
Abstract
Recent advances in molecular biology and biotechnology have opened the way for the development of new imaging and treatment modalities based on the biological properties of tissues. In oncology, the major advances have been achieved using peptide and antibody targeting molecules. This approach is based on: (1) the identification of new target structures, and (2) the application of new techniques for the development of new biocompatible molecules. These techniques rely on the identification of lead compounds followed by the screening of various derivatives of these compounds. For this purpose, high-throughput methods have been applied that generate a vast library of possible binders which can be used to screen the whole population for the few variants that show the property of interest. An attractive feature of this strategy is the huge number of candidate molecules that can be used for further evaluation, which consists of the characterization of structure–function relationships and the further improvement by rational design of corresponding analogs.
Similar content being viewed by others

Introduction
In oncology, targeted therapy is an alternative therapeutic approach to conventional chemo- and radiotherapy and may increase the efficiency of antitumor treatment of non-operable tumors and metastases.
Chemotherapeutic drugs or radioactive isotopes (endoradiotherapy) are delivered to the tumor using, primarily, antibodies or peptides as carrier molecules that specifically bind, respectively, to antigens and surface-associated proteins predominantly expressed on tumor cells. In endoradiotherapy, beta- or alpha-emitting radionuclides are coupled to the carrier molecules which selectively accumulate in the tumor but not in healthy tissues, thereby decreasing radiation toxicity to the peripheral organs [1–3]. Alternatively, instead of therapeutically useful alpha or beta isotopes, target-specific carrier molecules may be coupled with either a positron-emitting or gamma isotope and used for the identification of cancerous lesions in the patient.
Because of their high affinity for the target structure, antibodies, primarily, have been used for the delivery of the therapeutic agent to the tumor. However, due to their size, tissue penetration may be limited, especially in tumors typically characterized by high pressure in the intercellular space. Furthermore, antibodies often associate with plasma proteins, leading to prolonged circulation and, therefore, to a high background and a high level of bone marrow exposure. Small molecules, especially peptides, are, instead, an attractive alternative for imaging and endoradiotherapy, because they usually show high affinity for the tumor target and better tissue penetration, leading to rapid clearance. In addition, peptides, compared with antibodies, show a lower immunogenicity and are easily synthesized.
Several types of cancer are characterized by overexpression of specific receptor proteins which can be used as targets for therapy and imaging. Somatostatin receptor imaging and therapy represent the clinical paradigm for receptor targeting using peptides. The high-binding affinity of the peptide for its receptor and the internalization of the receptor–peptide complex facilitate retention of the tracer in the tumor, whereas its relatively small size results in rapid clearance from the blood. Somatostatin receptors are expressed in a variety of tumors including neuroendocrine tumors of the pancreas and the intestine, pituitary adenoma, pheochromocytoma, paraganglioma, small cell lung cancer, neuroblastoma, medullary thyroid carcinoma and meningioma. Due to their high receptor density, gastroenteropancreatic neuroendocrine tumors, pheochromocytomas, paragangliomas and bronchial carcinoids are promising candidates for therapy with radiolabeled peptides [4–7].
Rational design and black box approaches in the development of new ligands
The development and evaluation of new molecules suitable for imaging and/or therapy may be realized using either a rational design or a black box approach [8]. The rational design depends on the information obtained from the study of structure–function relationships and the conformational properties of the molecules involved. This approach relies on the identification of lead compounds followed by the screening of various derivatives of these compounds. Recently, small molecules have been developed for the diagnosis and therapy of prostate cancer. Almost all prostate cancers overexpress the prostate-specific membrane antigen (PSMA), a type II transmembrane protein with glutamate-carboxypeptidase activity. The expression of PSMA in most prostate cancers, its large extracellular domain, and the fact that ligands are internalized after binding make it an outstanding target for nuclear medicine. Early work in the development of inhibitors of the enzyme identified a number of small molecule inhibitors which have been modified for application as radiopharmaceuticals [9–13]. The first application of these high-affinity small-molecule inhibitors of PSMA in patients with prostate cancer demonstrated their ability to rapidly detect lesions in soft tissue, bone, and the prostate gland using SPECT/CT or PET as early as 1–4 h after injection [9–13]. In particular SPECT/CT with iodinated inhibitors of PSMA, 123I-MIP-1072 and 123I-MIP-1095 detected tumor lesions in soft tissue, bone, and the prostate gland as early as 1–4 h after tracer injection [9, 10]. In preclinical studies, 123I-MIP-1072 was evaluated in a chemotherapy model of prostate cancer and tumor uptake was shown to be directly proportional to viable tumor mass. This finding suggests that it may also be useful for monitoring response to therapy. A 68Ga PSMA ligand (Glu-NH-CO-NH-Lys(Ahx)-HBED-CC) was compared to fluoro-methyl choline and proved to be superior in most cases [14]. In Figs. 1 and 2, PET/CT scans using a 68Ga-labeled PMSA ligand demonstrate successful detection of prostate cancer (Fig. 1) and bone metastases (Figs. 2, 3).

