary Table 2.
Repaglinide Case Study—Model-Based Prediction of Insignificant DDI Effect to Support Appropriate Dosing Recommendations
As a clinically relevant probe substrate, repaglinide is commonly used to assess the DDI risk for CYP2C8 inhibitors. Repaglinide is an antidiabetic drug whose metabolism is mediated by CYP2C8, CYP3A4, and to a lesser extent UGT enzymes [35, 36]. For assessing the DDI risk, therefore, assigning the appropriate fm(CYP2C8) value for repaglinide is of great importance given the sensitivity of predicted AUC ratios to fm values [37]. CYP2C8 and CYP3A4 have been reported to contribute equally to the in vitro metabolism of repaglinide, ∼50% [36]. However, an alternative fm(CYP2C8) value of 0.83 has been proposed based on meta-analysis of in vivo data [38]. As these two fm> values would result in very different maximal repaglinide DDI effects assuming complete enzyme inhibition (2.4 and 5.9 for fm values of 0.59 and 0.83, respectively, following oral administration), it was important to consider both possibilities in the DDI assessment.
A number of clinically relevant DDIs with repaglinide have been reported (Table 2). These DDIs include interactions with inhibitors of CYP2C8, CYP3A4, and OATP1B1/3 as well as compounds which interact via multiple mechanisms. The extent of clinical DDIs with repaglinide may be assessed as (1) large extent (≥5-fold AUC change) due to inhibition of multiple processes or TDI of CYP2C8 [39, 40] and (2) a substantially lower risk can be anticipated for inhibition of a single process, <2.5-fold AUC change for competitive CYP3A4 or CYP2C8 inhibitors [41, 42].
Alectinib (Alecensa®) is a small molecule kinase inhibitor which has received FDA accelerated approval for the treatment of patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib treatment [43]. Alectinib has shown weak competitive and time-dependent inhibition of CYP3A4 in vitro which has not translated in vivo [44]. Alectinib is also a competitive inhibitor of CYP2C8 with an unbound in vitro K i value of 0.147 μM [45]. DDI predictions with repaglinide were performed using a PBPK modeling approach to evaluate the clinical relevance of the CYP2C8 liability. The measured fu(plasma) and blood to plasma concentration ratio used in the PBPK simulations were 0.003 and 2.64 (consequently the fu(blood) was 0.0011). In the PBPK assessment of repaglinide, DDI potential fm(CYP2C8) values of both 0.59 and 0.83 were used. In order to investigate the sensitivity of the DDI simulations to the in vitro K i value of alectinib, the following scenarios were tested for both repaglinide models: true in vivo K i = 1×, 1/3×, 1/10×, and 1/30× of the in vitro K i value [46].
Based on the simulations, no significant interaction (>25% change of AUC) is anticipated regardless of the assumptions around the in vivo fm(CYP2C8) value of repaglinide (0.59 or 0.83). A sensitivity analysis revealed that a risk for an AUC change of greater than 25% can only be expected in case that the in vivo inhibitory potency of alectinib is considerably higher than anticipated from in vitro data and the in vivo fm(CYP2C8) of repaglinide is 0.83. This model-based assessment for characterization of clinical DDI between alectinib and CYP2C8 substrates was accepted in lieu of a clinical DDI study with repaglinide and justified the product labeling text “No clinical meaningful effect on the exposure of … repaglinide (sensitive CYP2C8 substrate) is expected following co-administration with ALESENSA” [43].
Alectinib Efficacy Case Study—Translation of DDI Effects Into Pharmacodynamic Effects: Relevance and Contribution of a Major Active Metabolite to Analysis and Interpretation of a Clinical DDI
Human metabolites are usually considered in terms of safety when formed at greater than 10% of total drug-related systemic exposure at steady state [47]. In terms of drug-drug interactions, metabolites formed in vivo and reaching significant exposures (e.g., ≥25% of parent drug exposure) have been recommended to be characterized further in terms of metabolism, transport, and for potential drug-drug interactions [48]. A metabolite may bind to on- or off-target receptors and thus can be considered active and contribute to intended and/or unintended effects [49,50,51].
Alectinib is metabolized by CYP3A4 and to a smaller extent by other enzymes to generate a number of metabolites including a major metabolite M4 [52]. Population PK analysis of the pivotal phase 2 studies showed that the geometric mean M4 metabolite/parent (M/P) ratio in plasma was 0.4 with an effective elimination half-life (t 1/2) of approximately 33 and 31 h for alectinib and M4, respectively [53]. In vitro pharmacology studies demonstrated that both alectinib and M4 are potent inhibitors of the target ALK with similar potency (IC50 of 1.9 and 1.2 nM, for alectinib and M4, respectively, in biochemical assays) and exhibit similar plasma protein binding (>99% protein bound) [52].
