
Abstract
Acute myeloid leukemia (AML) is a complex disease characterized by genetic and clinical heterogeneity and high mortality. After 40 years during which the standard of care for patients evolved very little, the therapeutic landscape has recently seen rapid changes, with the approval of eight new drugs by the Food and Drug Administration (FDA) within the last 2 years, providing new opportunities, as well as new challenges, for treating clinicians. These therapies include FLT3 inhibitors midostaurin and gilteritinib, CPX-351 (liposomal cytarabine and daunorubicin), gemtuzumab ozogamicin (GO, anti-CD33 monoclonal antibody conjugated with calicheamicin), IDH1/IDH2 inhibitors ivosidenib and enasidenib, Hedgehog inhibitor glasdegib, and BCL-2 inhibitor venetoclax. In this review, we summarize currently available data on these new drugs and discuss the rapidly evolving therapeutic armamentarium for AML, focusing on targeted therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
After no substantial innovations in 40 years, the scenario of treatment in acute myeloid leukemia (AML) has recently seen a number of changes, with FDA approval of eight new non-cytostatic compounds. |
The new FDA-approved targeted therapies are midostaurin, gilteritinib, glasdegib, ivosidenib, enasidenib, venetoclax, and gemtuzumab ozogamicin. |
RATIFY is the first randomized trial to show that the combination of targeted therapy with standard chemotherapy significantly improves survival in AML. |
Phase II trials are currently evaluating new drugs targeting EZH2, DOT1L, MLL, BET, and LSD1. |
Background
The so-called 7+3 schedule (cytarabine 100–200 mg/m2 on days 1–7 and daunorubicin 60 mg/m2 on days 1–3) has been the standard first-line induction therapy in adults with acute myeloid leukemia (AML) for 40 years. However, in recent years, the scenario of treatment in AML has changed substantially, with approval by the US Food and Drug Administration (FDA) of eight non-cytostatic compounds, able to interact with specific targets of different AML subtypes. Leukemogenesis is characterized by multiple somatically acquired mutations that affect genes of different functional categories. Mutations in genes encoding epigenetic modifiers, such as DNMT3A, ASXL1, TET2, IDH1, and IDH2, are commonly acquired early and can be found in the founding clone; such mutations may persist after therapy, lead to clonal expansion during hematological remission, and eventually lead to relapse. In contrast, mutations involving NPM1 or signaling molecules (e.g., FLT3, RAS) are typical secondary events that occur later during leukemogenesis; in many cases their selective inhibition may involve only a leukemia subclone, sparing other clones, and these clonal relationships need to be taken into account when designing clinical trials with molecular-targeted agents.
Immunotherapy with GO was the first targeted therapy explored and the last to be approved by the FDA, due to the non-hematological toxicity observed in patients treated with higher-dose regimens. Unfortunately, the sensitivity of calicheamicin to the drug extrusion mechanism, mediated by P-glycoprotein (PgP), strongly limited its efficacy in AML patients with unfavorable cytogenetics, and experimental studies with second-generation anti-CD33 antibodies were interrupted because of hematological toxicity.
In a seminal pivotal study [1], 1540 AML patients were studied extensively, and six patterns of co-occurrence and mutual exclusivity of genetic changes were identified:
-
AML with balanced rearrangements
-
AML with NPM1 mutation
-
AML with mutation of genes that regulate chromatin (ASXL1, STAG2, BCOR, KMT2A PTD [partial tandem duplication], EZH2, and PHF6), RNA splicing (SRSF2, SF3B1, U2AF1, and ZRSR2), or both, or transcription (RUNX1)
-
AML with IDH1 or IDH2 mutation
-
AML with TP53 mutation, chromosomal aneuploidy, or both
Thanks to these achievements, AML now represents an important field for investigation of new drugs such as tyrosine kinase inhibitors (TKI), epigenetic modulators, immune checkpoint inhibitors, mitochondrial inhibitors, and molecules targeting specific oncogenic proteins and the AML microenvironment; among them, the FLT3 inhibitors midostaurin and gilteritinib, the anti-Hedgehog pathway glasdegib, the anti-IDH1 ivosidenib, the anti-IDH2 enasidenib, and the anti-BCL2 venetoclax were recently approved by the FDA for AML treatment [2].
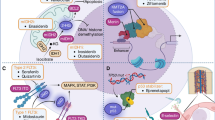
A schematic view of the mechanism of action of recently FDA-approved AML-targeted therapies is shown in Fig. 1 (FLT3 inhibitors), Fig. 2 (glasdegib, ivosidenib, and enasidenib), and Fig. 3 (venetoclax and GO).

Mechanism of action of FLT3 inhibitors

Mechanism of action of glasdegib, ivosidenib, and enasidenib

Mechanism of action of GO and venetoclax
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
FLT3 Inhibitors
The class III receptor tyrosine kinase (RTK) FMS-like tyrosine kinase 3 (FLT3) plays a key role in myelopoiesis. Two different types of FLT3 mutations have been detected in about 30% of AML patients: the internal tandem duplication (ITD) in the juxtamembrane region, and point mutations in the tyrosine kinase domain (TKD), mainly involving codons D835 and I836.
These mutations lead to ligand-independent activation of the receptor promoting proliferation, survival, and resistance to apoptosis of leukemic stem cells [3].
Several type I (binding the gatekeeper domain) or type II (binding the activation loop) tyrosine kinase inhibitors have been investigated, with variable pharmacokinetics, selectivity for FLT3, and in vitro efficacy [4].
First-generation FLT3 inhibitors (midostaurin, lestaurtinib, sorafenib) are multi-targeted kinase inhibitors, showing potent in vitro inhibition of mutant FLT3. Clinically, they have shown insufficient activity as single agents [5] but interesting synergy with chemotherapy [6, 7].