PET/CT with a 68Ga-labeled PSMA ligand (Glu-NH-CO-NH-Lys(Ahx)-HBED-CC) visualizes prostate cancer in a patient with a PSA value of 17 (color figure online)

PET/CT with Glu-NH-CO-NH-Lys(Ahx)-HBED-CC: visualization of multiple bone metastases (color figure online)

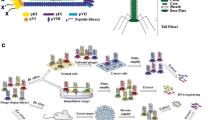
Principle of biopanning with phage and ribosome display. The libraries are exposed to the target, non-binders removed and binders amplified for the next round of exposure (color figure online)
Furthermore, bioinformatics can be applied to work out the information encoded in the human genome by screening the sequence for identity and similarity with known gene families, using a range of sequence-search methods or by comparing sequences between different organisms. All sequence-search methods use sequence similarity as a basis for identifying homologs. It is relatively easy to identify homologs that share a sequence identity of 30 % or more using pairwise sequence-search methods, but the success rate decreases dramatically in the case of homologs with a sequence identity in the range of 20–30 % or lower. Comparative genomics evaluates similarities and differences between several organisms. It has been speculated that approximately 40 % of human genes have unknown biological functions. This pool of genes may contain families of genes which are associated with diseases and could serve as possible biological targets [1, 15]. Furthermore, gene expression data may be used to identify genes which are overexpressed in diseased states as compared to the normal state. These may serve as target structures for the development of new diagnostic or therapeutic approaches. However, since protein levels often do not reflect mRNA levels, gene profiling data have to be confirmed at the post-translational level.
In contrast to the rational design approach, the central idea of combinatorial chemistry is to design vast libraries of possible variants of the molecule of interest and to screen the whole population for the few variants that show the desired property. The appeal of this concept lies in the huge number of candidate molecules that can be used for further evaluation. For example, the diversity of a hexapeptide library randomizing the 20 natural amino acids at each position will amount to 64 million sequence variants. The key to success is to develop sensitive and efficient high-throughput screening techniques capable of rapidly selecting molecules with a desired property. Following characterization of the structure–function relationships of the lead compounds identified through this process, the approach can be further improved through rational design of analogs.
There are two ways of creating high-diversity molecular libraries: the chemical way, which is based on random or directed synthesis of compounds typically displayed on solid supports such as activated beads, pins or batch arrays, and the biological way, which uses recombinant techniques for the development of genetically encoded and expressed systems to generate diverse ribonucleic acid (RNA), deoxyribonucleic acid (DNA), protein and peptide libraries [16]. These recombinant techniques include display techniques such as phage display, ribosome display and mRNA display. The resulting molecules can be further improved by subjecting them to mutation and amplification in order to create a new pool of diverse molecules for further screening rounds. The iterative process of mutation, selection and amplification to evolve the best-fit molecule is known as ‘directed evolution’.
The structure–function relationships and conformational properties of, for example, a peptide structure may provide the lead for the design of non-peptide analogs with equal or often enhanced bioactivity. Strategies in the development of peptidomimetics include peptide backbone modifications, β-turn-based architecture and constrained cyclic structures. For example, peptidomimetic ligands have been developed on the basis of the sequence of target proteins for SH2 and SH3 domains, which are involved in the initiation of the signal transduction of activated tyrosine kinase. These compounds are intended to be used for the treatment of acute lymphocytic leukemias or HER2/neu-expressing tumors such as breast and ovarian cancer [15, 17].