As both alectinib and M4 are substrates of CYP3A, dedicated clinical pharmacology studies were undertaken to evaluate the effect of a strong CYP3A inhibitor (posaconazole) and strong CYP3A inducer (rifampin) on the pharmacokinetics of alectinib and M4 [44]. Notably, the results from the clinical DDI study with posaconazole showed that its co-administration increased alectinib exposure and decreased M4 exposure while results from the rifampin DDI study showed that its co-administration decreased alectinib exposure and increased M4 exposure [44] (Fig. 2). As both alectinib and M4 are similarly active against ALK and exhibit similar protein binding, it is expected that both substances contribute to overall alectinib efficacy and safety. Therefore, to support clinical dosing recommendations in the presence of CYP3A inhibitors and inducers, changes in the combined molar exposure of alectinib and M4 (i.e., molar sum of alectinib + M4) were evaluated (Fig. 2). The minor effects seen on the combined exposure supported the statement “no dosage adjustment required with co-administered CYP3A inhibitors or inducers” in US prescribing information for Alecensa® [43].

Forrest plot of the drug-drug interaction potential between alectinib and the potent CYP3A inhibitor, posaconazole, or the potent CYP3A inducer, rifampin [48]
The alectinib case represents an approach to the understanding of drug-drug interaction potential by utilization of integrated non-clinical and clinical data of a parent molecule and its major active metabolite. The knowledge of clinical pharmacology attributes of both the parent and metabolite enabled dosing recommendations based on the changes occurring to both substances. To support this, characterization of both alectinib and M4 was undertaken throughout the development process from preclinical safety and drug metabolism/pharmacokinetic testing through to clinical exposure-response evaluation of alectinib [54, 55]. Indeed, clinical exposure-response analyses evaluated the relationship between key efficacy and safety endpoints emerging from alectinib pivotal studies and the combined exposure of alectinib and M4 [53]. Thus, while the changes seen in the alectinib exposure when co-administered with posaconazole or rifampin may have potentially warranted dosage adjustments, consideration of the combined changes suggested that no dosage adjustments were needed. This approach to consideration of parent and metabolite contributions to clinical DDI or exposure-response interpretation has been successfully applied previously for other small molecules with active metabolites (e.g., regorafenib, ezetimibe, ruxolitinib, dabrafenib, and sunitinib) [56,57,58,59,60,61]. Cumulatively, the alectinib case illustrates the relevance and contribution of a major active metabolite to clinical DDI analyses and interpretation.
Current Frontiers in DDI Prediction From In Vitro
Enhanced DDI Predictions From Time-Dependent Inhibition Measurements Using Human Hepatocytes Suspended in Full Plasma
Preclinical prediction of CYP inhibition-mediated DDIs has been performed conventionally using the well-characterized and intensively studied human liver microsomal (HLM) assay, which shows high detection sensitivity and low likelihood of false-negative predictions [62]. An in vitro assay using human hepatocytes (hHEPs) suspended in whole human plasma (plasma hHEPs) has been reported to give more accurate prediction of the extent of clinical relevant effect due to CYP inhibition [63,64,65,66]. Advantages of assessing DDI in human hepatocytes supplemented with 100% plasma include (1) inherent accounting for plasma protein and microsomal/hepatocyte binding of a drug, (2) compound is available to enzyme in its native environment within the cell, i.e., more a physiologically relevant condition, (3) metabolism of the compound by both CYP and non-CYP pathways is possible, and (4) transporter-mediated uptake into hepatocytes may occur.
An elegant study recently published by Mao et al. [67••] compared side-by-side DDI prediction due to CYP3A inhibition from the plasma hHEP assay with three other assays: (a) HLM, (b) plated hHEPs, and (c) hHEPs suspended in Dulbecco’s modified Eagle’s medium (DMEM) for 12 marketed drugs (10 protein kinase inhibitors and 2 prototypical CYP3A time-dependent inhibitors). Kinetic parameters were generated for the apparent reversible inhibition constant (K i,app) and/or TDI (K I,app and k inact) and directly used for quantitative prediction of the fold-increase in midazolam AUC0–inf (AUCR) following co-administration with CYP3A inhibitors based on a static mechanistic model and the total average systemic plasma concentration without correction for free drug fraction (fu). The result from this study demonstrated that the plasma hHEP assay offered a clear enhancement of DDI prediction (95% accuracy) with no false-negative or false-positive outcomes. The accuracies for the other three assays were 58, 84, and 74% for HLM, plated hHEPs, and DMEM hHEPs, respectively.
In this study [67••], a number of drugs were shown to give both reversible inhibition and TDI for CYP3A in the HLM assay (for example erlotinib, nilotinib, and pazopanib) but interestingly, these drugs were not inhibitory in the plasma HEP assay. While the clinical data confirmed low DDI due to CYP3A inhibition for these drugs as predicted by the plasma HEP assay, a more complete mechanistic understanding for the discrepancy between the two systems would be helpful when considering the differential sensitivities of the test systems. The traditional HLM TDI assay is robust, sensitive, and backed by a substantial body of published data [12, 68, 69] which can be used to rank and to some extent to predict CYP-mediated DDI during the discovery stage. It is however suggested to consider using the plasma hHEP TDI assay for an enhanced assessment of the potential DDI during the candidate selection and early stages of drug development as a de-risking approach for TDI-positive candidate compounds.
