
Abstract
Serving as a proof of concept, additive manufacturing and electrophoretic deposition are leveraged in this work to enable structural lithium-ion batteries with load-bearing and energy storage dual functionality. The preparation steps of a complex 3D printed copper current collector, involving the formulation of a photocurable resin formulation, as well as the vat photopolymerization process followed by a precursors-based solution soaking step and thermal post-processing are presented. Compression and microhardness testing onto the resulting 3D printed copper current collector are shown to demonstrate adequate mechanical performance. Electrophoretic deposition of graphite as a negative electrode active material and other additives was then performed onto the 3D printed copper collector, with the intention to demonstrate energy storage functionality. Half-cell electrochemical cycling of the 3D multi-material current collector/negative electrode versus lithium metal finally demonstrates that structural battery components can be successfully obtained through this approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Improvement in energy density of lithium metal and lithium-ion batteries used in transportation and commercial electronic applications via active material optimization (e.g., anodes and cathodes) has been an important technological focus over the past two decades [1]. In electric vehicles, for instance, batteries with an energy capacity of ~100 kWh represent approximately 1/4 of the total weight [2,3,4]. To further increase the overall energy density, structural batteries, serving both as energy storage and structural support, have been recently proposed [5,6,7,8]. Approaches include structural power composites aimed to provide lightweight energy storage for electrically powered systems such as carbon fibers within a structural electrolyte matrix material [8,9,10,11], and thin films deposited onto composite laminates capable of sustaining high mechanical loads [12,13,14]. The majority of structural batteries reported to date are planar and maintain the unidimensional lithium-ion diffusion characteristic of commercial batteries. Alternatively, shape-conformable batteries that can be produced through additive manufacturing, also known as 3D printing, stand to be optimized to provide both high energy density and load-bearing functionality [15,16,17,18].
In this context, recent interest in batteries with 3D internal architectures fabricated using a variety of 3D printing techniques has increased [17, 19,20,21,22]. Among the different 3D printing technologies, vat photopolymerization (VPP) appears to be a promising technology for producing conformal batteries due to the high manufacturing resolution down to a few tens of nanometers for two-photon polymerization (2PP) and comparatively low cost and high surface quality [23, 24]. Of particular interest is the manufacturing process of copper current collector structures to be employed as structural battery components. VPP relies on layer-by-layer photopolymerization of a UV-sensitive resin composed of a mixture of monomers, oligomers, photoinitiator, light absorbers and photosensitizers. While 3D printing of copper has been achieved via powder bed fusion [25,26,27] and material extrusion [28,29,30], these techniques are complex, expensive, or limited in resolution [31]. On the other hand, VPP of copper is less common with the generally-understood requirement of addition of copper particles within the resin, thus increasing viscosity and decreasing the photopolymerization efficiency due to sedimentation issues leading to a high light scattering. The viscosity issue can be resolved by increasing the resin temperature during printing [32], using smaller filler particles [33], and/or adding dispersant and diluent compounds [34]. The sedimentation issue and photopolymerization efficiency, however, are difficult to control, requiring the use of either smaller copper particles or an adapted printer allowing a continuous recirculation of the resin. Recently, the Greer group demonstrated an unconventional printing method in which 3D copper structures were obtained without the addition of solid particles [35, 36]. The process consisted of printing a resin containing water-soluble salts, followed by their dissolution and finally soaking of the 3D structure within a copper precursor solution. The final copper piece is obtained after thermal post-processing. This novel approach for fabricating metal structures may lead to a dramatic increase in the spatial resolution and intricate detail of geometrically-complex metallic structures, which could in turn enable new applications in terahertz electromagnetics, thermal management, printed electronics, and in the case of the current work, energy storage. The present work investigates a new metal fabrication approach with additional electrophoretic deposition to manufacture battery electrodes. To obtain complex structures of pure copper, a base resin (with no additional water-soluble salts) was 3D printed via VPP, followed by a dehydration step of the printed items, a subsequent soaking step within a copper precursor solution, and finally, thermal post-processing. This method facilitates the uptake of the copper precursor salt by the dehydrated printed items. This research presents an optimization of the thermal post-processing steps, based on the analysis of the thermogravimetric data, followed by mechanical testing to appraise the use of the 3D printed copper current collectors in structural batteries. Finally, the electrophoretic deposition of graphite, a common negative electrode material for lithium-ion batteries, and other additives onto the printed current collector enabled evaluation of the electrochemical performance of the resulting structure in half-cell configuration.
2 Materials and methods
2.1 Base resin preparation, 3D printing, and precursor uptake
The base resin was prepared by mixing poly(ethylene glycol) diacrylate (PEGDA avg. Mn 575, (C2H4O)nC6H6O3, Sigma Aldrich) and diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide (TPO, 97%, Sigma Aldrich) as a photoinitiator in a mass ratio PEGDA:TPO of 1:0.005 2 and magnetically stirring the mixture for two hours. After this period, water was added in a mass ratio PEGDA:H2O of 1:1.12, and the mixture was stirred for one hour. The base resin was kept inside a UV-blocking container at 10 °C until used for 3D printing.