Second-generation FLT3 inhibitors, such as quizartinib, crenolanib, and gilteritinib, are more selective and more potent than midostaurin and are currently in clinical development.
Results in Upfront Therapy
Midostaurin is a multi-kinase inhibitor, initially developed as a protein kinase C inhibitor and later as an inhibitor of VEGF/angiogenesis [8] and FLT3 [9, 10].
The CALGB 10603/RATIFY trial, a multicenter phase III study, enrolled 717 adult patients (18–59 years of age) with newly diagnosed AML and FLT3 ITD or TKD mutation [11]; patients were randomized to receive standard induction chemotherapy ± midostaurin 50 mg orally b.i.d., given on days 8–22, and four courses of high-dose cytarabine (HDAC) ± midostaurin as consolidation. The trial also included a 12-month maintenance phase of midostaurin/placebo.
Patients were allowed to receive post-consolidation with allogeneic hematopoietic stem cell transplantation (HSCT), but in this case maintenance with midostaurin was not permitted.
The rate of complete remission (CR) was similar between the two arms (58.9% vs. 53.5%; p = 0.15), but the overall survival (OS), the study primary endpoint, was 74.7 months (median) in the midostaurin group, significantly better than 25.6 months in the placebo group (HR = 0.78, p = 0.009). Also, both event-free survival (EFS) and disease-free survival (DFS) were significantly better in the midostaurin arm (8.2 vs. 3.0 months, p = 0.002; 26.7 vs. 15.5 months, p = 0.01, respectively), with a 21.6% lower risk of relapse (HR = 0.78, p = 0.002).
The better outcome was not related to the FLT3 mutational site (ITD vs. TKD) or the FLT3-ITD/wild-type (WT) ratio; 28.1% of AML patients treated with midostaurin underwent HSCT during their first CR, versus 22.7% in the placebo arm (p = 0.10). Patients in the midostaurin arm receiving transplants demonstrated a trend toward improved 4-year OS in comparison with those receiving placebo (63.7% vs. 55.7%; p = 0.08); the benefit of midostaurin was observed only in patients who received HSCT during the first remission and not at later time points.
Most of the adverse events were similar between the two arms, and no significant treatment-related adverse events (TRAE) of grade ≥ 3 were reported in the midostaurin arm, with only a slightly increased incidence of rash, nausea, and anemia.
RATIFY was the first randomized trial to show that the combination of a targeted therapy with standard chemotherapy significantly improved survival in AML.
Based on this pivotal trial, on April 28, 2017, the FDA approved midostaurin in combination with intensive induction and consolidation therapy for patients with FLT3-mutant, newly diagnosed AML, while in Europe it was approved for induction, consolidation, and maintenance (Table 1).
Sorafenib is an oral multikinase inhibitor of RAF-1, VEGF, c-KIT, PDGFR, ERK, and FLT3. Currently, sorafenib is approved for hepatocellular carcinoma and renal cell carcinoma, but also has a potent anti-leukemic effect on FLT3-mutated AML. In a previous study, sorafenib in combination with intensive chemotherapy failed to increase OS [12], but in a subsequent phase III trial, sorafenib prolonged OS and relapse-free survival (RFS) when administered as maintenance after HSCT [13].
Quizartinib is a selective second-generation inhibitor of FLT3-WT and FLT3-ITD, without activity on FLT3-TKD. A phase III trial in which it is being administered with standard induction chemotherapy in younger adults with newly diagnosed FLT3-ITD-mutated AML is still ongoing (NCT02668653).
Crenolanib is a type-1 FLT3 inhibitor active against both FLT3-ITD- and FLT3-TKD-mutant AML, originally developed as a selective inhibitor of the platelet-derived growth factor receptors (PDGFR). It is also a potent inhibitor of mutated FLT3, particularly the secondary mutation D835 [14], which is one of the mechanisms of resistance to FLT3 inhibitors [15]. The addition of crenolanib (100 mg, three times/day) to standard 7+3 induction chemotherapy resulted in CR/incomplete count recovery (CRi) rates of 24/25 (96%) among patients with FLT3-mutant AML, and was able to overcome the poor prognostic impact of co-occurring driver mutations such as FLT3-ITD, NPM1, and DNMT3A [16, 17].
Gilteritinib, a pyrazinecarboxamide derivative also known as ASP-2215, is a selective and potent inhibitor of FLT3 [18]; when administered at doses ≥ 80 mg/day in combination with induction and consolidation chemotherapy, gilteritinib achieved CR/CRi rates of 89% in a phase I study [19].
Results in Relapsed/Refractory (R/R) AML
Gilteritinib and quizartinib have demonstrated a survival benefit compared with chemotherapy in prospective randomized trials in R/R patients: the ADMIRAL phase III trial (NCT02421939) randomized 138 adults with R/R AML with FLT3 ITD, D835, or I836 mutations to oral gilteritinib 120 mg daily versus investigators’ choice of low-dose cytarabine (LDAC), azacitidine, or second-line therapy [mitoxantrone, etoposide, and cytarabine (MEC), or fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA)]. The median OS in the gilteritinib arm was 9.3 months, compared with 5.6 months in patients who received standard chemotherapy (SC) [hazard ratio = 0.637 (95% CI 0.490, 0.830), p = 0.0007)]; 1-year OS was 37% in the gilteritinib arm versus 17% in the SC arm. The CR/CRh = CR with incomplete hematologic recovery: All CR criteria except for residual neutropenia (< 1.0 x 10e3/L) or thrombocytopenia (100 x 10e9/L) rates for gilteritinib and SC were 34% and 15.3%, respectively (p = 0.0001); CR rates were 21.1% and 10.5% (two-sided p = 0.0106). Median EFS was 2.8 months and 0.7 months in the gilteritinib and SC arms, respectively (HR 0.793, p = 0.0830) [20]. Based on the interim data [21], in November 2018 the FDA approved gilteritinib for the secondary treatment of AML in adults with a FLT3 mutation.
