
Abstract
Gene therapies have emerged as promising treatments in clinical development for various retinal disorders, offering hope to patients with inherited degenerative eye conditions. Several gene therapies have already shown remarkable success in clinical trials, with significant improvements observed in visual acuity and the preservation of retinal function. A multitude of gene therapies have now been delivered safely in human clinical trials for a wide range of inherited retinal disorders but there are some gaps in the reported trial data. Some of the most exciting treatment options are not under peer review and information is only available in press release form. Whilst many trials appear to have delivered good outcomes of safety, others have failed to meet primary endpoints and therefore not proceeded to phase III. Despite this, such trials have enabled researchers to learn how best to assess and monitor patient outcomes, which will guide future trials to greater success. In this review, we consider recent and ongoing clinical trials for a variety of potential retinal gene therapy treatments and discuss the positive and negative issues related to these trials. We discuss the treatment potential following clinical trials as well as the potential risks of some treatments under investigation. As these therapies continue to advance through rigorous testing and regulatory approval processes, they hold the potential to revolutionise the landscape of retinal disorder treatments, providing renewed vision and enhancing the quality of life for countless individuals worldwide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
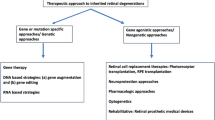
Gene therapy development for inherited retinal disease is a highly active area of research. |
Delivery of gene therapies to the retina is well tolerated across multiple human trials. |
Completed trials can guide future trial planning with a better understandining of appropriate endpoints. |
1 Introduction
Inherited retinal diseases (IRDs) arise in various forms with causative mutations reported in 289 genes (RetNet, accessed May 2024). Despite the varied genetic origins, gene therapy development and application for IRDs has been at the forefront of translational medicine. In the early 2000s, recombinant adeno-associated vectors were developed to enable efficient gene delivery with serotype and transgene variants showing encouraging pre-clinical results in animal models [1]. Autosomal recessive and X-linked disorders were identified as being ripe for gene supplementation therapy and over the last two decades these have advanced to clinical trials with one now available as an approved treatment. Therapeutic approaches have since evolved to encompass more complex strategies than single gene supplementation, such as expression of factors to stimulate retinal health, optogenetic activation to convert cells to become light sensitive, and the most modern strategy of genome editing. In this review, we discuss the current state of gene therapies for IRDs and consider their future potential to become approved treatments. We have included as up-to-date information as possible at the time of writing but are aware this is an ever-advancing field. To the best of our knowledge, all relevant ongoing trials as of May 2024 are discussed.
2 Gene Supplementation
2.1 Rod-Cone Dystrophies
Retinitis pigmentosa (RP) worldwide prevalence is estimated at 1 in 4000–5000 and describes a condition in which visual function is lost in the peripheral retina, also referred to as rod-cone dystrophy. The mechanism of this loss is dependent on the genetic origin, which is highly varied with numerous genes and mutation types responsible for this condition. Roughly 30% of autosomal dominant RP cases are caused by dominant mutations in the rhodopsin (RHO) gene. More than 150 dominant RHO mutations have been identified and implicated in autosomal dominant RP and these mutations can be classified into eight different groups depending on the molecular pathogenesis [2]. Photoreceptor toxicity can be induced by misfolded proteins that result in excessive endoplasmic reticulum stress and eventual apoptosis following activation of the unfolded protein response [3]. Consequently, it is insufficient to simply deliver the wild-type coding sequence: the existing deleterious protein and its toxic effects need to be addressed. Moreover, gene supplementation therapies (particularly in dominant diseases) can lead to protein levels exceeding physiological values, resulting in additional toxic gain-of-function mechanisms [4]. Rhodopsin mutations are therefore challenging to treat and have yet to be addressed in clinical trials but approval is being sought, for example, by Opus Genetics who acquired the rights from Iveric Bio in 2023 to develop an AAV.RHO vector that knocks down native rhodopsin and provides replacement. Gene therapies for autosomal recessive RP caused by mutations in other genes have been investigated in trials summarised below.
2.1.1 CPK850 (Novartis Pharmaceuticals)
This drug delivers the coding sequence for retinaldehyde binding protein 1, RLBP1, a crucial component of the visual cycle in rod and cone photoreceptors. A self-complementary transgene in AAV8 (scAAV8-RLBP1) is being investigated using a short RLBP1 promoter to drive RLBP1 expression in an ongoing phase I/II clinical trial (NCT03374657) to identify the maximum tolerated dose of CPK850 in addition to assessing the impact on visual function. No data are currently available on this trial.