Furthermore, the metabolic pathways for the synthesis of natural biopolymers can be manipulated, shuffled and recombined in vitro. The application of combinatorial chemistry to metabolic engineering results in structurally diverse libraries of molecules with novel functional properties. In this respect, engineered biosynthesis of the polyketide pathway has been used to design therapeutically active compounds, such as antibiotics and anticancer agents, by variation of the start unit and chain length as well as the degree of reduction and the type of elongation [18].
Display systems
The identification of useful new molecules from huge libraries is achieved through the use of high-throughput methods. Display systems are applied to selected molecules from libraries in which peptides or proteins (phenotype) are physically linked to their corresponding encoding sequences (genotype). In addition, these systems can be used to modify the biophysical properties of the displayed molecules by evolution through cycles of mutation, selection and replication. Essentially, two types of display system are available: cell-based systems such as phage display or cell surface display, and cell-free systems such as ribosome display and mRNA display.
Phage surfaces can be modified without losing their ability to infect bacteria. In contrast to lytic bacteriophages such as T4, filamentous phages replicate and assemble without killing their host. Therefore, they are good candidates for application in biopanning. The term ‘biopanning’ refers to the process of displaying peptide or protein libraries to targets with the aim of screening for high-affinity binders. Phages can be used after modification of their genome so that they present library-encoded peptides on their surface. The common principle is the fusion of peptide libraries with the carboxy-terminal domain of the minor coat protein, pIII [1–3]. There are five copies of pIII at each end of the phage virion; pIII, together with the minor coat surface protein pVI, is involved in bacterial cell binding and termination of phage particle assembly during self-assembly of the virions in the host cell. The resulting fusion protein is displayed on the phage surface. In order to obtain correct folding of the presented peptide, a spacer usually comprising three glycines is inserted between the minor coat protein and the peptide library. Basically there are two phage library variants: polyvalent and monovalent [3]. Polyvalent phage display is usually used for libraries encoding small peptides. Each capsid protein is able to display the polypeptide without loss of function for phage replication and assembly. The viral vector is modified by insertion of a library connected to the minor coat protein. In monovalent phage display, the encoded polypeptides are too large to be displayed by all five capsid proteins. Therefore, a mosaic of recombinant and wild-type capsid proteins is produced by the phagemid vector and a helper phage. The helper phage provides all the proteins required for phagemid replication, ssDNA production and packaging and escape from the host.
The affinity constants of the molecules identified by phage display are usually in the micromolar to nanomolar range. Polyvalent phage display, presenting multiple copies of displayed peptides, generally detects lower-affinity binding compared with monovalent phage display. In order to optimize the properties of the identified molecules, affinity maturation for low-range binders can be performed individually by generation of secondary libraries covering mutated peptides [19].
Besides phage display with the M13 phage, considerable efforts are being made to develop alternative phage display systems. One disadvantage of filamentous phages is that proteins fold properly only in the periplasm of the host bacterium. Filamentous phages are assembled in the cytoplasm and secreted without lysis of the host. Some displayed peptides may not be able to cross the lipid bilayer of the inner membrane. Therefore, lytic bacteriophages have been used which assemble in the cytoplasm and are released through cell lysis so that the correct transfer of the hybrid capsid protein is guaranteed [20].
The phage display method is used for various applications, such as mapping and mimicking of epitopes, identifying new receptors and natural ligands, identifying high-affinity antibodies and analogs, isolating specific antigens binding to bioactive compounds, producing novel enzyme inhibitors and DNA-binding proteins, and probing cellular and tissue-specific processes. Phage display has successfully been applied for the identification of novel peptides with high specificity for target proteins overexpressed in the tumor neovasculature, such as delta-like ligand 4 (Dll4), or for human lung carcinoma, mammary carcinoma, prostate carcinoma and neuroblastoma cells [1, 2, 21–26].