Quizartinib achieved positive results in the phase III QuANTUM-R trial, which randomized (2:1) 367 patients with FLT3-ITD allelic burden ≥ 3% to either single-agent quizartinib (n = 245; 60 mg, with a 30-mg lead-in of 15 days) or investigators’ choice chemotherapy (total = 122). Median OS improved from 4.7 to 6.2 months (HR 0.76; 95% CI 0.58–0.98; p = 0.0177) in favor of quizartinib. A higher response rate (composite CR 48% vs. 27%) was reported in the quizartinib arm, lasting a median of 12.1 months. The subsequent SCT rate was higher for patients receiving quizartinib (32 vs. 12%). Significant non-hematological grade ≥ 3 TRAEs were limited to QTc prolongation (10%) and reversible gastrointestinal symptoms [22]. However, the internal FDA analysis could not confirm a significant EFS benefit with quizartinib versus chemotherapy: median EFS was 6.0 weeks in the quizartinib arm (95% CI 0.1–8.3) versus 3.7 in the control arm (95% CI 0.4–6.0), respectively (HR 0.9; 95% CI 0.71–1.16; p = 0.114). Moreover, the FDA focused on cardiac toxicity as a key concern with quizartinib. Additional safety issues identified in the FDA analysis included potentially fatal differentiation syndrome, acute febrile neutrophilic dermatosis, and prolonged cytopenia [23].
A phase II study in relapsed/refractory FLT3-mutated AML showed that crenolanib at 200 mg three times/day continuously (28-day cycles) achieved 23% CR with CRi in naive patients, and 5% CR in patients previously treated with other FLT3 inhibitors [24].
IDH1 and IDH2 inhibitors
The isocitrate dehydrogenase (IDH) family is involved in the cellular energy pathway, by catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate. While IDH1 is localized in peroxisomes in the cytosol, IDH2 resides in mitochondria [25, 26].
IDH mutations occur in approximately 20% of AML patients [IDH1 (8%) and IDH2 (12%)], and are more common in the elderly (25–28%). They are usually associated with intermediate-risk cytogenetic, FLT3, and NPM1 mutations [27,28,29]; Bose et al. [30].
Somatic mutations in catalytically active arginine residues decrease their enzymatic activity and also confer a gain in functional activity, leading to the production of the oncometabolite 2-hydroxyglutarate (2-HG) instead of alpha-ketoglutarate (α-KG).
2-HG competitively inhibits the function of αKG-dependent oxygenases involved in DNA or histone demethylation, resulting in global DNA hypermethylation of regulatory genes and arrested myeloid differentiation. The mutation also increases reactive oxygen species (ROS) output and cell-cycle transition through the activation of MAPK (mitogen-activated protein kinase) signaling and the repression of cyclin-dependent kinase inhibitors Cdkn2a and Cdkn2b, resulting in metabolic changes leading to upregulation of NF-Kb and BCL-2 proteins [31,32,33,34,35,36].
Ivosidenib and enasidenib are two orally available selective inhibitors of mutant IDH1 and IDH2, and have been shown to decrease cellular 2-HG production by more than 90%, thus reducing histone and DNA hypermethylation and inducing myeloid differentiation [37, 38].
Results in R/R AML
Enasidenib, formerly known as AG-221, binds to mutated IDH2, reducing its affinity to NADPH, leading to impaired catalytic activity. It was administered at 100 mg/day in a phase I/II trial [39] in 239 patients (median age 67 years, range 19–100) with mutant IDH2 and advanced myeloid malignancies. The maximum tolerated dose was not reached. The same dose (FDA-approved) was designated for the expansion phase (n = 126) based on favorable pharmacokinetic and pharmacodynamic profiles.
Among patients with R/R AML (n = 176), the ORR was 40.3%, while complete clinical remission (cCR) and CR rates were 26% and 19%, respectively. The median time to first response was 1.9 months (range 0.5–9.4 months) and median time to CR was 3.7 months (range 0.7–11.2). The median response duration was 5.6 months (range 3.8–9.7). Among those patients achieving CR, the median response duration was 8.8 months (range 5.3–not reached).
Over a median follow-up of 7.7 months, the median OS was 9.3 months (95% CI 8.2–10.9), with an estimated 1-year survival of 39%.
In patients achieving CR, the median OS was 19.7 months (95% CI 11.6–not reached).
Moreover, 10% of patients proceeded to transplantation, suggesting that enasidenib could be a bridge to curative treatment. Approximately 35–43% of patients became transfusion-independent, including those with non-CR/CRi responses.
Enasidenib showed an acceptable tolerability profile; nevertheless, the overall TRAE incidence was 82%, although most of these were mild: 46% nausea (5% grades 3/4), 45% hyperbilirubinemia (18% grades 3/4), and 40% fatigue (8% grades 3/4) or diarrhea (4% grades 3/4). Thrombocytopenia was seen in 27% of patients (23% grades 3/4), anemia in 27% (19% grades 3/4), and IDH differentiation syndrome (IDH-DS) in 10% (6% grades 3/4). Median time to onset of IDH-DS was 48 days (range 10–340), significantly longer than that of DS induced by all-trans retinoic acid, which usually occurs within 1–2 weeks. IDH-DS was managed with temporary drug interruption, dexamethasone 10 mg orally every 12 h for 3 days or until improvement, and hydroxyurea 2–4 g/day. Permanent drug discontinuation was not required in any patients.