2.1.2 CTx-PDE6B/HORA-PDE6B (Coave Therapeutics)
Rod cGMP phosphodiesterase 6B (PDE6B) provides the beta subunit for a specialised complex important in the light response of rod photoreceptor cells. Coave Therapeutics have developed an AAV5 vector to deliver the PDE6B coding sequence under the photoreceptor-specific rhodopsin kinase (GRK1) promoter and are providing this to adults aged 18 years and older in a phase I/II clinical trial (NCT03328130). A press release in May 2023 [5] and data presented at ARVO 2024 indicated that at 12 months, the gene therapy was well tolerated in 17 patients. It was highlighted that in a subgroup of six patients with less advanced disease that microperimetry sensitivity in the central four loci improved compared with the untreated eyes.
2.1.3 AAV.PDE6A (STZ eyetrial)
Retinitis pigmentosa can also result from mutations in the alpha subunit of the rod phosphodiesterase complex and the phase I/II PIGMENT trial (NCT04611503) has provided supplementary PDE6A under the GRK1 promoter in an AAV8 vector by subretinal delivery. Preliminary data of safety and vision outcomes were presented at ARVO 2023 [6], indicating that whilst the gene therapy was generally well tolerated, in two patients (one receiving 1 × 1011 and the other receiving 5 × 1011 vector genomes), a severe loss of visual acuity occurred that did not resolve 12 months post-injection.
2.1.4 rAAV2-VMD2-hMERTK (STZ eyetrial)
The MER proto-oncogene tyrosine kinase (MERTK) is expressed in the retinal pigment epithelium, but the loss of activity leads to incorrect processing of photoreceptor outer segments, which causes RP. A phase I study (NCT01482195) was initiated in 2011, in which an AAV2 vector was provided by a subretinal injection to provide supplementation of MERTK under the control of the RPE-specific vitelliform macular dystrophy 2 (VMD2) promoter. The data from six patients have been published [7] and whilst indicating no severe adverse events, the results have not led to a follow-up study.
2.2 X-Linked RP (XLRP)
X-linked RP (XLRP) is one of the most severe forms of inherited retinal degeneration, characterised by the progressive loss of function of photoreceptors that begins with night vision and peripheral vision loss and leads to legal blindness. The major contributor to this aggressive form of RP is the RP GTPase regulator (RPGR) gene, whose pathogenic variants are responsible for approximately 70% of all XLRP cases [8]. RPGR-related retinal degeneration is a genetically and clinically heterogeneous disease that affects male individuals, although female carriers can also be severely affected because of skewed X chromosome inactivation. The RPGR clinical phenotype reflects the type and severity of the photoreceptor class affected and, depending on the location of the pathogenic variant within the gene, can be manifested as a classic RP in which rods are primarily affected or a cone-dystrophy phenotype restricted to cones [9].
There is currently no approved treatment for RPGR-related retinal disease and the clinical management of patients remains supportive. Several gene therapy candidates are being assessed in clinical trials to study the safety and efficacy of delivering a functioning copy of the retina-specific isoform, RPGRORF15, to supplement the mutant gene in surviving photoreceptors.
2.2.1 Botaretigene Sparoparvovec (MeiraGTx and Janssen Pharmaceuticals, Inc.)
Botaretigene sparoparvovec (AAV5-RPGR) is an investigational gene therapy that consists of a shortened form of the human RPGRORF15 coding sequence, expressed under the control of the GRK1 promoter, packaged in AAV5. The human RPGRORF15 sequence administered with this vector contains an in-frame deletion of 378 base pairs in the ORF15 region, which results in the absence of 126 amino acids and two point mutations in the distal end of the gene: c.3396C>T (p.N1132N) and c.3430G>A (p.V1144I) [10]. The deletion within ORF15 makes the sequence less prone to mutations during the DNA manipulation and the generation of the gene therapy vector. However, an additional truncated form of the RPGR protein is expressed from this vector [11], probably due to abnormal splicing. The long-term effects of a gene replacement therapy using a human RPGRORF15 sequence containing a deletion that generates two truncated proteins unable to be completely glutamylated are unpredictable [12]. To date, there is no rationale as to why an RPGR protein with such a large deletion in a critical region for post-translational modification should be used when the full length wild-type protein can be stably expressed from a codon-optimised transgene.