Challenges of DDI Prediction in Cases of Metabolism by “Unusual” CYP Enzymes or Non-CYP Enzymes
Despite advances in in vitro enzymology technologies, there continues to be much to learn about enzymes which, while unimportant in the metabolism of drugs in general, are key contributors to the metabolism of particular drug compounds. For example, the SGLT2 inhibitor tofogliflozin is metabolized by CYPs 2C18, 4A11, and 4F3B [70], and the multiple sclerosis drug fingolimod is metabolized by CYP4F enzymes [71]. These enzymes are usually regarded as “minor” CYP isoforms and would not routinely be included in enzyme phenotyping screens [10•, 72]. This raises the question of how one is to know that an important pathway is “missed” in initial in vitro assessments. Due to the availability of well-characterized and selective inhibitors for CYP isoforms, it may be apparent should the activities of routinely tested CYP enzymes not account for the majority of metabolism in vitro. In such cases, additional in vitro work using recombinantly expressed enzymes and (semi-)selective CYP inhibitors may be performed to try to obtain more clarity on enzyme contributions to metabolism, although this may prove challenging. DDI risks could then be addressed through screening of potential co-medicant substances either as inhibitors of the involved metabolic enzyme or, more empirically, as inhibitors of turnover of the drug in development itself. Due to a lack of system information (enzyme expression and activity levels, polymorphism status, effect of disease, ontogeny), it is unlikely that special population or polymorphism risk assessments can be made at this time.
The situation is even more challenging when “unusual” non-CYP enzymes are involved. In one recent example, an investigational trace amine-associated receptor antagonist RO5263397 was found to be principally cleared by UGT2B10 [73••]. At the time of compound selection, UGT2B10 was not considered an important enzyme in drug metabolism and was not commercially available for testing, and no selective inhibitors were characterized [74,75,76,77]. Co-administration with potent UGT2B10 inhibitors could potentially mimic the UGT2B10 poor metabolizer phenotype which resulted in a 136-fold higher AUC for one individual after a single 10 mg dose in a phase I clinical study [73••].
Such cases also provide substantial learning opportunities. As a result of this observation, a new splice site polymorphism was identified (prevalent in individuals of African origin but almost absent in Caucasians). This is relevant for clearance of other UGT2B10 substrates [78, 79]. In addition, increased understanding of the enzyme system and in vitro tools to assess UGT2B10 contribution to metabolism have been developed which can be rapidly employed in the future. In this way, UGT2B10 illustrates the process by which an enzyme not previously considered in drug metabolism testing progresses from being an “essentially uncharacterized” to a “largely characterized” metabolic enzyme system [80, 81]. A similar experience had been reported by Wang et al. for a Merck development compound MK-7246 which is cleared by polymorphic UGT2B17 [82]. It is likely that such learning experiences will be repeated as drug development continues to move into areas of novel chemical space in pursuit of new drug targets and further examples are discovered where previously little studied enzymes are important for individual drug clearance.
Future Prospects for DDI Prediction
To date, most in vitro systems used in DDI prediction have employed short timescale incubations to generate mechanistic parameters which can then be used to build up long-term model predictions of DDIs in vivo. Short timescale incubations cannot however address issues such as enzyme inactivation by highly metabolically stable compounds or the interplay of enzyme inactivation and induction which will drive the effective steady-state change in metabolic enzyme capacity. Although the sensitivity of short-term plated human hepatocytes to inhibition and induction has been demonstrated [83], such systems are unlikely to reflect steady-state conditions due to the transient nature of the cell cultures used. The advent of long-term hepatocyte culture systems may allow effective in vitro pharmacokinetic assessments to be made which will better reflect the clinical situation. The potential of long-term hepatocyte cultures has initially been demonstrated for clearance assessment of metabolically stable compounds [84,85,86,87••]. Their application to more sophisticated ADME assessments, such as induction [88], the effect of active uptake on apparent induction potency [89•], metabolism profiling/cross-species comparison [90], and to a limited extent for drug-drug interactions [85] have also been demonstrated. New long-term hepatocyte systems may therefore offer a completely new opportunity to simultaneously study multiple processes involved in drug-drug interactions which were not previously possible in vitro, especially for highly metabolically stable compounds. The development of long-term hepatocyte systems may also be seen as a first step in the direction of functional in vitro test systems with cells from multiple organs such as the liver, intestine, kidney, skin, and brain [91, 92], within a single test system (“chip”). When validated, data from the new experimental systems will quickly be incorporated into PBPK-based modeling tools further enhancing prediction of clinical DDIs.
Conclusions
This review has drawn upon personal experiences and recent literature reports to highlight achievements and ongoing challenges in the rapidly developing areas of metabolic DDI assessment, prediction, interpretation, and drug product labeling. Examples have been shown of how a model-based approach to understanding DDIs has progressed from data integration (bitopertin) to being accepted for interpolative (ibrutinib) and increasingly extrapolative DDI predictions (eliglustat and alectinib). Scientific confidence in and regulatory acceptance of PBPK modeling have increased with growing knowledge of DDIs, availability and robustness of in vitro test systems, and experience in DDI prediction. Predictions from well-executed analyses using validated models have enabled explicit dosing recommendations in product labels for clinical DDIs based on PBPK modeling in lieu of dedicated clinical DDI studies. Modeling approaches may indeed offer the only way to explore some potential DDIs where clinical investigation is unfeasible due to ethical considerations or the inability to recruit suitable study subjects.
The impact of characterizing major active metabolites during drug development has also been exemplified in the case of alectinib. This has been shown to be critical in the interpretation of clinical DDIs where exposure changes occur to both the parent and an active metabolite and are relevant to clinical efficacy and safety. Understanding the pharmacological, pharmacokinetic, and disposition properties of a metabolite using in vitro and in vivo studies can allow for estimation of its contribution in clinical DDI interpretation and subsequently its potential impact on clinical efficacy and safety in support of appropriate dosing recommendations.