Several mechanisms of resistance leading to late relapse have already been proposed, including acquisition of IDH1-mutated subclones or additional non-catalytic second-site mutations of IDH2 [40, 41].
Ivosidenib, formerly known as AG-120, a selective inhibitor of mutant IDH1, was explored in a phase I trial and in an expanded study including 258 patients with IDH1-mutated hematologic malignancies [42]; when administered at 50 mg/day in 125 R/R AML patients, ivosidenib achieved ORR, cCR, and CR rates of 41%, 30%, and 22%, respectively. Median time to cCR was 2.7 months and median duration of response was 6.5 months (8.2 months for patients with CR/CRi). During a median follow-up of 14.8 months, the median OS was 8.8 months, and in patients achieving cCR, the 18-month OS was 50%. IDH1 mutational clearance was observed in 21% of patients with CR or CRi.
Ivosidenib was well tolerated, with QTc prolongation (7% grade ≥ 3) and IDH-DS (4.7% grade ≥ 3) the main toxicities, and no dose-limiting toxicity. Similar to enasidenib, patients with a high co-mutational burden were less likely to respond to ivosidenib; however, in contrast to enasidenib, RAS mutations did not affect the clinical response to ivosidenib.
Based on these non-randomized studies, both enasidenib (August 2017) and ivosidenib (July 2018) were approved by the FDA as a single agent for relapsed AML with IDH2 and IDH1 mutations, respectively.
Results in Untreated AML
In the frontline AML setting, monotherapy with enasidenib and ivosidenib achieved CR/CRi rates of 21–43% [43,44,45] and 41% [46], respectively. IDH inhibitors have also been tested in combination with intensive chemotherapy (7+3 schedule) for induction, achieving an ORR of 93% and 73% in the ivosidenib and enasidenib arms, respectively, with mutational clearance of 41% and 30%, respectively [44].
A summary of clinical trials with ivosidenib and enasidenib is shown in Tables 2 and 3.
Gemtuzumab Ozogamicin and Anti-CD33 Antibodies
In recent years, a number of antigen-specific immunotherapies, including antibodies against both leukemic myeloid antigens (CD33, CD123) and more recently against some leukemia stem cell markers (CD123, CD25, CD44, CD96, CD47, CD32), have been tested in preclinical studies [4, 47].
Gemtuzumab ozogamicin is a recombinant humanized anti-CD33 antibody coupled with the cytotoxic drug calicheamicin, delivered to CD33-expressing leukemic cells after internalization and intracellular release.
GO was approved in 2000 for relapsed non-chemo-eligible CD33+ AML, based on non-randomized phase 2 trials in which it was administered at a dose of 9 mg/m2 every 2 weeks, yielding a 26% CR rate [48, 49].
Unfortunately, the phase III SWOG (Southwest Oncology Group) study, S0106, comparing GO at 6 mg/m2 coupled with conventional induction therapy versus conventional induction therapy alone in newly diagnosed AML patients (< 60 years of age), failed to demonstrate any advantage in either ORR or OS, and showed significantly higher mortality in the GO arm (5.5% vs. 1.4%) [50]. Of note, an increase in both hematological and liver toxicity, including high rates of veno-occlusive disease (VOD), especially after HSCT, have been reported. The lack of clinical benefits, as well as the emerging safety concerns raised by the interim analysis of the SWOG S0106 study, led to an early termination of this trial and the withdrawal of GO from the market in 2010.
Over the next 8 years, several clinical trials, including ALFA-0701, AML-19, and MyloFrance-1, exploring different schedules of these drugs in order to reduce the toxicity, contributed to the favorable reassessment of GO (Table 4).
In the GIMEMA randomized trial, GO monotherapy was tested in patients aged > 60 years who were ineligible for induction chemotherapy. The study population was randomized to GO (either 3 mg/m2 on days 1, 4, and 7 or 6 mg/m2 on day 1 and 3 mg/m2 on day 8) versus best supportive care. An ORR (CR + CRi) of 27% was achieved in the GO arm, with median OS of 4.9 vs. 3.6 months in favor of the GO arm [51]. This study showed that even lower doses of GO could achieve meaningful responses with high saturation of the CD33 sites. Moreover, the rapid re-expression of CD33 molecules on the cell surface after a first exposure to the drug suggested that the administration of fractionated doses could be beneficial [52].
Furthermore, the AML 17 trial confirmed that single doses higher than 3 mg/m2 should not be employed because of increased incidence of VOD and early mortality [53].
In the ALFA-0701 phase III study, patients aged 50–70 with de novo AML were randomized to receive standard induction with or without GO, administered in three fractionated doses of 3 mg/m2 on days 1, 4, and 7. Patients who achieved a CR/CRi underwent two consolidation cycles with intermediate-dose cytarabine with or without GO, based on the initial randomization. The CR rate did not differ between the two arms (81% GO arm vs. 75% control arm; OR 1.46, 95% CI 0.20–2.59; p = 0.25).
Nevertheless, GO was associated with significantly longer EFS (median 15.6 vs. 9.7 months; 2-year EFS 40.8% vs. 17.1%) and RFS (median 28.1 vs. 11.4 months; 2-year RFS: 50.3% vs. 22.7%) and better OS (median 34 vs. 19 months; 2-year OS 53.2% vs. 41.9%) [54]. A post hoc analysis showed that EFS was improved only in patients with high levels of AML blast with CD33+ expression > 70% (49% in GO arm vs. 17% in control arm; HR, 0.56; 95% CI 0.37–0.85; p = 0.0051) [55].