Human antibody fragment libraries displayed on phages are obtained by cloning of V H and V L domains of human antibodies in filamentous phage DNA and displaying them on the surface coat of phages as single-chain Fv or Fab fragments. This results in the isolation of high-affinity human antibodies of many different antigens. For the production of recombinant single-chain Fv (scFv) fragments, the V H and V L domains are linked together with a polypeptide bridge of about 15 hydrophilic amino acid residues and screened by phage display. However, these monovalent antibody fragments are cleared rapidly from the blood as a result of their small molecular size and because of their monodenticity. Since intact antibodies are generally polyvalent molecules, Fab or scFv molecules have been combined into dimers or higher multimers such as diabodies, triabodies and minibodies to produce highly functional reagents of 60–120 kDa in size. Diabodies and triabodies show rapid tumor penetration, are relatively flexible with respect to the orientation of antibody-binding sites, and reveal a higher functional affinity with reduced kidney clearance rates. Minibodies have proven to be efficient in localizing tumor xenografts in mice and show high retention in tumor cells [15].
In vitro selection systems such as ribosome display and mRNA display, although technically demanding, have two important advantages: the ability to handle very large libraries (1013–1014 different sequences) and the possibility of using polymerase chain reaction (PCR) amplification steps to introduce further diversity into the system, which may be used to evolve proteins through an iteration of random mutagenesis and selection (affinity maturation) [1–3, 27]. In phage display, the efficiency of transformations is always limited and, therefore, the diversity of the library is lower than theoretically possible. For example, the diversity of the commercially available library PH.D.12™ is diminished from the theoretical value of 4.1 × 1015 to 109 [27]. Both ribosome display and mRNA display have been used to select linear peptides or single-chain antibodies that bind to protein targets with low picomolar affinities.
As with phage display, ribosome display is based on physical phenotype–genotype linkage of the peptides, miniproteins or antibodies encoded by the library. In ribosome display the phenotype, a nascent peptide, is linked to its genotype, the corresponding mRNA, through the formation of stable peptide–ribosome–mRNA complexes. The mRNAs in the library used have no stop codon, therefore, the scanning process of the ribosome is extended to the end of the mRNA molecule. The corresponding polypeptide emerges from the ribosome while its end is still fixed within the ribosomal tunnel, and its last amino acid is connected to the peptidyl-tRNA. The absence of stop codons prevents the binding of release factors, which normally catalyze the release of the polypeptide from the ribosome. This leads to the formation of a protein–ribosome–RNA complex, which connects the phenotype to the genotype [3, 28]. The different steps of ribosome display are: construction of a large DNA library encoding the polypeptide of interest fused in frame to a C-terminal spacer, in vitro transcription of the library into mRNA, and in vitro translation. Thereafter, the complexes consisting of protein, mRNA and ribosomes are exposed to the target structure; this phase includes washing steps to remove non-binders, which leads to enrichment of binding molecules. After reverse transcription of the RNA, amplification is performed by PCR followed by the next round of transcription, translation and exposure. During this amplification step diversity may be further increased using error-prone PCR. Identification of high-affinity binders is usually obtained after three to six rounds [28]. Figure 4 shows visualization of a neuroendocrine tumor in a rat model with an iodine-labeled peptide directed against Dll4 which was identified by ribosome display [26].

Scintigraphy of a rat bearing a neuroendocrine tumor using a Dll4-specific peptide identified by ribosome display (color figure online)
Random libraries were used to select peptides for binding to several targets such as an antibody against dynorphin B, the prostate-specific antigen, streptavidin and lysozyme, or to identify the main antigenic polypeptides of Staphylococcus aureus. The affinity of the peptides ranged from 7.2 to 140 nM. Similar results were obtained for scFv fragments using targets such as the GCN4 leucine zipper, DNA structures in eukaryotic telomeres, progesterone and fluorescein. In this setting all selected scFv fragments acquired genetic mutations during the cycles of ribosome display (due to errors introduced by the DNA polymerase), which led to significant improvement (by up to 40-fold) in their affinities for the antigen. The best scFv fragments selected showed affinities in the low picomolar range and could be further improved by off-rate selection and error-prone PCR [29].