The phase I/II MGT009 clinical trial (NCT03252847) provided botaretigene sparopavovec to adults and children with XLRP caused by variants in the RPGR gene. Patients received a subretinal injection of varying bleb size (0.3–0.8 mL) in the worse-seeing eye of either: 2 × 1011 vector genomes per millilitre (vg/mL), 4 × 1011 vg/mL, or 8 × 1011 vg/mL. Results obtained at 6 months were presented at the American Academy of Ophthalmology Annual and ARVO Meetings in 2022 [13, 14] and indicated an acceptable safety profile. No differences in efficacy were observed between patients who received the low or intermediate dose—data pooled from both cohorts showed an improvement in mean retinal sensitivity, measured by static perimetry in the central 10-degree area of the retina, in treated eyes compared with untreated eyes. Functional vision, assessed using a visual mobility maze without obstacles and maintaining the same course in the baseline and after treatment tests, seemed to increase with AAV5-RPGR as the walking time improved in treated eyes of the low-dose and intermediate-dose cohorts compared with the untreated eyes in the control arm [13]. It should be noted however that some patients were excluded from this analysis and no peer-reviewed publication is yet forthcoming.
Further development of this therapy has continued with the phase III LUMEOS trial (NCT04671433). Botaretigene sparoparvovec has been granted Fast Track and Orphan Drug designations by the US Food and Drug Administration and PRIority MEdicines (PRIME), and Advanced Therapy Medicinal Product and Orphan designations by the European Medicines Agency.
2.2.2 AGTC-501 (AGTC, Now Beacon Therapeutics)
AGTC-501 (AAV2tYF-coRPGR) carries a codon-optimised human RPGRORF15 coding sequence that maintains the amino acid sequence of the protein, expressed under the control of the GRK1 promoter and packaged in an AAV2 capsid variant with three tyrosine to phenylalanine mutations (AAV2tYF). The phase I/II HORIZON trial (NCT03316560) began in April 2018 with 29 male adults receiving a single dose of AGTC-501 in one eye. The interim analysis at month 24, presented in the 23rd EURETINA Congress [15], showed that AGTC-501 has a favourable safety profile, with no serious adverse event linked to the investigational product. Subretinal administration of AGTC-501 resulted in a significant treatment effect in visual sensitivity, an improvement in retinal sensitivity assessed by microperimetry, which continued to correlate with improvements in the retinal structure, as observed in the spectral domain optical coherence tomography. Best-corrected visual acuity data indicated evidence of a biological response. These positive and promising results led to the phase II SKYLINE trial in which two doses of AGTC-501 are being tested in paediatric and adult male patients. AGTC-501 is now the lead clinical asset of Beacon Therapeutics, an ophthalmic gene therapy company that is planning a phase II/III VISTA multi-centre trial (NCT04850118), evaluating the safety, efficacy and tolerability of two doses of AGTC-501 compared to an untreated control group.
2.2.3 Cotoretigene Toliparvovec (BIIB112) [Nightstar/Biogen]
Cotoretigene toliparvovec (BIIB112/AAV8-coRPGR) was developed by Nightstar Therapeutics Ltd. (later acquired by Biogen Inc.) and the University of Oxford and was the first gene therapy candidate to enter clinical trials for RPGR-associated RP (NCT03116113). The gene therapy combined an AAV8 vector with a stable transgene containing the GRK1 promoter and a codon-optimised version of the human RPGRORF15 [16]. As with the codon-optimised vector developed by AGTC, silent substitutions were introduced in the RPGRORF15 coding sequence to stabilise the sequence whilst additionally preventing synthesis of truncated RPGR proteins, or RPGR proteins with large in-frame deletions.
The XIRIUS study (NCT03116113) combined dose escalation (phase I) and dose expansion (phase I/II) evaluating the safety, tolerability and efficacy of a single subretinal injection of BIIB112 in male adults with mutations in the RPGR gene. Promising results were achieved, including no notable safety concerns, and visual field improvements in treated eyes beginning at month 1 and sustained at month 6 of follow-up [17] but this study did not meet its primary endpoint [18]. A post-hoc analysis of 18 participants in the XIRIUS study, and 103 participants in the Natural History of the Progression of XLRP (XOLARIS) trial, showed an early and sustained improvement in retinal sensitivity among the 12 participants who received the four highest doses of BIIB112 compared with untreated individuals in the XOLARIS trial. No dose-limiting toxic effects were observed [19].
2.2.4 4D-125 (4D Molecular Therapeutics)
4D Molecular Therapeutics also developed a vector carrying a codon-optimised version of the human RPGRORF15. This optimised transgene was packaged into an engineered capsid, R100, designed to reduce immunogenicity and to increase transgene expression [20]. Unlike the other studies discussed above, 4D-125 is administered through an intravitreal route. There is however no data in the literature to support the notion that intravitreal AAV can transduce primate photoreceptors of the outer retina effectively. To do this, the vector would need to penetrate the full retinal thickness and potential barrier of the internal limiting membrane as well as diffuse against the gradient of the fluid flow of aqueous from the ciliary body. Vector transduction will be dependent on the concentration in contact with receptors on the cell surface. Even excluding the retinal barriers, a simple application of Fick’s Law, in which diffusion is inversely proportional to concentration, suggests that a typical subretinal volume of 40–50 μL of vector will be two log units more efficacious than when diluted into the 4.2-ml volume of the vitreous cavity. Achieving therapeutic levels is unlikely to be achieved without significant inflammation.