3D models were designed with Fusion 360 (Autodesk, USA) and nTopology softwares (nTopology, USA). The architectures consisted of six discs (16 mm diameter and 3 mm thick) exhibiting various infill patterns (octet, fluorite, splitP, cylindrical gyroid, classical gyroid, and diamond) with 30% infill density. The models in Standard Tessellation Language (.stl files) were sliced into 50 μm thick 2D slices using Tethonware software (Tethon 3D, USA). Printing of the 3D structures was performed at 25 °C using a Bison 1000 Digital Light Processing (DLP) 3D printer (Tethon 3D, USA) after loading the 100 mL of resin. An initial light exposure time of 25 s was set for the 20 first printed layers to ensure good printing quality. Subsequent layers only required a light exposure of 2.5 s and a dwell time of 45 s between each layer. Brightness was set to 700 mW/cm2. Prior to printing, the resin was magnetically stirred for 30 min, and the bed platform and tank were cleaned with 2-propanol.
3D printed items were stored in an oven for 12 h at 60 °C under air to promote gentle dehydration. The copper precursor was subsequently infiltrated by soaking the dehydrated pieces in an aqueous 4.5 mol/L solution of copper II nitrate hemipentahydrate (Cu(NO3)3·2.5H2O) for four days.
2.2 Thermal post-processing
Precursor-soaked 3D structures were placed in an alumina boat for the thermal treatment inside a GSL-1700X tubular furnace (MTI Corporation, USA). The thermal post-processing profile was deduced from experimental thermogravimetric analysis. The purpose of the first step of the thermal post-process performed under flowing air at a pressure of 0.025 MPa is to eliminate the polymeric resin and components and produce sintered copper oxide. This step is followed by reduction of the copper oxide to yield metallic copper by heating the sample at 950 °C for 5 h under flowing 5 mol% of H2 in argon.
2.3 Materials characterization
Thermogravimetric analysis - mass spectrometry (TGA/MS) was achieved by means of a STA 449F3 instrument (NETZSCH, Germany) placed inside an argon-filled glove box (O2 and H2O < 0.1 ppm, Jacomex). The experiments were performed from 25 °C to 900 °C at a rate of 5 °C/min under an argon flow rate of 40 mL/min.
X-ray powder diffraction (XRD) patterns were acquired with an Empyrean-2 X-ray diffractometer (Malvern Panalytical, UK) using Cu Kα radiation (λ = 0.154 18 nm), 45 kV of accelerating voltage, and a current of 40 mA. Data were recorded from 10° to 90° 2θ with a step size of 0.013° and a scan rate of 8°/min.
Secondary electron or backscattered electron images of the 3D printed items were obtained by means of an S-4800 (Hitachi, Japan) field emission scanning electron microscope (SEM) operating in high vacuum mode and 15 kV of acceleration voltage.
Quasistatic compression testing was performed using Instron, a universal mechanical testing machine with a Berkovich indenter. The testing and reporting were based on ISO 13314. A NANOVEA M1 Hardness tester was used to perform the microhardness testing. The software associated within the instrument is defaulted to follow the ASTM E2546 standard to calculate hardness and elastic modulus [37]. The area function for a Berkovich indenter is shown in Eq. (1), where hc is the contact depth and calculated using Eq. (2). Here, hmax, Pmax and S are the maximum contact depth, the maximum load and the load slope, respectively. The hardness is calculated using Eq. (3).
A Keyence VHX 7000 optical microscope was used to conduct the surface roughness analysis on the surface and struts of the printed lattice.
The resistance of the item was measured at room temperature using a Keysight Truevolt digital multimeter. The electronic conductivity σ was calculated using Eq. (4), where R is the resistance; A is the contact area of the sample; and t is the thickness.
2.4 Electrophoretic deposition (EPD)
For the EPD bath, 230 mg of graphite (Timcal TIMREX SLS, ~1.5 m2/g), 10 mg of conductive carbon black Timcal Super C45 (BET = 45 m2/g and 20 nm particle size, MSE Supplies), 10 mg of polyvinylidene difluoride (Sigma Aldrich), 0.2 mL of Triton-X100 (Sigma Aldrich) and 10 mg of iodine (≥99.8% Sigma Aldrich) were magnetically stirred with 50 mL of acetone (ACS reagent ≥99.5%) for 5 min and sonicated for an additional 10 min. Selected copper current collectors (planar foil or 3D printed) were used (one at a time) as substrates, and a platinum wire ~ 8 mm apart was used as a counter electrode. A voltage difference of 65 V and a current of 0.1 A were applied between the electrodes for 5 min to promote the deposition of the EPD bath components. The resulting electrodes were washed lightly with acetone to eliminate unbonded residues, and dried overnight at 70 °C.
2.5 Electrochemical characterization
After EPD, the 3D electrode structures or the planar ones were employed as working electrodes within a Swagelok®-type half-cell. The counter and reference electrode was a lithium foil (0.38 mm thick ribbon, 99.9% purity, Sigma Aldrich), and as a separator, a fiberglass (Whatman GE Healthcare) impregnated with 200 µL of 1 mol/L LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (EC:DMC 1:1 wt, Sigma Aldrich) was used. Prior to cycling, the cells were left to rest for 10 h to allow complete electrolyte impregnation. Cells were galvanostatically charged and discharged at C/20 (18.6 mA/g), C/10 (37.2 mA/g), C/5 (74.4 mA/g) and again at C/20, in a potential window of 0.01−2.5 V vs Li/Li+, and at 25 °C by means of a LBT galvanostat (Arbin Instruments, USA).