In mRNA display, a complex between mRNA and the polypeptide encoded by the mRNA is applied in a specific selection process [30]. The procedure differs from that of ribosome display in the covalent nature of the linkage between the mRNA and the protein in the mRNA–protein complex, which is achieved by linkage of the two molecules through a small adaptor molecule, typically puromycin. The different steps include transcription into mRNA of a large DNA library encoding the molecules of interest, and free of stop codons, ligation of an adaptor molecule to the 3′-ends, and in vitro translation [1]. This procedure results in a peptide bond between the adaptor molecule and the C-terminal amino acid residue in the polypeptide chain. After introduction of cDNA chains by reverse transcription for stabilization and ease of recovery of the genetic information, the cDNA/mRNA protein library is exposed to the target. Isolation of binders is obtained by affinity chromatography or immunoprecipitation of the target structure. The cDNA is then used for further amplification and the following panning rounds. Four to ten selection rounds are usually necessary to select proteins with nanomolar affinity for a given target, as in the case of engineered libraries of linear peptides, constrained peptides, single-domain antibody mimics, and variable heavy domains of antibodies and single-chain antibodies. Using targets such as an anti-c-Myc antibody or streptavidin, binders with a K D down to 2.5 nM were found [1, 31].
Scaffold structures
The advantage of biomolecules derived from constrained peptides compared to linear peptides lies in their complex structure, which is similar to that of globular proteins, and their smaller loss of entropy upon target binding [28, 32]. Binding surfaces for proteins have been engineered on the surfaces of helical bundles or similar molecules [28, 32]. Table 1 provides an overview of the most commonly used scaffolds. Some scaffolds, such as those based on lipocalins, the tenth type III fibronectin domain or helical bundles such as affibodies which are derived from the Z-domain of S. aureus protein A, have been used for the selection of high-affinity binders [33]. The fibronectin type III domain, which has an immunoglobulin-like fold but is smaller than a V H domain, was used to generate mRNA display libraries where the diversity was concentrated in three exposed loops by randomizing the 21 residues in these loops analogously to antibody complementary-determining regions. Selection against the tumor necrosis factor α (TNF-α) followed by affinity maturation with error-prone PCR resulted in Fn3-like domains with affinities down to 20 pM [34]. Similar results were obtained with several other human targets, including vascular endothelial growth factor receptor two (VEGF-R2) or specific conformational states of the estrogen receptor ERα, which were induced by the application of agonists, antagonists or ERα modulators. Furthermore, a disulphide-constrained library based on the knottin trypsin inhibitor EETI-II was designed by randomizing the six residues of the trypsin-binding site. mRNA display selected new trypsin-binding peptides, which were found to be highly homologous to wild-type EETI-II [28, 32].
Affibodies are three-helix-bundle 58-amino acid length proteins derived from the Z-domain of S. aureus protein A, which binds to the Fc domain of IgG antibodies. A phage display library has been constructed by random mutagenesis of residues from the Z-domain binding surface and binders were isolated for a variety of target structures such as human insulin, Taq DNA polymerase, HIV-1 gp120, human apolipoprotein A-1, amyloid β peptides and HER2 [32, 35, 36]. Many studies have been done with an anti-HER2 affibody (ZHER2:342) that shows highly selective uptake in HER2-expressing tumors with excellent image contrast. However, the small size (7 kDa) of the molecule caused rapid glomerular filtration and high kidney uptake. Therefore, a dimeric affibody was fused to albumin thereby reducing renal uptake by a factor of 25. Treatment of a tumor with high HER2 expression (SKOV-3) with 17 or 22 MBq of a 177Lu-labeled compound (177Lu-CHX-A″-DTPA-ABD-(ZHER2:342)2) completely prevented the growth of tumors. Even in xenografts with low HER2 expression (LS174T), therapy with this molecule caused a small but significant increase in survival [36].