A sometimes underemphasized factor affecting DDI predictions is the availability of good quality clinical DDI data with which can be used for validation purposes. This is especially the case for drugs predominantly metabolized by “unusual” CYP enzymes or non-CYP enzymes. Examples where CYP4F enzymes or UGT2B10 catalyze drug clearance have been discussed. We can expect that the continued development of experimental techniques and the increases in knowledge of enzymology and DDI will be reflected in increased DDI prediction confidence for such drugs.
Use of a whole plasma human hepatocyte TDI assay has shown to improve in vitro-in vivo extrapolation. Further TDI assay developments are anticipated using long-term hepatocyte culture systems and other “organs on a chip” technologies. These offer the promise of in vitro systems where an integrated assessment of enzyme inhibition, inactivation, and induction can be made. The ability to use the same modeling approaches to understand such “in vitro pharmacokinetics/DDI” experiments and then directly transfer this understanding to the human DDI situation may allow a further step forward in DDI prediction to be made in the near future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–9. doi:10.1007/s40495-016-0059-9.
Yu J, Ritchie TK, Mulgaonkar A, Ragueneau-Majlessi I. Drug disposition and drug-drug interaction data in 2013 FDA new drug applications: a systematic review. Drug Metab Dispos. 2014;42(12):1991–2001. doi:10.1124/dmd.114.060392.
Yu J, Ritchie TK, Zhou Z, Ragueneau-Majlessi I. Key findings from preclinical and clinical drug interaction studies presented in new drug and biological license applications approved by the Food and Drug Administration in 2014. Drug Metab Dispos. 2016;44(1):83–101. doi:10.1124/dmd.115.066720.
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43(11):1823–37. doi:10.1124/dmd.115.065920.
•• Yu J, Zhou Z, Owens KH, Ritchie TK, Ragueneau-Majlessi I. What can be learned from recent new drug applications? A systematic review of drug interaction data for drugs approved by the US FDA in 2015. Drug Metab Dispos. 2017;45(1):86–108. doi:10.1124/dmd.116.073411. Highly informative overview of NDA information from 2015, covering many data which are not published as journal articles.
• Rowland M, Matin SB. Kinetics of drug-drug interactions. J Pharmacokinet Biopharm. 1973;1(6):553–67. A classic paper and relevant reminder of where the static mechanistic modelling came from and its original basis in half-life change estimation.
Boulenc X, Nicolas O, Hermabessiere S, Zobouyan I, Martin V, Donazzolo Y, et al. CYP3A4-based drug-drug interaction: CYP3A4 substrates’ pharmacokinetic properties and ketoconazole dose regimen effect. Eur J Drug Metab Pharmacokinet. 2016;41(1):45–54. doi:10.1007/s13318-014-0235-4.
Obach RS. Predicting drug-drug interactions from in vitro drug metabolism data: challenges and recent advances. Curr Opin Drug Discov Devel. 2009;12(1):81–9.
Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, et al. Comparison of different algorithms for predicting clinical drug-drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37(8):1658–66. doi:10.1124/dmd.108.026252.
• Bohnert T, Patel A, Templeton I, Chen Y, Lu C, Lai G, et al. Evaluation of a new molecular entity as a victim of metabolic drug-drug interactions—an industry perspective. Drug Metab Dispos. 2016;44(8):1399–423. doi:10.1124/dmd.115.069096. An excellent overview of current practices for metabolic enzyme phenotyping work.
Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi:10.1146/annurev-pharmtox-010510-100540.
Grimm SW, Einolf HJ, Hall SD, He K, Lim HK, Ling KH, et al. The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America. Drug Metab Dispos. 2009;37(7):1355–70. doi:10.1124/dmd.109.026716.
Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277(1):423–31.
Eagling VA, Back DJ, Barry MG. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J Clin Pharmacol. 1997;44(2):190–4.
Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, et al. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab Dispos. 1998;26(6):552–61.
Kharasch ED, Mitchell D, Coles R, Blanco R. Rapid clinical induction of hepatic cytochrome P4502B6 activity by ritonavir. Antimicrob Agents Chemother. 2008;52(5):1663–9. doi:10.1128/AAC.01600-07.
Alberati D, Moreau JL, Lengyel J, Hauser N, Mory R, Borroni E, et al. Glycine reuptake inhibitor RG1678: a pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology. 2012;62(2):1152–61. doi:10.1016/j.neuropharm.2011.11.008.
Winter M, Funk J, Korner A, Alberati D, Christen F, Schmitt G, et al. Effects of GlyT1 inhibition on erythropoiesis and iron homeostasis in rats. Exp Hematol. 2016;44(10):964–74. e4 doi:10.1016/j.exphem.2016.07.003.
• Boetsch C, Parrott N, Fowler S, Poirier A, Hainzl D, Banken L, et al. Effects of cytochrome P450 3A4 inhibitors-ketoconazole and erythromycin-on bitopertin pharmacokinetics and comparison with physiologically based modelling predictions. Clin Pharmacokinet. 2016;55(2):237–47. doi:10.1007/s40262-015-0312-0. The publication detailing bitopertin DDIs for readers wanting to simulate a classical DDI using the data given in the Supplementary information.