3 Results and discussion
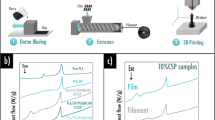
One of the challenges associated with copper 3D printing via VPP is the high viscosity of the resin when loaded with copper powder, and the associated sedimentation of the metallic particles. In this context, the use of a precursor approach involving a photocurable resin loaded with soluble metallic salts followed by printing and additional thermal post-processing to obtain pure metals represents an attractive manufacturing process [38]. However, this approach needs to be adapted for copper, as Cu2+ quenches UV-generated radicals [39] thus preventing the photopolymerization process. The fabrication process that was followed in this work is displayed in Fig. 1. The first step consisted in 3D printing the PEGDA/photoinitiator/H2O base resin by means of a DLP 3D printer. For demonstration purposes, a variety of intricate lattice structures were printed (see Fig. S.1). Among the printed items, discs with different infill patterns were specifically designed to fit within a Swagelok®-type cell for subsequent half-cell electrochemical testing. The second step was performed immediately after printing and consisted in a dehydration step to remove moisture from the printed samples. The third step consisted in soaking the 3D printed structure into a solution containing copper precursors (4.5 mol/L). Thanks to the previous dehydration step, the absorption of the aqueous copper precursor solution into the polymer lattice structure was promoted. It is important to note that among the different air-stable water-soluble precursor salts CuCl2, CuSO4·5H2O, and Cu(NO3)3·2.5H2O that were considered, the latter was selected due to its greater water solubility (138 g/100 mL at 0°C [37]) and due to its thermal decomposition into mostly harmless gaseous products. To transform the copper precursors into pure copper, the next step was to subject the structures to two consecutive thermal post-processing treatments: the first under flowing air, and the second under flowing Ar/H2. During thermal processing under air (fourth step displayed in Fig. 1), the polymer matrix is eliminated, while at the same time the copper salts contained within the polymer matrix turn into copper oxide. Subsequently, during the thermal post processing step under a reductive atmosphere of Ar/H2 (fifth step displayed in Fig. 1), the copper oxide turned into pure copper metal. Finally, the last step consisted in performing EPD of the negative electrode material in order to obtain a current collector-negative electrode system.

Optical images of the fabrication steps leading to a negative electrode coated onto a VPP 3D printed current collector
TGA/MS of the 3D printed structures with and without copper precursors were performed in order to establish an adequate thermal post-processing profile. The decomposition of the base resin without copper precursor (serving here as reference) showed thermal degradation up to 600 °C (see Fig. 2a). The first decomposition step started at 205 °C with a DTG peak centered at 410 °C, along with the production of CO2 and H2O. Then, a second CO2 production occurred one hundred degrees higher, with a DTG peak centered at 516 °C. This suggests a two-step decomposition process of the polymeric matrix PEGDA. Interestingly, when the copper precursor is contained within the 3D printed piece, the combustion of PEGDA was confirmed to occur below 400 °C (see Fig. 2b). A previous analysis by Martinez et al. [38] based on a similar study proposed that the decomposition temperature lowered at least one hundred degrees in the presence of metal nitrate salts catalyzing the PEGDA combustion.

TGA/MS analyses under air flow of a a dehydrated 3D printed polymer structure, and b the same structure after copper precursor solution infiltration
Indeed, around 58% of weight loss was observed in the item containing copper precursors below 130 °C, which according to the mass spectrometry data corresponds to gasses with m/z 18, 44, 30, and an almost undetectable 46 (see Fig. 2b). These gasses correspond to H2O, CO2, NO, and NO2, respectively. The water lost at this temperature corresponds mainly to water from the precursor solution, and also from the combustion of PEGDA that is accompanied by CO2 release (see Eq. (5)). As for the detected NO and NO2, they are both products of the decomposition of copper nitrate in air. Copper is multivalent, so during its decomposition it may form CuO, Cu4O3 and Cu2O, the final chemical composition depending on the oxygen concentration and temperature. Under the conditions used in this work, CuO was the most favorable because copper maintains its oxidation state (see Eqs. (6) and (7)) [40]. From 130 °C to 400 °C, with a DTG peak centered at 251 °C, another weight loss accounting for ~27.5 % of the total weight was recorded. Mass spectrometry data registered the production of CO2 and H2O, which corresponded to the thermal decomposition of the polymeric resin up to 500 °C.
From these observations, the following thermal post-processing profile was established (see Fig. 3a). The profile consists of heating at 1 °C/min until a plateau at 120 °C to allow water-removal and polymer combustion, a slow heating step at 0.2 °C/min until 250 °C to favor the production of CuO, and heating at the same rate until a debinding plateau 450 °C to completely combust the polymer. Then, heating at 2 °C/min up to a plateau 750 °C was implemented to fuse together the CuO particles, followed by a cooling step at the same rate. Finally, heating at 4 °C/min until a plateau of 900 °C was performed for 5 h under a flowing reducing atmosphere in order to yield pure copper. When comparing the dimensions of the 3D printed polymer structure to the pure 3D printed copper current collector, a shrinkage of 65% was witnessed along the X and Y axes while a shrinkage of 41% was observed along the Z axis. In terms of surface area, the 3D printed copper current collector with a diameter of 5.6 mm exhibited a calculated surface area of 186.81 mm2 (not considering the rugosity that adds up to the available surface). In comparison, a conventional planar current collector with the same diameter would only present a surface area of 24.63 mm2.