A higher incidence of grade ≥ 3 hemorrhage (22.9% vs. 9.5%) and a longer time to platelet recovery were observed with GO, but the incidence of VOD (two cases out of 139 patients in the GO group) was low. A subsequent study suggests that delaying HSCT 90 days after GO administration may reduce the risk of this serious complication [56].
Finally, a meta-analysis of five randomized studies including 3325 patients with AML showed that combining GO with intensive chemotherapy in the treatment-naive AML patients was associated with significantly better OS, due to a reduced risk of relapse. The CR rates were similar with and without GO, but an improved 5-year OS was observed in patients treated with GO [34.6% vs. 30.7%; HR 0.90 (95% CI 0.82-0.98), p = 0.01]. A survival benefit was observed in patients with core binding factor (CBF) AML [6-year OS 75.5% vs. 545.8%; HR 0.47 (0.31–0.73), p = 0.0006] and in those with normal cytogenetics, while no benefit was seen in patients with adverse cytogenetics [57]. Finally, the results of a pediatric study (AAML0531), suggesting that CD33-splicing single-nucleotide polymorphism (SNP) affects the response to GO, were not confirmed in the UK MRC/NCRI AML15 trials in young AML patients [58, 59].
Based on this background, including other favorable experiences [60,61,62], GO, at a fractionated dosing schedule, was re-approved by the FDA on September 1, 2017, for treatment of newly diagnosed and R/R CD33-positive AML in combination with daunorubicin and cytarabine or as a stand-alone treatment.
The Hedgehog Inhibitor Glasdegib
The Hedgehog (Hh) signaling pathway is vital for embryogenesis and fetal development. Aberrant signaling in this pathway affects the proliferation of leukemic stem cells, and its upregulation has been suggested as an important mechanism of chemoresistance in AML cell lines [63,64,65,66].
The Hh pathway is tightly regulated by two transmembrane proteins, patched (PTCH), which is a negative regulator, and smoothened (SMO), a positive regulator [67, 68].
Glasdegib is an oral agent that inhibits the Hh pathway by interacting with smoothened protein [69]. In vitro and in vivo studies with this agent showed that it inhibited the growth of AML cell lines and human leukemia stem cells [70].
In two phase I trials in adult patients with myeloid malignancies, glasdegib was well tolerated and was associated with an ORR of up to 49% [71, 72], with muscle spasms, dysgeusia, and alopecia being the most common TRAEs. The dose of 100 mg daily was recommended for phase II studies.
A separate phase II study, designed to evaluate the combination of glasdegib given at 100 mg daily for 28 days with standard 7+3 induction in patients over 55 years of age with de novo AML, demonstrated a CR rate of 46.4% [73], with median OS in this older population of 14.9 months.
In the randomized phase II BRIGHT AML 1003 study, 115 patients with newly diagnosed AML who were either ≥ 75 years of age or unfit for intensive chemotherapy were randomized to LDAC 20 mg subcutaneously b.i.d. on days 1–10 plus glasdegib 100 mg daily (n = 77) versus LDAC alone (n = 38) [74]. Despite the low CR rate (18.2% in patients receiving glasdegib, compared with 2.3% for LDAC alone), median OS was significantly better: 4.9 vs. 8.8 months (HR 0.51, 95% CI 0.39–0.67) (Table 5). Glasdegib was well tolerated: the most common AEs occurring at higher rates in the glasdegib/LDAC arm were cytopenia and gastrointestinal symptoms (mostly grade 1–2); cytopenia was not associated with an increased incidence of sepsis or bleeding as compared with LDAC.
Based on these results, in November 2018, glasdegib was approved by the FDA for use in combination with LDAC for AML patients either aged ≥ 75 years or with comorbidities precluding intensive induction chemotherapy.
Venetoclax and Other Pro-Apoptotic Agents
B-cell leukemia/lymphoma-2 (BCL2) is an antiapoptotic protein that promotes leukemic cell survival through regulation of the mitochondrial apoptotic pathway. An increased level of BCL-2 expression relative to the pro-apoptotic BAX protein is associated with poor outcome in patients receiving intensive chemotherapy for AML [76].
Sensitizer BCL-2 homology 3 (BH3) proteins are antagonists of these antiapoptotic proteins and activate downstream BAX and BAK, which induce the release of mitochondrial intermembrane molecules such as cytochrome c, resulting in activation of the caspase cascade and, finally, apoptosis [77].
Venetoclax is an oral, selective BH3-mimetic that binds to and inhibits the BH3 domain of BCL2 proteins and thereby dislodges proapoptotic factors, such as BIM, from their BCL2 binding site, reactivating the mitochondrial apoptotic pathway [78]. It was previously approved by the FDA (April 2016) for the treatment of chronic lymphocytic leukemia [79].
In a phase II study in high-risk R/R AML patients (n = 32), treatment with venetoclax at 800 mg/day as a single agent achieved an ORR of 19%; interestingly, patients with IDH mutations showed a higher ORR (33%), as IDH-mutant AML cells depend on BCL-2 for survival. Eighty-one percent of the patients experienced grade 3/4 adverse events, including febrile neutropenia (31%) and hypokalemia (22%) [80].
Preclinical studies have shown that resistance to venetoclax on a molecular basis is mediated by the antiapoptotic proteins Bcl-xL and MCL1, which can be overcome by combination therapy with hypomethylating agents (HMAs) [81, 82], anthracyclines (idarubicin and daunorubicin) [83], and the MDM2 antagonist idasanutlin [84,85,86,87].
Nevertheless, these preliminary results, considering the potent proapoptotic activity of this drug in the presence of cytotoxic stress, led to a phase Ib-II trial in which venetoclax at 400 mg/day was combined with HMA (azacitidine or decitabine based on institutional preference) as upfront treatment in 145 AML patients ≥ 65 years of age who were unfit for intensive chemotherapy; this treatment was well tolerated, yielding an impressive 61% CRi rate [88].