Parrott N, Hainzl D, Alberati D, Hofmann C, Robson R, Boutouyrie B, et al. Physiologically based pharmacokinetic modelling to predict single- and multiple-dose human pharmacokinetics of bitopertin. Clin Pharmacokinet. 2013;52(8):673–83. doi:10.1007/s40262-013-0061-x.
Parrott N, Hainzl D, Scheubel E, Krimmer S, Boetsch C, Guerini E, et al. Physiologically based absorption modelling to predict the impact of drug properties on pharmacokinetics of bitopertin. AAPS J. 2014;16(5):1077–84. doi:10.1208/s12248-014-9639-y.
Grillo JA, Zhao P, Bullock J, Booth BP, Lu M, Robie-Suh K, et al. Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos. 2012;33(2):99–110. doi:10.1002/bdd.1771.
Huang SM, Abernethy DR, Wang Y, Zhao P, Zineh I. The utility of modeling and simulation in drug development and regulatory review. J Pharm Sci. 2013;102(9):2912–23. doi:10.1002/jps.23570.
Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism, and excretion of oral (1)(4)C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43(2):289–97. doi:10.1124/dmd.114.060061.
de Vries R, Smit JW, Hellemans P, Jiao J, Murphy J, Skee D, et al. Stable isotope-labelled intravenous microdose for absolute bioavailability and effect of grapefruit juice on ibrutinib in healthy adults. Br J Clin Pharmacol. 2016;81(2):235–45. doi:10.1111/bcp.12787.
de Zwart L, Snoeys J, De Jong J, Sukbuntherng J, Mannaert E, Monshouwer M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Ther. 2016;100(5):548–57. doi:10.1002/cpt.419.
NDA 205552 (Ibrutinib): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205552Orig2s000ClinPharmR.pdf. Accessed: 28 Nov 2016
de Jong J, Skee D, Murphy J, Sukbuntherng J, Hellemans P, Smit J, et al. Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol Res Perspect. 2015;3(4):e00156. doi:10.1002/prp2.156.
IMBRUVICA™: Prescribing information. U. S. Food and Drug Administration. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205552Orig2lbl.pdf. Accessed: 28 Nov 2016
IMBRUVICA™: Summary basis of decision. Health Canada. 2015. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2015_imbruvica_174029-eng.php. Accessed: 28 Nov 2016
IMBRUVICA™: EPAR—product information. European Medicines Agency. 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003791/WC500177775.pdf. Accessed: 28 Nov 2016
IMBRUVICA™: Regulatory decision [in Japanese]. Ministry of Health, Labour and Welfare, Pharmaceuticals and Medical Devices Agency. 2016. Available from: http://www.pmda.go.jp/drugs/2016/P20160404001/800155000_22800AMX00387_A100_1.pdf Accessed: 28 Nov 2016
NDA 205494 (CERDELGA®): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205494Orig1s000ClinPharmR.pdf. Accessed: 28 Nov 2016
CERDELGA™: Prescribing information. U. S. Food and Drug Administration. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205494Orig1s000lbl.pdf. Accessed: 28 Nov 2016
Gan J, Chen W, Shen H, Gao L, Hong Y, Tian Y, et al. Repaglinide-gemfibrozil drug interaction: inhibition of repaglinide glucuronidation as a potential additional contributing mechanism. Br J Clin Pharmacol. 2010;70(6):870–80. doi:10.1111/j.1365-2125.2010.03772.x.
Sall C, Houston JB, Galetin A. A comprehensive assessment of repaglinide metabolic pathways: impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab Dispos. 2012;40(7):1279–89. doi:10.1124/dmd.112.045286.
Ito K, Hallifax D, Obach RS, Houston JB. Impact of parallel pathways of drug elimination and multiple cytochrome P450 involvement on drug-drug interactions: CYP2D6 paradigm. Drug Metab Dispos. 2005;33(6):837–44. doi:10.1124/dmd.104.003715.
Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Dose-dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab Dispos. 2011;39(10):1977–86. doi:10.1124/dmd.111.040931.
Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46(3):347–51. doi:10.1007/s00125-003-1034-7.
Tornio A, Niemi M, Neuvonen M, Laitila J, Kalliokoski A, Neuvonen PJ, et al. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h after the dose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther. 2008;84(3):403–11. doi:10.1038/clpt.2008.34.
Niemi M, Kajosaari LI, Neuvonen M, Backman JT, Neuvonen PJ. The CYP2C8 inhibitor trimethoprim increases the plasma concentrations of repaglinide in healthy subjects. Br J Clin Pharmacol. 2004;57(4):441–7. doi:10.1046/j.1365-2125.2003.02027.x.
Kajosaari LI, Niemi M, Backman JT, Neuvonen PJ. Telithromycin, but not montelukast, increases the plasma concentrations and effects of the cytochrome P450 3A4 and 2C8 substrate repaglinide. Clin Pharmacol Ther. 2006;79(3):231–42. doi:10.1016/j.clpt.2005.11.002.
ALECENSA®: Prescribing information. U. S. Food and Drug Administration. 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed: 28 Nov 2016
Morcos PN, Cleary Y, Guerini E, Dall G, Bogman K, De Petris L, et al. Clinical drug-drug interactions through cytochrome P450 3A (CYP3A) for the selective ALK inhibitor alectinib. Clin Pharmacol Drug Dev. 2016; doi:10.1002/cpdd.298.