a Thermal post-processing profile established from TGA/MS analysis, b XRD of the printed pieces after thermal treatments under air and H2/Ar atmospheres
XRD analysis of the 3D printed pieces after the first step of thermal post-processing under air revealed the presence of CuO, Cu2O and metallic Cu (see Fig. 3b, black color data). Although the formation of metallic Cu from Cu(NO3)2 is theoretically possible through a two-step decomposition process reducing intermediary Cu2O above 900 °C (see Eqs. (8) and (9) [41], this reaction is not favorable as the maximum temperature employed in this work is 750 °C. Instead, we propose that a carbothermal reaction took place, as already reported for copper oxides starting around 700 °C [42]. This reaction implies that Cu(II)O can be reduced at high temperature in the presence of a carbon source into Cu(0) with CO as a byproduct (see Eq. (10)). Alternatively, CO could react in gaseous form with the oxide to form Cu(0) and CO2 (see Eq. (11)), as proposed by L’vov [42]. Both options are equally probable, and discernment is for now not possible at this stage because the mass spectrometry data show that both CO and CO2 are produced in equal and minimal quantities above 700 °C (zoom beyond 600 °C displayed in Fig. S.2).
Following the thermal treatment of the CuO structures under a reducing atmosphere, the metallic copper composition was confirmed by XRD (see Fig. 3b, orange color data). No oxide impurity was found, only copper indexed to a face centered cubic lattice (JCPDS No. 040836) with 2θ positions at 44.2°, 51.0°, and 74.8°, corresponding to the (111), (200) and (220) planes, respectively.
To appraise the potential of the 3D printed copper current collectors as load-bearing current collectors within a structural battery, compression and microhardness tests were performed onto gyroid copper lattice. The compression tests were conducted based on ISO 13314 at a rate of 1 mm/min and showed that the copper lattice displayed a compressive strength of 19.39 MPa. Based on the standard, the plateau stress was calculated to be the arithmetic mean of compressive stress between 20% and 40% of compressive strain. Copper being ductile and malleable, all the compressive forces subjected onto the struts of the gyroid during the testing had resulted in a permanent deformation and the part post testing resembled a thin coin. Even though there were cracks on the struts of the lattice before the test, it was observed that they did not have significantly impacted the results. This can be attributed to the properties of copper and the loading rate. For the microhardness testing, a Berkovich microindenter was used. The specimen area at the indentation site, the load-depth curves and the summary of the results are shown in Figs. 4a and b and Table S.1. The calculated microhardness value of 0.272 ± 0.039 GPa for the 3D printed copper current collector is about 2 times lower to what was reported by McLaughlin and Clegg [43] in their work on the copper single crystal as 0.622 GPa. In a related work, Saccone et al. [36] measured the hardness of hydrogel-infusion-manufactured copper to be 1.81 ± 0.37 GPa employing an Agilent G200 Nano Indenter and a Berkovich tip. Further optimization to enhance the density and mechanical performance of these printed current collectors is currently underway.

Microhardness results a the sample at one of the indentation sites, b load-depth curves
The electrical conductivity of the printed gyroid disc was measured in order to determine if this item is sufficiently electronically conductive to undergo EPD of the battery material and other additives. The obtained value was 3.09 × 103 S/m, which appeared low in comparison with the electrical conductivity of annealed copper of 5.8 × 107 S/m [44]. However, it should be considered that the contact area used in the electrical conductivity formula is always significantly lower for a 3D printed sample due to the surface rugosity caused by the manufacturing process. To evaluate the extent of the surface rugosity, Figs. 5a and b show the surface analysis of the 3D printed copper current collector. The selected area displayed an arithmetical mean height of the surface value (Sa) of 10 µm and the maximum height value (Sz) of 30 µm. Since the Sz value is defined as the sum of the largest peak height and the largest pit depth within the selected area, it can be concluded that the layered pattern indeed provides considerable rugosity that affects considerably electrical conductivity measurements. Furthermore, a copper gyroid cube was 3D printed and sintered from a resin containing copper particles in view to obtain a conductivity reference from 3D printed copper (see Fig. S.3). The resulting value was 1.25 × 103 S/m, which confirms that the value obtained previously seems low due to the surface rugosity conferred by 3D printing, but is sufficiently high to perform EPD experiments. It is important to note that the electrical conductivity measurement can also be affected by the purity of copper and the material imperfections such as cracks created as a consequence of shrinkage during thermal post-processing [45, 46], but those are not extensively evaluated in this work.