The phase I/II trial (NCT04517149) is a multi-centre study with two parallel parts: an observational natural history cohort and an interventional trial in male individuals with RPGR-associated retinal dystrophy. The safety, tolerability and efficacy of a single intravitreal injection of 4D-125 at two dose levels (3 × 1011 vg/eye and 1 × 1012 vg/eye) in one or both eyes are being evaluated. At the 2021 American Society of Retina Specialists meeting, interim data were presented indicating 4D-125 was well tolerated and a dose expansion with the 1 × 1012 vg/eye ongoing [21]. 4D-125 was granted Fast Track Designation by the Food and Drug Administration.
2.2.5 FT-002 (Frontera Therapeutics)
FT-002 is a recombinant AAV designed to deliver RPGR currently being tested in patients in China (NCT05874310). At the time of writing this review, no details about the gene therapy vector or the design of the clinical trial are available.
2.3 Leber Congenital Amaurosis
The retinal pigment epithelium supports the visual cycle and mutations in the retinal pigment epithelium‐specific 65‐kDa protein (RPE65) lead to Leber congenital amaurosis (LCA). Clinical trials assessing gene supplementation of RPE65 have successfully led to an approved treatment.
2.3.1 Luxturna/Voretigene Neparvovec (Spark Therapeutics)
Luxturna® (voretigene neparvovec), authorised for patients with confirmed biallelic loss-of-function RPE65 mutation-associated retinal dystrophy, is currently the only approved ocular gene therapy. Food and Drug Administration approval was granted on 19 December, 2017 and on 23 November, 2018, approval was granted for use in all European Union member states. In a landmark phase III clinical trial conducted by Spark Therapeutics using a dose of 1.5 × 1011 vector genomes, 31 patients were treated with the AAV2 vector containing the RPE65 coding sequence under the control of the ubiquitous hybrid promoter containing cytomegalovirus enhancer and chicken beta actin promoter [22]. Patients were aged between 4 and 44 years and the worse-seeing eye was treated first, followed by the fellow eye 6–18 days later. A novel primary endpoint was chosen, the multi-luminance mobility test [23], in an effort to generate a single quantifiable test to be utilised to assess visual function changes in patients with rod-dominant inherited retinal dystrophies.
Voretigene neparvovec treatment has resulted in significant visual function improvement and no serious adverse events after 1 year and durability of improvement after 3–4 years follow-up [22, 24, 25]. Improvements in ambulatory navigation, light sensitivity and visual fields were consistent in both intervention groups. Average sensitivity changes of <− 2 log units (cd.s/m2) (i.e. around a 100-fold improvement in sensitivity), nearly maximal by 30 days, were maintained for at least 4 years. The safety profile was consistent with pars plana vitrectomy and the subretinal injection procedure and was similar between intervention groups, with no product-related serious adverse events or deleterious immune responses.
However, in 2022, in addition to the chorioretinal atrophy around the retinotomy site, it was reported that a subset of patients undergoing a subretinal voretigene neparvovec injection developed progressive perifoveal chorioretinal atrophy after surgery despite a good functional outcome [26]. Although an immunological cause has been postulated, clinical examinations at follow-ups have not revealed any distinct clinical signs of inflammation. A second hypothesis put forward is that photoreceptor rescue in a degenerate tissue with decreased metabolic activity may result in a sudden increase in metabolic and oxygenation demand, leading to cell death. The case may be that RPE-related gene therapies are more difficult to fine tune compared with photoreceptor-related gene therapies. Long-term observations of patients treated with voretigene neparvovec will help elucidate the reactive processes that occur in the retina in response to newly increased metabolic activity, and aid in the improvement of gene therapies for the RPE-related genes.
2.3.2 AAV-miniCEP290 (Iveric Bio)
In addition to RPE65, mutations in the cilia-centrosomal protein CEP290 are frequently observed in LCA cases, accounting for approximately a quarter of patients with LCA [27]. CEP290 is located in the ciliary transition zone of photoreceptors and is vital for the formation and stability of primary cilia. It acts as a molecular gatekeeper, regulating the ciliary trafficking of proteins and lipids from the photoreceptor inner segment to the outer segment [28]. At ~ 7.4 kb, CEP290 cDNA is larger than the maximum cargo capacity of AAV, therefore providing the complete coding sequence is not possible.