Lipocalins are small polypeptides (160–180 residues) involved in the storage or transport of metabolites that fold into an eight-stranded antiparallel β barrel with a cavity formed from four loops. Modulation of these proteins by randomization of up to 17 residues in the loops was used to select binders (anticalins) for digoxigenin and fluorescein [33, 37]. Novel strategies for the creation of non-natural receptors are based on repeat proteins [28, 38]. These consist of repeating structural units of 20–40 residues, which stack together to form elongated protein domains with a continuous target-binding surface. Natural repeat proteins, such as ankyrin or leucine-rich repeat proteins, which are involved in a wide range of biological processes, have been used as scaffolds. The modularity of these proteins allows the development of novel evolutionary strategies, such as module shuffling, module insertions or module deletions. Ankyrin repeat protein libraries have been used in selections for binding against several globular proteins with affinities in the low nanomolar range [1, 29]. For example, selection from libraries of two or three repeats flanked by capping repeats, expressed in a ribosome display format, allowed the isolation of binders with nanomolar affinity for the maltose-binding protein and two eukaryotic kinases [1, 29]. In general, small size and robust structure are promising features of scaffolds for the creation of libraries of binders.
Ligands based on nucleic acids
DNA and RNA are basically involved in storage and expression of genetic information and may function similarly to proteins. Nucleic acids, given that they are folded into more globular structures, which usually include both helical structures and long-distance tertiary contacts, are able to bind to a variety of target structures. Examples of these structured nucleic acid molecules are aptamers, ribozymes, deoxyribozymes and riboswitches, which show a wide range of target specificities and affinities. In many bacteria, RNA aptamers and riboswitches have been shown to be involved in metabolite sensing and regulation of gene expression.
Due to their capacity to target molecules with high affinity, natural or engineered aptamers have been considered promising candidates for therapeutic applications in vivo [1, 2].
Engineered aptamers may be created using directed evolution techniques followed by selective amplification in order to enrich the population with variants that bind to a particular target [39, 40]. The functional characteristics of these molecules, for instance, an anti-VEGF (vascular endothelial growth factor) aptamer and two anti-clotting aptamers, are similar to those of antibodies specifically recognizing proteins or small molecule ligands at target concentrations in the nanomolar or picomolar range. However, deficient delivery into the target cells and degradation by nuclease might limit the use of aptamers for therapeutic applications. In order to avoid enzymatic degradation, chemical modifications of the backbone (such as phosphorothioate linkages or modifications at the ribose moiety) could be introduced, resulting in partial nuclease resistance. Furthermore, employing the same production protocol, mirror-image aptamers, called Spiegelmers, can be synthesized as stable receptors for their corresponding ligands [39, 40]. The chiral configuration (L-RNA) of Spiegelmers represents a mirror image of the naturally occurring D-RNA. Although the spontaneous degradation caused by the inherent chemical instability of RNA should theoretically remain unchanged, Spiegelmers have been reported to be resistant to degradation by most nucleases.
Besides their important role as promising therapeutic agents, aptamers could also function as biosensors. Binding to ligands is usually accompanied by a conformational change of aptamers. Therefore, following the introduction of fluorescent tags into the aptamer molecule in solution or immobilized on surfaces, specific proteins or small molecules could be identified by the change in fluorescence that occurs upon ligand binding.
In vitro evolution may also be used to create new ribozymes that catalyze RNA cleavage as well as many other chemical reactions, or ribozymes that covalently attach to specific proteins. Thus designer ribozymes could be created which selectively couple to many different potential therapeutic or diagnostic protein targets.
Conclusion
In recent years, a huge amount of information about tumor biology has been obtained by means of molecular biology techniques. These data can be used to develop new molecular structures that may serve as potential diagnostic or drug discovery targets. In addition, novel biotechnology methods have opened up a huge segment of sequence space that can be scanned for biomolecules applicable to these ends. Furthermore, there are many known scaffold structures which may be used to improve the molecule in terms of its binding affinity and stability. Taken together these techniques represent promising tools of great interest for the establishment of new diagnostic and therapeutic procedures.