Sekiguchi N, Nagao S, Takanashi K, Kato M, Kaneko A, Morita K, et al. Preclinical evaluation of the potential for cytochrome P450 inhibition and induction of the selective ALK inhibitor, alectinib. Xenobiotica. 2016:1–10. doi:10.1080/00498254.2016.1261308.
Cleary Y, Gertz M, Morcos P, Youdim K, Fowler S, Yu L et al. Physiologically-based pharmacokinetic (PBPK) modelling to evaluate drug-drug interaction (DDI) risk of alectinib. American Society for Clinical Pharmacology and Therapeutics - 118th Annual Meeting 2017.
Guidance for Industry: Safety testing of drug metabolites. Revision 1. U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2016. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079266.pdf. Accessed: 06 Jan 2017
Guidance for Industry: Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations [draft guidance]. U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2012. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed: 28 Nov 2016
Guidance for Industry: Safety testing of drug metabolites U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2008. Available from: http://www.fda.gov/OHRMS/DOCKETS/98fr/FDA-2008-D-0065-GDL.pdf. Accessed: 28 Nov 2016
Guidance for Industry M3(R2): Nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. Revision 1. U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2010. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073246.pdf. Accessed: 28 Nov 2016
Guidance for Industry M3(R2): Nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. Questions and Answers (R2). Revision 1. U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2013. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292340.pdf. Access Ibrutinibed: 28 Nov 2016
Nakagawa T, Takanashi K, Hoshino-Yoshino A, Yamauchi T, Kawashima K, Tachibana Y et al. In vitro metabolism of alectinib, a novel highly potent ALK inhibitor: contribution of CYP3A enzymes in microsomes and hepatocytes. International Society for the Study of Xenobiotics (ISSX). 2016; Busan, South Korea 2016.
Hsu J, Carnac R, Henschel V, Bogman K, Martin-Facklam M, Guerini E et al. Population pharmacokinetics (popPK) and exposure-response (ER) analyses to confirm alectinib 600 mg BID dose selection in a crizotinib-progressed or intolerant population. J Clin Oncol 2016;34:(suppl; abstr e20598).
NDA 208434 (ALECENSA®): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208434Orig1s000ClinPharmR.pdf. Accessed: 28 Nov 2016
NDA 208434 (ALECENSA®): Pharmacology review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208434Orig1s000PharmR.pdf. Accessed: 28 Nov 2016
NDA 203085 (STIVARGA®): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2012. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000ClinPharmR.pdf. Accessed: 28 Nov 2016
NDA 202806 (TAFINLAR®): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/202806Orig1s000ClinPharmR.pdf. Accessed: 28 Nov 2016
NDA 21-938/21-968 (SUTENTTM): Clinical pharmacology and biopharmaceutics review(s). U. S. Food and Drug Administration, Center for Drug Evaluation and Research. 2006. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021938_S000_Sutent_BioPharmR.pdf. Accessed: 28 Nov 2016
Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66(2):357–71. doi:10.1007/s00280-009-1170-y.
Shi JG, Chen X, Emm T, Scherle PA, McGee RF, Lo Y, et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J Clin Pharmacol. 2012;52(6):809–18. doi:10.1177/0091270011405663.
Kosoglou T, Statkevich P, Johnson-Levonas AO, Paolini JF, Bergman AJ, Alton KB. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44(5):467–94.
Fowler S, Zhang H. In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: current status on methodologies and their utility for predicting drug-drug interactions. AAPS J. 2008;10(2):410–24. doi:10.1208/s12248-008-9042-7.
Mao J, Johnson TR, Shen Z, Yamazaki S. Prediction of crizotinib-midazolam interaction using the Simcyp population-based simulator: comparison of CYP3A time-dependent inhibition between human liver microsomes versus hepatocytes. Drug Metab Dispos. 2013;41(2):343–52. doi:10.1124/dmd.112.049114.
Mao J, Mohutsky MA, Harrelson JP, Wrighton SA, Hall SD. Prediction of CYP3A-mediated drug-drug interactions using human hepatocytes suspended in human plasma. Drug Metab Dispos. 2011;39(4):591–602. doi:10.1124/dmd.110.036400.
Mao J, Mohutsky MA, Harrelson JP, Wrighton SA, Hall SD. Predictions of cytochrome P450-mediated drug-drug interactions using cryopreserved human hepatocytes: comparison of plasma and protein-free media incubation conditions. Drug Metab Dispos. 2012;40(4):706–16. doi:10.1124/dmd.111.043158.
Lu C, Miwa GT, Prakash SR, Gan LS, Balani SK. A novel model for the prediction of drug-drug interactions in humans based on in vitro cytochrome p450 phenotypic data. Drug Metab Dispos. 2007;35(1):79–85. doi:10.1124/dmd.106.011346.
•• Mao J, Tay S, Khojasteh CS, Chen Y, Hop CE, Kenny JR. Evaluation of time dependent inhibition assays for marketed oncology drugs: comparison of human hepatocytes and liver microsomes in the presence and absence of human plasma. Pharm Res. 2016;33(5):1204–19. doi:10.1007/s11095-016-1865-9. Development of good in vitro–in vivo correlations using the “full plasma” short-term suspension culture CYP3A4 TDI assay system.