a Optical image of the copper 3D current collector where the surface analysis was performed, b depth view of the valley and hills on the selected area
EPD was employed to deposit the components of a lithium-ion negative electrode onto the conductive 3D printed copper gyroid disc in order to explore its potential use as a current collector. The EPD process has been studied in the past to produce battery cathodes and anodes [47,48,49,50], however, it has never been adopted for scalable manufacturing of complex 3D battery architectures due to the existence of substrate defects in spite of the fast deposition rates, outstanding throwing power, and cost-effectiveness [51]. The EPD process was chosen in this work due to its ability to coat conductive or insulating materials onto a conductive substrate, even when the substrate was geometrically complex [52, 53]. The EPD bath was composed of an optimized mixture of graphite as electroactive material, C45 as conductive carbon, polyvinylidene difluoride as binder, iodine as charging agent, Triton-X100 as dispersant agent, and acetone as solvent, in the amounts specified in the Materials and Methods section. The stability of the coating was ensured by the addition of a small amount of binder and dispersant that did not compromise the electrochemical performance [52, 54]. In regards to the role of iodine, the mechanism widely accepted considers the formation of iodoacetone, suggesting that the surface of suspended graphite and carbon particles become positive as a consequence of the proton formation during acetone iodination ((CH3)2CO + I2 → CH3COCH2I + I− + H+) [55]. After deposition, the 3D gyroid electrode was washed and dried. These conditions promoted thin and homogeneous coverage, with a calculated loading of 36 mgEPDcomponents/gcurrent collector. Note that under different conditions of voltage, temperature and time, the coating thickness and mass can be tuned.
The microstructure of the negative electrode EPD coating was evaluated using SEM analysis and compared to the bare 3D printed copper current collector as reference (see Figs. 6a−f). As shown in Figs. 6a and d, the macroporosity and printed features created by 3D printing were maintained after the EPD process, denoting a thin coating of the EPD bath components of about 10 μm (see Figs. 6g−i). Interestingly, before EPD, the bare 3D printed copper current collector presents pixel-like features (see Fig. 6b) that are intrinsic to the UV projector of the DLP 3D printer. It also exhibits micro-cracks (about 30−35 μm wide × 200 μm long, as shown in Fig. S.4) that were created during the debinding step of the thermal post-processing. As shown in Figs. S.4b and c, the micro-porosity throughout the copper structure consists of holes of about 10 μm × 15 μm, or stripes of about 20 μm long × 2−6 μm wide. After EPD, these features are no longer visible (see Fig. 6e), and it seems that the micro-cracks and surface defects created during the thermal post-processing steps did not affect the adhesion of the EPD coating. Upon closer examination of the lateral side of both samples (see Figs. 6c and f), it can be observed that the features related to the stacking of the successive layers of ~20 µm (intrinsic to the 3D printing process) are no longer visible after EPD of the electrode materials. Homogeneity of the coating was finally confirmed as no uncovered spots were found throughout different areas of the sample.

SEM images of the 3D printed current collector before (a, b and c) and after the EPD process (d, e and f), cross-sectional view of the 3D current collector/negative electrode after EPD (g, h and i)
The energy storage capability of the 3D multi-material current collector/negative electrode was finally demonstrated by performing electrochemical testing in a half-cell configuration. All electrochemical testing was done inside Swagelok®-type cells, where lithium foil was used both as a counter and reference electrode. The electrochemical testing started with ten charge-discharge cycles at C/20 (18.6 mA/g) (see Fig. 7a). The initial discharge capacity was 371 mAh/g, very close to the theoretical capacity value for graphite of 372 mAh/g with an irreversible capacity loss in the first cycle of 13.2% due to solid electrolyte interface (SEI) formation [56]. According to the capacity vs voltage plot of the first discharge (see Fig. 7b), different plateaus were observed at around 0.80 (SEI formation), and at 0.35, 0.18, and 0.10 V vs Li/Li+, attributed to the typical graphite intercalation stages [57, 58]. Also note that since there is no copper oxide, no conversion reaction exists between 1.3 V and−1.5 V vs Li/Li+ [59]. After ten cycles, the rate was changed to C/10 (37.2 mA/g), and the discharge capacity was observed to drop to 153 mAh/g in the 11th cycle and to 108 mAh/g in the 20th cycle (see Fig. 7a). At C/5 (74.4 mA/g), the discharge capacity remained for ten cycles at values between 75 mA/g and 60 mA/g. When coming back to C/20 the capacity slightly recovered to values of 150 mA/g. The gradual capacity loss upon cycling (see Fig. 7b) could be related to the loss of mobile Li+ through side reactions that occur with the electrolyte to form compounds which trap free lithium, reducing the number of Li+ that could shuttle between the electrodes. The loss of mobile ions reduces the maximum capacity that can be achieved by the battery. Another potential explanation is related to the electrode structure as well as electrode adhesion to the copper current collector backbone being damaged upon repetitive Li+ intercalation and deintercalation taking place during cycling. These structural disorders and adhesion issues could potentially arise during cycling as a result of the movement of Li+ in and out of the electrodes, thus reducing the number of Li+ that the electrode can accept into its structure, thus reducing the battery capacity. In future studies, tuning the EPD bath composition could be envisioned. Moreover, this trend can also be partially attributed to the non-optimized geometry of our test cell that uses a planar counter electrode, instead of a complementary 3D electrode.

Electrochemical testing of a 3D printed (a, b) and a planar (c, d) current collector containing graphite and other additives; the corresponding specific capacity vs voltage plots are shown in b and d
For comparison purposes, a planar copper foil was used as a substrate in the same EPD procedure to create a planar graphite electrode. The resulting electrochemical results are shown in Figs. 7c and d. The planar EPD electrode showed equivalent excellent coulombic efficiencies and better cycling stability along the 40 cycles at different C-rates, in comparison with the 3D multi-material current collector/negative electrode. These results indicate that the non-optimized geometry of our test cell is an important cause for capacity fading when cycling the 3D electrode (see Fig. 7a), because the cell geometry impacts the SEI formation across the 3D surfaces of the electrode and the battery kinetics upon cycling. Nonetheless, it is important to mention that 3D printing appears as a solution to manufacture unique and intricate shape-conformable electrode geometries. These geometries can be tailored on demand to maximize the energy storage capability within the final application (eg., smartwatch, drone) while also providing the load-bearing capability. Future studies regarding structural batteries should envisage designing complementary electrodes that incorporate a solid or gel printed electrolyte that allows optimal compatibility.