References
Haberkorn U, Eisenhut M, Altmann A, Mier W (2008) End radiotherapy with peptides: status and future development. Curr Med Chem 15:219–234
Haberkorn U, Markert A, Eisenhut M, Mier W, Altmann A (2011) Development of molecular techniques for imaging and treatment of tumors. Q J Nucl Med Mol Imaging 55:655–670
Marr A, Markert A, Altmann A, Askoxylakis V, Haberkorn U (2011) Biotechnology techniques for the development of new tumor specific peptides. Methods 55:215–222
Maecke HR, Hofmann M, Haberkorn U (2005) Gallium-68 labeled peptides in tumor imaging. J Nucl Med 46:S172–S178
Waldherr C, Pless M, Maecke HR, Haldemann A, Mueller-Brand J (2001) The clinical value of [90Y-DOTA]-D-Phe1-Tyr3-octreotide (90Y-DOTATOC) in the treatment of neuroendocrine tumours: a clinical phase II study. Ann Oncol 12:941–945
Waldherr C, Pless M, Maecke HR, Schumacher T, Crazzolara A, Nitzsche EU, Haldemann A, Mueller-Brand J (2002) Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq (90)Y-DOTATOC. J Nucl Med 43:610–616
Bodei L, Cremonesi M, Zoboli S, Grana C, Mazzetta C, Rocca P, Caracciolo M, Mäcke HR, Chinol M, Paganelli G (2003) Receptor-mediated radionuclide therapy with 90Y-DOTATOC in association with amino acid infusion: a phase I study. Eur J Nucl Med Mol Imaging 30:207–216
Haberkorn U, Altmann A, Eisenhut M (2002) Functional genomics and proteomics: the role of nuclear medicine. Eur J Nucl Med Mol Imaging 29:115–132
Maresca KP, Hillier SM, Femia FJ, Keith D, Barone C, Joyal JL, Zimmerman CN, Kozikowski AP, Barrett JA, Eckelman WC, Babich JW et al (2009) A series of halogenated heterodimeric inhibitors of prostate-specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer. J Med Chem 52:347–357
Hillier SM, Maresca KP, Femia FJ, Marquis JC, Foss CA, Nguyen N, Zimmerman CN, Barrett JA, Eckelman WC, Pomper MG, Joyal JL, Babich JW (2009) Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res 69:6932–6940
Eder M, Schäfer M, Bauder-Wuest U, Hull WE, Wängler C, Mier W, Haberkorn U, Eisenhut M (2012) 68 Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug Chem 23:688–697
Afshar-Oromieh A, Malcher A, Eder M, Eisenhut M, Linhart HG, Hadaschik BA, Holland-Letz T, Giesel FL, Kratochwil C, Haufe S, Haberkorn U, Zechmann CM (2013) PET imaging with a [68 Ga] gallium-labelled PSMA ligand for the diagnosis of prostate cancer: biodistribution in humans and first evaluation of tumour lesions. Eur J Nucl Med Mol Imaging 40:486–495
Eder M, Eisenhut M, Babich J, Haberkorn U (2013) PSMA as a target for radiolabelled small molecules. Eur J Nucl Med Mol Imaging 40:819–823
Afshar-Oromieh A, Zechmann CM, Malcher A, Eder M, Eisenhut M, Linhart HG, Holland-Letz T, Hadaschik BA, Giesel FL, Debus J, Haberkorn U (2014) Comparison of PET imaging with a 68 Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur J Nucl Med Mol Imaging 41:11–20
RyuDD Nam DH (2000) Recent progress in biomolecular engineering. Biotechnol Prog 16:2–16
Wong D, Robertson A (2004) Applying combinatorial chemistry and biology to food research. J Agric Food Chem 52:7187–7198
Smithgall TE (1995) SH2 and SH3 domains: potential targets for anti-cancer drug design. J Pharmacol Toxicol Methods 34:125–132
Gaisser S, Kellenberger L, Kaja A et al (2003) Direct production of ivermectin-like drugs after domain exchange in the avermectinpolyketide synthase of Streptomyces avermitilis ATCC31272. Org Biomol Chem 1:2840–2847
Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH (2006) Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods 313:129–139
Castagnoli L, Zucconi A, Quondam M, Rossi M, Vaccaro P, Panni S, Paoluzi S, Santonico E, Dente L, Cesareni G (2001) Alternative bacteriophage display systems. Comb Chem High Throughput Screen 4:121–133
Zitzmann S, Mier W, Schad A, Kinscherf R, Askoxylakis V, Altmann A, Krämer S, Eisenhut M, Haberkorn U (2005) A new prostate carcinoma binding peptide (DUP-1) for tumor imaging and therapy. Clin Cancer Res 11:139–146