Zimmerlin A, Trunzer M, Faller B. CYP3A time-dependent inhibition risk assessment validated with 400 reference drugs. Drug Metab Dispos. 2011;39(6):1039–46. doi:10.1124/dmd.110.037911.
Orr ST, Ripp SL, Ballard TE, Henderson JL, Scott DO, Obach RS, et al. Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J Med Chem. 2012;55(11):4896–933. doi:10.1021/jm300065h.
Yamane M, Kawashima K, Yamaguchi K, Nagao S, Sato M, Suzuki M, et al. In vitro profiling of the metabolism and drug-drug interaction of tofogliflozin, a potent and highly specific sodium-glucose co-transporter 2 inhibitor, using human liver microsomes, human hepatocytes, and recombinant human CYP. Xenobiotica. 2015;45(3):230–8. doi:10.3109/00498254.2014.976296.
Jin Y, Zollinger M, Borell H, Zimmerlin A, Patten CJ. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug Metab Dispos. 2011;39(2):191–8. doi:10.1124/dmd.110.035378.
Zientek MA, Youdim K. Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos. 2015;43(1):163–81. doi:10.1124/dmd.114.058750.
•• Fowler S, Kletzl H, Finel M, Manevski N, Schmid P, Tuerck D, et al. A UGT2B10 splicing polymorphism common in African populations may greatly increase drug exposure. J Pharmacol Exp Ther. 2015;352(2):358–67. doi:10.1124/jpet.114.220194. The largest so far reported clinical effect of a UGT polymorphism on exposure. Also highlights high ethnic variation in polymorphism frequencies and the need to consider this in clinical trials.
Kaivosaari S, Toivonen P, Aitio O, Sipila J, Koskinen M, Salonen JS, et al. Regio- and stereospecific N-glucuronidation of medetomidine: the differences between UDP glucuronosyltransferase (UGT) 1A4 and UGT2B10 account for the complex kinetics of human liver microsomes. Drug Metab Dispos. 2008;36(8):1529–37. doi:10.1124/dmd.108.021709.
Kaivosaari S, Toivonen P, Hesse LM, Koskinen M, Court MH, Finel M. Nicotine glucuronidation and the human UDP-glucuronosyltransferase UGT2B10. Mol Pharmacol. 2007;72(3):761–8. doi:10.1124/mol.107.037093.
Kazmi F, Yerino P, Barbara JE, Parkinson A. Further characterization of the metabolism of desloratadine and its cytochrome P450 and UDP-glucuronosyltransferase inhibition potential: identification of desloratadine as a relatively selective UGT2B10 inhibitor. Drug Metab Dispos. 2015;43(9):1294–302. doi:10.1124/dmd.115.065011.
Pattanawongsa A, Nair PC, Rowland A, Miners JO. Human UDP-glucuronosyltransferase (UGT) 2B10: validation of cotinine as a selective probe substrate, inhibition by UGT enzyme-selective inhibitors and antidepressant and antipsychotic drugs, and structural determinants of enzyme inhibition. Drug Metab Dispos. 2016;44(3):378–88. doi:10.1124/dmd.115.068213.
Kaivosaari S, Finel M, Koskinen M. N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica. 2011;41(8):652–69. doi:10.3109/00498254.2011.563327.
Kato Y, Izukawa T, Oda S, Fukami T, Finel M, Yokoi T, et al. Human UDP-glucuronosyltransferase (UGT) 2B10 in drug N-glucuronidation: substrate screening and comparison with UGT1A3 and UGT1A4. Drug Metab Dispos. 2013;41(7):1389–97. doi:10.1124/dmd.113.051565.
Kazmi F, Barbara JE, Yerino P, Parkinson A. A long-standing mystery solved: the formation of 3-hydroxydesloratadine is catalyzed by CYP2C8 but prior glucuronidation of desloratadine by UDP-glucuronosyltransferase 2B10 is an obligatory requirement. Drug Metab Dispos. 2015;43(4):523–33. doi:10.1124/dmd.114.062620.
Gradinaru J, Romand S, Geiser L, Carrupt PA, Spaggiari D, Rudaz S. Inhibition screening method of microsomal UGTs using the cocktail approach. Eur J Pharm Sci. 2015;71:35–45. doi:10.1016/j.ejps.2015.02.001.
Wang YH, Trucksis M, McElwee JJ, Wong PH, Maciolek C, Thompson CD, et al. UGT2B17 genetic polymorphisms dramatically affect the pharmacokinetics of MK-7246 in healthy subjects in a first-in-human study. Clin Pharmacol Ther. 2012;92(1):96–102. doi:10.1038/clpt.2012.20.
Beumer JH, Pillai VC, Parise RA, Christner SM, Kiesel BF, Rudek MA, et al. Human hepatocyte assessment of imatinib drug-drug interactions—complexities in clinical translation. Br J Clin Pharmacol. 2015;80(5):1097–108. doi:10.1111/bcp.12723.
Bonn B, Svanberg P, Janefeldt A, Hultman I, Grime K. Determination of human hepatocyte intrinsic clearance for slowly metabolized compounds: comparison of a primary hepatocyte/stromal cell co-culture with plated primary hepatocytes and HepaRG. Drug Metab Dispos. 2016;44(4):527–33. doi:10.1124/dmd.115.067769.