The phase II data showed a CR + CRi rate of 73%, with median OS of 17.5 months; 46% of the total population was alive at 2 years [89]. Venetoclax showed a good safety profile, with the main TRAEs being neutropenia and nausea.
Given these results, the combination of venetoclax with HMAs is a candidate as the new standard of care for elderly AML patients unfit for standard chemotherapy.
A phase I/II trial with LDAC plus venetoclax [90] showed similar results in 71 untreated, unfit AML patients. Based on these response rates, far exceeding the historical outcome observed with azacitidine alone (composite CR: 28%, median OS: 10.4 months) [91], venetoclax, in combination with azacitidine, decitabine, or LDAC, was granted breakthrough designation by the FDA on November 21, 2018, for previously untreated patients with AML who were older than 75 years or unfit for intensive chemotherapy.
Phase 3 randomized placebo-controlled registration studies in unfit elderly AML patients are currently ongoing, in order to confirm the benefit of venetoclax plus azacitidine versus azacitidine alone (NCT02993523) or venetoclax plus LDAC versus LDAC alone (NCT03069352).
Given these encouraging data, a number of studies have been proposed combining venetoclax with other agents, including 7+3 induction (NCT03709758), multi-CDK inhibitor dinaciclib (NCT03484520), gilteritinib (NCT03625505), 10-day schedule decitabine (NCT03404193), and the Mcl-1 inhibitor S64315 (NCT03672695).
In the R/R AML setting, HMAs + venetoclax achieved an ORR of up to 76% in small retrospective series, including cases with previous HMA exposure [92]. In a retrospective study in heavily pretreated patients, venetoclax plus either HMAs or LDAC yielded a response rate of 21%, with median OS of 3.0 months [93].
A list of completed clinical trials with venetoclax in AML is shown in Table 6.
Conclusions and Perspectives
AML is a biologically and clinically heterogeneous disease. Although advances in supportive care and prognostic risk stratification have optimized the performance of standard established therapies, overall long-term survival remains poor. Elderly patients, who represent the majority of AML cases, are more likely to have more aggressive disease, often with adverse cytogenetic features. At the same time, the risk of treatment-related mortality and several toxicities often precludes this population from receiving intensive chemotherapy or stem cell transplantation. Novel targeted therapies combine effective anti-leukemic activity with reduced toxicity, but the multiple biological pathways involved in leukemogenesis hamper the development of a single “magic bullet” against this disease. In particular, the eradication of leukemic stem cells remains the main issue in the development of effective new drugs. Leukemic stem cells (LSC) are quiescent and therefore could be more sensitive to drugs affecting different targets such as mitochondrial activity, heat shock protein 90 (Hsp 90) and inflammasome, NF-Kb and proteasome activity, and epigenetic modulators, probably with selective susceptibility in comparison with normal stem cells [94]. It has been demonstrated that chemotherapy-resistant AML LSCs reside in the endosteal region of the bone marrow and adipose tissue, where microenvironmental factors alter the intracellular metabolism, leading to chemotherapy resistance [95, 96]. Modulation of niche interactions may increase the chemosensitivity of LSCs. Finally, there is also evidence that a proinflammatory state can influence LSC growth and survival [97].
Inhibition of proinflammatory factors may both reduce LSC activity and facilitate an environment more favorable for normal stem cells. This is particularly important in the context of post-chemotherapy treatment, where the need for suppression of residual disease and promotion of normal cell regeneration is perhaps most acute.
The future scenario of new experimental drugs is very intriguing and includes the epigenetic regulators targeting DNMT3A, IDH1/2, TET2, and ASXL1, interacting with enzymes involved in writing (EZH2, DOT1L, MLL), reading (BET, bromodomains), or erasing (LSD1) histone marks involved in activating or repressing gene expression and the disruptor of telomeric silencing 1-like (DOT1L) inhibiting MLL fusion genes [98].
Furthermore, in addition to mutated enzymes and upregulated pathways, the identification of unique cell surface markers can provide a therapeutic target for recombinant monoclonal antibodies or chimeric antigen receptors (CAR) [99].
The immunosuppression related to the disease represents another important mechanism of minimal residual disease and immune escape. Immune checkpoint inhibitors have been widely studied in solid tumors, and their migration to hematological malignancies has become more prominent since the success in Hodgkin’s lymphoma. So far, clinical studies on immune checkpoint inhibitors (nivolumab, pembrolizumab, and ipilimumab) have provided limited results in AML [100,101,102,103]. However, immune checkpoint inhibitors given in association with HMAs could represent an interesting strategy to enhance the activity of immunotherapies.
The last—but not least—new drug approved in this setting is CPX-351, a dual-drug liposomal encapsulation of cytarabine and daunorubicin with a synergistic 5:1 molar ratio of cytarabine and daunorubicin within the liposome. This formulation was aimed at increasing the release of drugs specifically in bone marrow, reducing extra-hematological toxicity and bypassing P-glycoprotein-based efflux pumps, which are important mediators of chemotherapy resistance [104].
Studies with CAR T-cell therapy are ongoing, with promising preliminary results [105, 106], but an optimal response requires a specific and stable myeloid leukemic antigen that has yet to be identified. Lastly, the development of well-tolerated oral therapies will improve both the feasibility of treatment and the quality of life in this very difficult-to-treat setting.
References
Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Kucukyurt S, Eskazan AE. New drugs approved for acute myeloid leukemia (AML) in 2018. Br J Clin Pharmacol. 2019. https://doi.org/10.1111/bcp.14105(epub ahead of print).