Askoxylakis V, Zitzmann S, Mier W, Graham K, Krämer S, von Wegner F, Fink RHA, Schwab M, Eisenhut M, Haberkorn U (2005) Preclinical evaluation of the breast cancer cell-binding peptide, p160. Clin Cancer Res 11:6705–6712
Askoxylakis V, Garcia-Boy R, Rana S, Krämer S, Hebling U, Mier W, Altmann A, Markert A, Debus J, Haberkorn U (2010) A new peptide ligand for targeting human carbonic anhydrase IX, identified through the phage display technology. PLoS ONE 5:e15962
Nothelfer EM, Zitzmann-Kolbe S, Garcia-Boy R, Krämer S, Herold-Mende C, Altmann A, Eisenhut M, Mier W, Haberkorn U (2009) Identification and characterization of a peptide with affinity to head and neck cancer. J Nucl Med 50:426–434
Zoller F, Markert A, Barthe P, Zhao W, Weichert W, Askoxylakis V, Altmann A, Mier W, Haberkorn U (2012) Combination of phage display and molecular grafting generates highly specific tumor-targeting miniproteins. Angew Chem Int Ed Engl 51:13136–13139
Zoller F, Markert A, Barthe P, Hebling U, Altmann A, Lindner T, Mier W, Haberkorn U (2013) A disulfide-constrained miniprotein with striking tumor-binding specificity developed by ribosome display. Angew Chem Int Ed Engl 52:11760–11764
Ph.D.™Phage Display Libraries, Instruction manual, version 1.0, 9/09, New England Biolabs
Lipovsek D, Plueckthun A (2004) In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods 290:51–67
Hanes J, Schaffitzel C, Knappik A, Plückthun A (2000) Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol 18:1287–1292
Roberts RW, Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci USA 94:12297–12302
Wilson DS, Keefe AD, Szostak JW (2001) The use of mRNA display to select high-affinity protein-binding peptides. Proc Natl Acad Sci USA 98:3750–3755
Nygren PA, Uhlén M (1997) Scaffolds for engineering novel binding sites in proteins. Curr Opin Struct Biol 7:463–469
Mathonet P, Fastrez J (2004) Engineering of non-natural receptors. Curr Opin Struct Biol 14:505–511
Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T et al (2002) Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol 9:933–942
Högbom M, Eklund M, Nygren PA, Nordlund P (2003) Structural basis for recognition by an in vitro evolved affibody. Proc Natl Acad Sci USA 100:3191–3196
Orlova A, Tolmachev V, Pehrson R, Galli J, Baastrup B, Andersson K, Sandström M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY (2007) Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res 67:2773–2782
Skerra A (2000) Lippocalins as a scaffold. Biochim Biophys Acta 1482:337–350
Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A (2003) Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332:489–503
Breaker RR (2004) Natural and engineered nucleic acids as tools to explore biology. Nature 432:838–845
Suess B, Fink B, Berens C, Stenz R, Hillen W (2004) A theophylline responsive riboswitch based on helix slipping controls gene expression in vivo. Nucleic Acids Res 32:1610–1614
Acknowledgments
Part of this work was funded by the Klaus-Tschirra-Stiftung, by the BMBF programs MOBIMED and MOBITECH and by the grant program of the National Centre for Tumor Diseases (NCT) Heidelberg.
Conflict of interest
U. Haberkorn, W. Mier, A. Dimitrakopoulou-Strauss, M. Eder, K. Kopka, A. Altmann declare no conflict of interest.
Ethical standard
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Color figures online at http://link.springer.com/article/10.1007/s40336-014-0048-0
Rights and permissions
About this article
Cite this article
Haberkorn, U., Mier, W., Dimitrakopoulou-Strauss, A. et al. Mechanistic and high-throughput approaches for the design of molecular imaging probes and targeted therapeutics. Clin Transl Imaging 2, 33–41 (2014). https://doi.org/10.1007/s40336-014-0048-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40336-014-0048-0