Lin C, Shi J, Moore A, Khetani SR. Prediction of drug clearance and drug-drug interactions in microscale cultures of human hepatocytes. Drug Metab Dispos. 2016;44(1):127–36. doi:10.1124/dmd.115.066027.
Schaefer M, Schanzle G, Bischoff D, Sussmuth RD. Upcyte human hepatocytes: a potent in vitro tool for the prediction of hepatic clearance of metabolically stable compounds. Drug Metab Dispos. 2016;44(3):435–44. doi:10.1124/dmd.115.067348.
•• Kratochwil N, Meille C, Fowler S, Klammers F, Ekiciler A, Molitor B et al. Metabolic profiling of human long-term liver models and hepatic clearance predictions from in vitro data using nonlinear mixed-effects modeling. AAPS J. 2016; in press. Most comprehensive survey to date of drug metabolizing enzyme activities in different long-term hepatocyte culture systems.
Dixit V, Moore A, Tsao H, Hariparsad N. Application of micropatterned cocultured hepatocytes to evaluate the inductive potential and degradation rate of major xenobiotic metabolizing enzymes. Drug Metab Dispos. 2016;44(2):250–61. doi:10.1124/dmd.115.067173.
• Moore A, Chothe PP, Tsao H, Hariparsad N. Evaluation of the interplay between uptake transport and CYP3A4 induction in micropatterned cocultured hepatocytes. Drug Metab Dispos. 2016;44(12):1910–9. doi:10.1124/dmd.116.072660. A very nice piece of work showing where the in vitro field is developing into the use of more sophisticated test systems which incorporate multiple DDI processes.
Ballard TE, Wang S, Cox LM, Moen MA, Krzyzewski S, Ukairo O, et al. Application of a micropatterned cocultured hepatocyte system to predict preclinical and human-specific drug metabolism. Drug Metab Dispos. 2016;44(2):172–9. doi:10.1124/dmd.115.066688.
Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip. 2015;15(12):2688–99. doi:10.1039/c5lc00392j.
Materne EM, Ramme AP, Terrasso AP, Serra M, Alves PM, Brito C, et al. A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing. J Biotechnol. 2015;205:36–46. doi:10.1016/j.jbiotec.2015.02.002.
Kalliokoski A, Backman JT, Kurkinen KJ, Neuvonen PJ, Niemi M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin Pharmacol Ther. 2008;84(4):488–96.
Backman JT, Honkalammi J, Neuvonen M, Kurkinen KJ, Tornio A, Niemi M, et al. CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: estimation of CYP2C8 half-life using repaglinide as an in vivo probe. Drug Metab Dispos. 2009;37(12):2359–66. doi:10.1124/dmd.109.029728.
Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Gemfibrozil is a strong inactivator of CYP2C8 in very small multiple doses. Clin Pharmacol Ther. 2012;91(5):846–55. doi:10.1038/clpt.2011.313.
Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Mechanism-based inactivation of CYP2C8 by gemfibrozil occurs rapidly in humans. Clin Pharmacol Ther. 2011;89(4):579–86. doi:10.1038/clpt.2010.358.
Tornio A, Filppula AM, Kailari O, Neuvonen M, Nyronen TH, Tapaninen T, et al. Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug-drug interactions. Clin Pharmacol Ther. 2014;96(4):498–507. doi:10.1038/clpt.2014.141.
Kajosaari LI, Niemi M, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin Pharmacol Ther. 2005;78(4):388–99. doi:10.1016/j.clpt.2005.07.005.
AUBAGIO®: Prescribing information. U. S. Food and Drug Administration. 2012. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf. Accessed: 07 Dec 2016
Niemi M, Neuvonen PJ, Kivisto KT. The cytochrome P4503A4 inhibitor clarithromycin increases the plasma concentrations and effects of repaglinide. Clin Pharmacol Ther. 2001;70(1):58–65. doi:10.1067/mcp.2001.116511.
Bidstrup TB, Damkier P, Olsen AK, Ekblom M, Karlsson A, Brosen K. The impact of CYP2C8 polymorphism and grapefruit juice on the pharmacokinetics of repaglinide. Br J Clin Pharmacol. 2006;61(1):49–57. doi:10.1111/j.1365-2125.2005.02516.x.
Hachad H, Ragueneau-Majlessi I, Levy RH. A useful tool for drug interaction evaluation: the University of Washington Metabolism and Transport Drug Interaction Database. Hum Genomics. 2010;5(1):61–72.
Acknowledgments
The authors thank Franz Schuler and Christoph Funk (Roche, Basel) for helpful suggestions and Alexander Nürnberg (Roche, Basel) for his diligent assistance in preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors are employees of F. Hoffmann-La Roche Ltd. There are no conflicts of interest to declare.
Human and Animal Rights and Informed Consent
This article does not contain previously unpublished studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Drug Disposition
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fowler, S., Morcos, P.N., Cleary, Y. et al. Progress in Prediction and Interpretation of Clinically Relevant Metabolic Drug-Drug Interactions: a Minireview Illustrating Recent Developments and Current Opportunities. Curr Pharmacol Rep 3, 36–49 (2017). https://doi.org/10.1007/s40495-017-0082-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-017-0082-5