4 Conclusions
This work demonstrates the 3D printing by means of the VPP process of intricate 3D current collector architectures that can be effectively used as substrates for subsequent EPD of battery electrode materials. Thanks to 3D printing, the development of intricate shape-conformable batteries that can be tailored on-demand can now be envisioned. Shape-conformable 3D batteries could potentially maximize the energy storage capability within the final object in comparison with conventional configurations restricted to stacking or rolling of planar components. The results shown in this paper elucidate that the pairing of 3D printing with electrophoretic deposition has the potential for structural battery manufacturing exhibiting dual functionality: load-bearing and energy storage capabilities. Our results also point to numerous opportunities for further optimization of the manufacturing process steps: on the formulation of the base resin, on the printing parameters, on the design of the printed pieces, on the thermal post-processing, and on the EPD process. Regarding the production of full structural batteries, we envision that the assembly of complementary current collectors containing deposited electroactive materials, as has been shown schematically before [17, 20, 38, 60,61,62,63,64], will allow us to produce full 3D batteries. The separator and electrolyte, as well as the cathode electrode are fertile areas for study and the subject of future work.
Equally significant, this work provides the basis for future 3D printing of complex copper current collector shapes otherwise unattainable by regular means, using a process that requires less energy in comparison with traditional metal 3D printing. Since our copper manufacturing process starts simply with the 3D printing of a photocurable resin, we believe that it could be successfully applied to VPP processes with much higher spatial resolution, such as the process based on two-photon photopolymerization. By leveraging the sub-100 nanometer scale resolution of this technique it is plausible to imagine printing microbattery components and other small consumer electronics such as biomedical implants and micro-UAVs.
References
Liu W, Placke T, Chau KT et al (2022) Overview of batteries and battery management for electric vehicles. Energy Rep 8:4058–4084
Berjoza D, Jurgena I (2017) Effects of change in the weight of electric vehicles on their performance characteristics. Agron Res 15:952–963
Shaffer B, Auffhammer M, Samaras C et al (2021) Make electric vehicles lighter to maximize climate and safety benefits. Nature 598:254–256
Hooftman N, Messagie M, Joint F et al (2018) In-life range modularity for electric vehicles: the environmental impact of a range-extender trailer system. NATO Adv Sci Inst Ser E Appl Sci 8:1016. https://doi.org/10.3390/app8071016
Fu W, Turcheniuk K, Naumov O et al (2021) Materials and technologies for multifunctional, flexible or integrated supercapacitors and batteries. Mater Today 48:176–197
Wu F, Liu M, Li Y et al (2021) High-mass-loading electrodes for advanced secondary batteries and supercapacitors. Electrochem Energy R 4:382–446
Danzi F, Salgado RM, Oliveira JE et al (2021) Structural batteries: a review. Molecules 26:2203. https://doi.org/10.3390/molecules26082203
Asp LE, Bouton K, Carlstedt D et al (2021) A structural battery and its multifunctional performance. Adv Energy Sustain Res 2:2000093. https://doi.org/10.1002/aesr.202000093
Snyder JF, Wong EL, Hubbard CW et al (2009) Evaluation of commercially available carbon fibers, fabrics, and papers for potential use in multifunctional energy storage applications. J Electrochem Soc 156:A215. https://doi.org/10.1149/1.3065070
Asp LE, Johansson M, Lindbergh G et al (2019) Structural battery composites: a review. Funct Compos Struct 1:042001. https://doi.org/10.1088/2631-6331/ab5571
Galos J, Pattarakunnan K, Best AS et al (2021) Energy storage structural composites with integrated lithium-ion batteries: a review. Adv Mater Technol 6:2001059. https://doi.org/10.1002/admt.202001059
Thomas JP, Qidwai MA (2005) The design and application of multifunctional structure-battery materials systems. JOM 57:18–24
Ladpli P, Nardari R, Kopsaftopoulos F et al (2019) Multifunctional energy storage composite structures with embedded lithium-ion batteries. J Power Sources 414:517–529