Stone RM. What FLT3 inhibitor holds the greatest promise? Best Pract Res Clin Haematol. 2018;31:401–4.
Assi R, Kantarjian H, Ravandi F, Daver N. Immune therapies in acute myeloid leukemia: a focus on monoclonal antibodies and immune checkpoint inhibitors. Curr Opin Hematol. 2018;25:136–45.
Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16:991–1001.
Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–50.
Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–503.
Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4):444–53.
Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–43.
Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, Cortes J, Kantarjian HM, DeAngelo DJ, Huntsman-Labed A, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26:2061–8.
Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377:454–64.
Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–9.
Chappell G, et al. Maintenance sorafenib in FLT3-ITD AML following allogeneic HCT favorably impacts relapse and overall survival. Bone Marrow Transplant. 2019;54:1518–20.
Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122:3607–15.
Weisberg E, Sattler M, Ray A, Griffin JD. Drug resistance in mutant FLT3-positive AML. Oncogene. 2010;29:5120–34.
Wang ES, Stone RM, Tallman MS, Walter RB, Eckardt JR, Collins R. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 mutant acute myeloid leukemia (AML). Blood. 2016;128(22):1071.
Goldberg AD, Collins RH, Stone RM, et al. Addition of crenolanib to induction chemotherapy overcomes the poor prognostic impact of co-occurring driver mutations in patients with newly diagnosed FLT3-mutated AML. Blood. 2018;132(Suppl 1):1436.
Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, Small D, Levis M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129:257–60.
Pratz KW, Cherry M, Altman JK, et al. Updated results from a phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in subjects with newly diagnosed acute myeloid leukemia (AML). Blood. 2018;132(Suppl 1):564.
Perl AE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381:1728–40. https://doi.org/10.1056/NEJMoa1902688.
Perl AE, et al. An open-label, randomized phase III study of gilteritinib versus salvage chemotherapy in relapsed or refractory FLT3 mutation-positive acute myeloid leukemia. J Clin Oncol. 2017;35(suppl; abstr #TPS7067). 2017 American Society of Clinical Oncology (ASCO) Annual Meeting, 2017 June 2–6; Chicago.
Cortes J, Perl AE, Döhner H, Kantarjian H, Martinelli G, Kovacsovics T, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19:889–903.
FDA Briefing Document Oncologic Drugs Advisory Committee (ODAC) Meeting May 14, 2019 NDA 212166 Quizartinib Applicant: Daiichi-Sankyo, Inc. 2019.
Randhawa JK, Kantarjian HM, Borthakur G, Thompson PA, Konopleva M, Daver N, Pemmaraju N, Jabbour E, Kadia TM, Estrov Z, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood. 2014;124:389.
Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018;34:186–95.
Ragon BK, DiNardo CD. Targeting IDH1 and IDH2 mutations in acute myeloid leukemia. Curr Hematol Malig Rep. 2017;12:537–46.
Shih AH, Shank KR, Meydan C, et al. AG-221, a small molecule mutant IDH2 inhibitor, remodels the epigenetic state of IDH2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood. 2014;124(21):437.
Kats LM, Vervoort SJ, Cole R, et al. A pharmacogenomic approach validates AG-221 as an effective and on-target therapy in IDH2 mutant AML. Leukemia. 2017;31(6):1466–70.
Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–6.
Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. 2017;18(3):17.
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207(2):339–44.
Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2hydroxyglutarate. Cancer Cell. 2010;17(3):225–34.
Chaturvedi A, Araujo Cruz MM, Jyotsana N, et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia. 2016;30(8):17081715.
Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Nassereddine S, Lap CJ, Haroun F, Tabbara I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. 2017;96:1983–91.
Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–41.
Hansen E, Quivoron C, Straley K, et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood. 2014;124(21):3734.
Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant-IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31.
Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559(7712):125–9.
Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med. 2018;24:1167–77.
DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Pollyea DA, Tallman MS, Botton S, et al. Enasidenib monotherapy is effective and well- tolerated in patients with previously untreated mutant-IDH2 (mIDH2) acute myeloid leukemia (AML). HemaSphere. 2018;2:S1 (S1561).
Stein EM, Shoben A, Borate U, et al. Enasidenib is highly active in previously untreated IDH2 mutant AML: early results from the Beat AML Master Trial. Blood. 2018;132(Suppl 1):287.
Stein EM, Garcia-Manero G, Rizzieri DA, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131:2661–9.
Roboz GJ, DiNardo CD, Stein EM, et al. Ivosidenib (AG-120) induced durable remissions and transfusion independence in patients with iDH1-mutant untreated AML: results from a phase 1 dose escalation and expansion study. Blood. 2018;132(Suppl 1):561.
Majeti R, et al. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30:1009–19.
Bross PF, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7:1490–6.
Larson RA, et al. Antibody-targeted chemotherapy of older patients with acute myeloid leukemia in first relapse using Mylotarg (gemtuzumab ozogamicin). Leukemia. 2002;16:1627–36.
Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854.
Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34:972–9.
Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–68.
Burnett AK, Cavenagh J, Russell N, Hills R, Kell J, Jones G, et al. Defining the dose of gemtuzumab ozogamicin in combination with induction chemotherapy in acute myeloid leukemia: a comparison of 3 mg/m2 with 6 mg/m2 in the NCRI AML17 trial. Haematologica. 2016;101:724–31.
Castaigne S, Pautas C, Terré C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379:1508–16.
Olombel G, Guerin E, Guy J, et al. The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood. 2016;127(17):2157–60.
Lambert J, Pautas C, Terré C, Raffoux E, Turlure P, Caillot D, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia:vfinal efficacy and safety updates from the open-label, phase 3 ALFA-0701 trial. Haematologica. 2018. https://doi.org/10.3324/haematol.2018.188888 (Epub).
Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986–96.
Lamba JK, Chauhan L, Shin M, et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2017;35(23):2674–82.
Gale RE, Popa T, Wright M, et al. No evidence that CD33 splicing SNP impacts the response to GO in younger adults with AML treated on UK MRC/NCRI trials. Blood. 2018;13:468–71.
Delaunay J, Recher C, Pigneux A, et al. Addition of gemtuzumab ozogamycin to chemotherapy improves event-free survival but not overall survival of AML patients with intermediate cytogenetics not eligible for allogeneic transplantation. Results of the GOELAMS AML 2006 I study. Blood. 2011;118(21):79.
Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–77.
Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012;30(32):3924–31.
Queiroz KCS, Ruela-de-Sousa RR, Fuhler GM, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. 2010;29:6314.
Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776.
Lin TL, Matsui W. Hedgehog pathway as a drug target: smoothened inhibitors in development. Onco Targets Ther. 2012;5:47–58.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–72.
Lim Y, Gondek L, Li L, et al. Integration of hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Transl Med. 2015;7:291ra296.
Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood. 2012;119:2196–204.
Munchhof MJ, Li Q, Shavnya A, et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett. 2012;3:106–11.
Sadarangani A, Pineda G, Lennon KM, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J Transl Med. 2015;13:98.
Martinelli G, Oehler VG, Papayannidis C, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015;2:e339–46.
Minami Y, Minami H, Miyamoto T, et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci. 2017;108:1628–33.
Cortes JE, Douglas Smith B, Wang ES, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: phase 2 study results. Am J Hematol. 2018;93:1301–10.
Cortes JE, Heidel FH, Heuser M, et al. A phase 2 randomized study of low dose Ara-C with or without glasdegib (PF-04449913) in untreated patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood. 2016;128:99.
Savona MR et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clinical Cancer Research. 2018;24(10):2294–303.
Del Poeta G, Venditti A, Del Principe MI, et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood. 2003;101(6):21252131.
Konopleva M, Letai A. BCL-2 inhibition in AML—an unexpected bonus? Blood. 2018;132(10):1007–12. https://doi.org/10.1182/blood-2018-03-828269.
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–83.
Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22.
Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6:1106–17.
Tsao T, Shi Y, Kornblau S, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. 2012;91(12):1861–70.
Bogenberger JM, Delman D, Hansen N, et al. Ex vivo activity of BCL-2 family inhibitor ABT199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma. 2015;56(1):226–9.
Teh TC, Nguyen NY, Moujalled DM, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32(2):303–12.
Lehmann C, Friess T, Birzele F, et al. Superior antitumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9:50.
Saiki AY, Caenepeel S, Yu D, et al. MDM2 antagonists synergize broadly and robustly with compounds targeting fundamental oncogenic signaling pathways. Oncotarget. 2014;5(8):2030–43.
Niu X, Zhao J, Ma J, et al. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22:4440–51.
Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. 2018;93(8):E202–5. https://doi.org/10.1002/ajh.25146.
DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19:216–28.
DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17.
Wei A, Strickland SA, Hou JZ, et al. Venetoclax with low-dose cytarabine induces rapid, deep, and durable responses in previously untreated older adults with AML ineligible for intensive chemotherapy. Blood. 2018;132:284.
Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with > 30% blasts. Blood. 2015;126:291–9.
Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Al Malki MM, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103:e404–7.
DiNardo CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93:401–7.
Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513–7.
Asada N, Katayama Y. Regulation of hematopoiesis in endosteal microenvironments. Int J Hematol. 2014;99(6):679–84.
Ye H, Adane B, Khan N, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19(1):23–37.
Mirantes C, Passegué E, Pietras EM. Proinflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. 2014;329(2):248–54.
Stein EM, DiNardo CD, Fathi AT, et al. Ivosidenib or enasidenib combined with induction and consolidation chemotherapy in patients with newly diagnosed AML with an IDH1 or IDH2 mutation is safe, effective, and leads to MRD-negative complete remissions. Blood. 2018;132(Suppl 1):560.
Assi R, Kantarjian HM, Daver NG, et al. Results of a phase 2, open-label study of idarubicin (I), cytarabine (A) and nivolumab (Nivo) in patients with newly diagnosed acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (MDS). Blood. 2018;132:905.
Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: choosing the best when the optimal does not exist. Am J Hematol. 2018;93:553–63.
Daver N, Basu S, Garcia-Manero G, et al. Phase IB/II study of nivolumab in combination with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML). Blood. 2016;128:763.
Kadia TM, Cortes JE, Ghorab A, et al. Nivolumab (Nivo) maintenance (maint) in high-risk (HR) acute myeloid leukemia (AML) patients. J Clin Oncol. 2018;36:7014.
Daver NG, Garcia-Manero G, Basu S, et al. Safety, efficacy, and biomarkers of response to azacitidine (AZA) with nivolumab (Nivo) and AZA with nivo and ipilimumab (Ipi) in relapsed/refractory acute myeloid leukemia: a nonrandomized, phase 2 study. Blood. 2018;132:906.
Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36:2684–92.
Liu F, Cao Y, Pinz K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. 2018;132(Suppl 1):901.
Budde L, Song JY, Kim Y, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood. 2017;130:811.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
The authors (Alessandro Fiorentini, Debora Capelli, Francesco Saraceni, Diego Menotti, and Attilio Olivieri) have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any study with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.11397897.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fiorentini, A., Capelli, D., Saraceni, F. et al. The Time Has Come for Targeted Therapies for AML: Lights and Shadows. Oncol Ther 8, 13–32 (2020). https://doi.org/10.1007/s40487-019-00108-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-019-00108-x