Liu P, Sherman E, Jacobsen A (2009) Design and fabrication of multifunctional structural batteries. J Power Sources 189:646–650
Kohlmeyer RR, Blake AJ, Hardin JO et al (2016) Composite batteries: a simple yet universal approach to 3D printable lithium-ion battery electrodes. J Mater Chem A Mater Energy Sustain 4:16856–16864
Maurel A, Grugeon S, Armand M et al (2020) Overview on lithium-Ion battery 3D-printing by means of material extrusion. ECS Trans 98:3. https://doi.org/10.1149/09813.0003ecst
Maurel A, Martinez AC, Grugeon S et al (2021) Toward high resolution 3D printing of shape-conformable batteries via vat photopolymerization: review and perspective. IEEE Access 9:140654–140666
Maurel A, Martinez AC, Dornbusch DA et al (2023) What would battery manufacturing look like on the moon and mars? ACS Energy Lett 8:1042–1049
Narita K, Saccone MA, Sun Y et al (2022) Additive manufacturing of 3D batteries: a perspective. J Mater Res 37:1535–1546
Maurel A, Pavone A, Stano G et al (2023) Manufacturing-oriented review on 3D printed lithium-ion batteries fabricated using material extrusion. Virtual Phys Prototyp 18:e2264281. https://doi.org/10.1080/17452759.2023.2264281
Martinez AC, Schiaffino EM, Aranzola AP et al (2023) Multiprocess 3D printing of sodium-ion batteries via vat photopolymerization and direct ink writing. J Phys Energy 5:045010. https://doi.org/10.1088/2515-7655/acf958
Maurel A, Martinez AC, Chavari SB et al (2023) 3D printed TiO2 negative electrodes for sodium-ion and lithium-ion batteries using vat photopolymerization. J Electrochem Soc 170:10. https://doi.org/10.1149/1945-7111/ad0420
Geng Q, Wang D, Chen P et al (2019) Ultrafast multi-focus 3-D nano-fabrication based on two-photon polymerization. Nat Commun 10:2179. https://doi.org/10.1038/s41467-019-10249-2
Nohut S, Schwentenwein M (2022) Vat photopolymerization additive manufacturing of functionally graded materials: a review. J Mater Process Manuf Sci 6:17. https://doi.org/10.3390/jmmp6010017
Lodes MA, Guschlbauer R, Körner C (2015) Process development for the manufacturing of 99.94% pure copper via selective electron beam melting. Mater Lett 143:298–301
Imai K, Ikeshoji TT, Sugitani Y et al (2020) Densification of pure copper by selective laser melting process. Mech Eng J 7:2. https://doi.org/10.1299/mej.19-00272
Ott J, Burghardt A, Britz D et al (2021) Microstructural analysis as a requirement for sinter-based additive manufacturing of highly conductive copper. Pract Metallogr 59:434–444
Bai Y, Williams CB (2015) An exploration of binder jetting of copper. Rapid Prototyping J 21:177–185
Mooraj S, Welborn SS, Jiang S et al (2020) Three-dimensional hierarchical nanoporous copper via direct ink writing and dealloying. Scr Mater 177:146–150
Maurel A, Kim H, Russo R et al (2021) Ag-coated Cu/polylactic acid composite filament for lithium and sodium-ion battery current collector three-dimensional printing via thermoplastic material extrusion. Front Energy Res 9:651041. https://doi.org/10.3389/fenrg.2021.651041
Jiang Q, Zhang P, Yu Z et al (2021) A review on additive manufacturing of pure copper. Coat World 11:740. https://doi.org/10.3390/coatings11060740
Steyrer B, Busetti B, Harakály G et al (2018) Hot lithography vs. room temperature DLP 3D-printing of a dimethacrylate. Addit Manuf 21:209–214
Yugang D, Yuan Z, Yiping T et al (2011) Nano-TiO2-modified photosensitive resin for RP. Rapid Prototyping J 17:247–252
Hinczewski C, Corbel S, Chartier T (1998) Ceramic suspensions suitable for stereolithography. J Eur Ceram Soc 18:583–590
Yee DW, Citrin MA, Taylor ZW et al (2021) Hydrogel-based additive manufacturing of lithium cobalt oxide. Adv Mater Technol 6(2):2000791. https://doi.org/10.1002/admt.202000791
Saccone MA, Gallivan RA, Narita K et al (2022) Additive manufacturing of micro-architected metals via hydrogel infusion. Nature 612(7941):685–690
Manogharan G, Yelamanchi B, Aman R et al (2016) Experimental study of disruption of columnar grains during rapid solidification in additive manufacturing. JOM 68:842–849
Martinez AC, Maurel A, Aranzola AP et al (2022) Additive manufacturing of LiNi1/3Mn1/3Co1/3O2 battery electrode material via vat photopolymerization precursor approach. Sci Rep 12:19010. https://doi.org/10.1038/s41598-022-22444-1
Liu S, Wang Y-M, Han J (2017) Fluorescent chemosensors for copper(II) ion: structure, mechanism and application. J Photochem Photobiol C Photochem Rev 32:78–103.
Keely WM, Maynor HW (1963) Thermal studies of nickel, cobalt, lron and copper oxides and nitrates. J Chem Eng Data 8:297–300
Yi F, DeLisio JB, Nguyen N et al (2017) High heating rate decomposition dynamics of copper oxide by nanocalorimetry-coupled time-of-flight mass spectrometry. Chem Phys Lett 689:26–29
L’vov BV (2000) Mechanism of carbothermal reduction of iron, cobalt, nickel and copper oxides. Thermochim Acta 360:109–120
McLaughlin KK, Clegg WJ (2008) Deformation underneath low-load indentations in copper. J Phys D Appl Phys 41:074007. https://doi.org/10.1088/0022-3727/41/7/074007
Ryspayeva A, Jones TDA, Esfahani MN et al (2019) Selective electroless copper deposition by using photolithographic polymer/ag nanocomposite. IEEE Trans Electron Devices 66:4. https://doi.org/10.1109/TED.2019.2897258
Timoshevskii V, Ke Y, Guo H et al (2008) The influence of surface roughness on electrical conductance of thin Cu films: an ab initio study. J Appl Phys 103:113705. https://doi.org/10.1063/1.2937188
Kong LW, Bai W, Guo AG (2012) Effects of cracks on the electrical conductivity of a fissured laterite: a combined experimental and statistical study. Geotech Test J 35:20120070. https://doi.org/10.1520/gtj20120070
Kanamura K, Goto A, Hamagami J et al (2000) Electrophoretic fabrication of positive electrodes for rechargeable lithium batteries. Electrochem Solid-State Lett 3(6):259. https://doi.org/10.1149/1.1391118
Dashairya L, Das D, Saha P (2020) Electrophoretic deposition of antimony/reduced graphite oxide hybrid nanostructure: a stable anode for lithium-ion batteries. Mater Today Commun 24:101189. https://doi.org/10.1016/j.mtcomm.2020.101189
Esper JD, Helmer A, Wu Y et al (2021) Electrophoretic deposition of out-of-plane oriented active material for lithium-ion batteries. Energy Technol 9(4):2000936. https://doi.org/10.1002/ente.202000936
Han Y, Ye L, Boateng B et al (2019) Direct electrophoretic deposition of an ultra-strong separator on an anode in a surfactant-free colloidal system for lithium ion batteries. J Mater Chem A Mater Energy Sustain 7(4):1410–1417
Boccaccini AR, Dickerson JH (2013) Electrophoretic deposition: fundamentals and applications. J Phys Chem B 117:1501. https://doi.org/10.1021/jp211212y
Mazor H, Golodnitsky D, Burstein L et al (2011) Electrophoretic deposition of lithium iron phosphate cathode for thin-film 3D-microbatteries. J Power Sources 198:264–272
Hajizadeh A, Shahalizade T, Riahifar R et al (2022) Electrophoretic deposition as a fabrication method for Li-ion battery electrodes and separators—a review. J Power Sources 535:231448. https://doi.org/10.1016/j.jpowsour.2022.231448
Martinez AC, Rigaud S, Grugeon S et al (2022) Chemical reactivity of lithium difluorophosphate as electrolyte additive in LiNi0.6Co0.2Mn0.2O2/graphite cells. Electrochim Acta 426:140765. https://doi.org/10.1016/j.electacta.2022.140765
Dusoulier L, Cloots R, Vertruyen B et al (2011) YBa2Cu3O7−x dispersion in iodine acetone for electrophoretic deposition: surface charging mechanism in a halogenated organic media. J Eur Ceram Soc 31:1075–1086
Lin YX, Liu Z, Leung K et al (2016) Connecting the irreversible capacity loss in Li-ion batteries with the electronic insulating properties of solid electrolyte interphase (SEI) components. J Power Sources 309:221–230
Asenbauer J, Eisenmann T, Kuenzel M et al (2020) The success story of graphite as a lithium-ion anode material—fundamentals, remaining challenges, and recent developments including silicon (oxide) composites. Sustain Energy Fuels 4(11):5387–5416
Maurel A (2020) Thermoplastic composite filaments formulation and 3D-printing of a lithium-ion battery via fused deposition modeling. Dissertation, Université de Picardie Jules Verne, Amiens
Grugeon S, Laruelle S, Herrera-Urbina R et al (2001) Particle size effects on the electrochemical performance of copper oxides toward lithium. J Electrochem Soc 148:A285. https://doi.org/10.1149/1.1353566
McKelvey K, Brunet Cabré M, Esmeraldo Paiva A (2020) Continuum simulations for microscale 3D batteries. Curr Opin Electrochem 21:76–83
Kühnelt H, Beutl A, Mastropierro F et al (2021) Structural batteries for aeronautic applications—state of the art, research gaps and technology development needs. Aerospace 9(1):7. https://doi.org/10.3390/aerospace9010007
Maurel A, Martinez Maciel AC, Panier S et al (2021) Lithium-Ion battery 3D printing: from thermoplastic material extrusion to vat photopolymerization process. Meet Abst 2:30. https://doi.org/10.1149/MA2021-02130mtgabs
Carlstedt D, Runesson K, Larsson F et al (2022) Computational modelling of structural batteries accounting for stress-assisted convection in the electrolyte. Int J Solids Struct 238:111343. https://doi.org/10.1016/j.ijsolstr.2021.111343
Martinez Maciel AC, Maurel A, Sreenivasan ST et al (2021) 3D printing of lthium-Ion battery components via vat photopolymerization. Meet Abstr 2:55. https://doi.org/10.1149/MA2021-02155mtgabs
Acknowledgements
This work was supported by the French Fulbright Program, the Murchison Chair from The University of Texas at El Paso, and the Friedman Endowment at Youngstown State University. The authors would like to thank Matthieu Courty for providing TGA/MS data, and Saqlain Zaman and Yirong Lin for their help on a thermal post-processing experiment. This work was also supported by the Laboratory Directed Research and Development program at Sandia National Laboratories, a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA-0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martinez, A.C., Maurel, A., Yelamanchi, B. et al. Combining 3D printing of copper current collectors and electrophoretic deposition of electrode materials for structural lithium-ion batteries. Adv. Manuf. (2024). https://doi.org/10.1007/s40436-024-00514-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40436-024-00514-z