In 2019, Iveric Bio announced a minigene programme to treat LCA10, with a truncated CEP290 transgene that could be packaged in AAV. To date, only pre-clinical data are available but an optimised transgene design achieved improved photoreceptor structure and function in a mouse model of the disease [29]. Developing a long-lasting and efficacious strategy for CEP290 gene therapy and other large gene disorders is a challenge in the field but minigene therapies may be a viable option, although as yet there are no clinical trials planned. Genome editing offers another strategy for the treatment of large gene disorders and has been applied to CEP290 mutations, discussed in a later section.
2.4 Choroideremia
Mutations in the gene encoding Rab escort protein 1 (REP1) lead to choroideremia, a rare X-linked disorder that presents at a young age and leads to progressive and severe chorioretinal degeneration. Trial data have been well reported but owing to a current lack of consensus in what constitutes appropriate outcomes of success, trials have failed to meet designated primary endpoints, although this does not reflect that, in many cases, positive outcomes were achieved.
2.5 Timrepigene Emparvovec (BIIB111/AAV2-REP1) [Biogen, acquired by Beacon Therapeutics in 2023]
This vector has been through phase I/II trials and, more recently, the phase III STAR clinical trial, which have confirmed this treatment (AAV2.CAG.REP1) to be well tolerated at 1 × 1011 vector genomes in 69 male individuals and 1 × 1010 vector genomes in 34 male individuals [30]. The primary endpoint of a 15-letter gain in best-corrected visual acuity from baseline was not achieved because there were only four responders in the treated groups (none in the control group). With regard to the secondary endpoint; nine of 65 high-dose patients and six of 35 low-dose patients achieved a ≥ 10-letter ETDRS improvement from baseline, compared with one of 62 in the control cohort (p < 0.007). Such outcomes highlight the need to re-consider primary endpoints that determine a trial’s ultimate success.
2.6 AAV2-REP1 (Spark Therapeutics)
Using an equivalent vector design to BIIB111, the phase I/II trial sponsored by Spark Therapeutics (NCT02341807) has also observed good tolerance of the vector with limited signs of improvement compared to untreated eyes at the 2-year follow-up [31].
2.7 AAV2-REP1 (University of Alberta)
A further AAV2 vector for delivering REP1 under the control of the CBA promoter by subfoveal delivery has been assessed in a phase I/II trial (NCT02077361), with 5-year results recently published [32]. Unlike the other REP1 trials that had larger cohorts, this trial only reported data on five patients and no impact of the treatment was evident in the patients recruited who all had late-stage CHM.
2.8 4D-110 (4D Molecular Therapeutics)
In contrast to the other CHM trials, the 4D Molecular Therapeutics approach delivers the treatment by an intravitreal injection using their novel AAV capsid variant 4D-R100. Press releases from the ongoing phase I/II trial (NCT04483440) have to date indicated the product is well tolerated in the low-dose cohort with severe adverse events occurring in the high-dose cohort [33]. This unfortunately is to be expected as intravitreal AAV above 1 × 1011 vg is likely to cause severe inflammation.
2.9 X-Linked Retinoschisis (XLRS)
X-linked retinoschisis (XLRS) affects 1 in 5000–20,000 male individuals and results from mutations in the RS1 gene, which encodes the 224-amino acid protein retinoschisin, expressed in bipolar and photoreceptor cells [34, 35]. RS1 is secreted into the extracellular space and considered to be important for cell-to-cell adhesion, maintaining retinal structural integrity, and for signal transduction between photoreceptors and bipolar cells [36]. Two gene therapy clinical trials have provided AAV by intravitreal delivery with a third alternative in the pipeline planning a subretinal injection.
2.9.1 rAAV2tYF-CB-hRS1 (AGTC, Acquired by Beacon Therapeutics in 2023)
A phase I/II dose escalation study (NCT02416622) sponsored by Applied Genetic Technologies Corporation used the triple mutant AAV2 capsid to deliver the RS1 coding sequence under the CBA promoter. Twenty-two adults and five children were enrolled and rAAV2tYF-CB-hRS1 was delivered at 1 × 1011, 3 × 1011, or 6 × 1011 vg/eye. One-year trial data revealed that the most common ocular adverse events were related to ocular inflammation that, except in three participants in the highest-dose group, responded to corticosteroid therapy [37]. No functional improvements (visual acuities, visual fields, microperimetry or full-field electroretinogram) were observed in any of the dose groups. Because of a lack of efficacy and inflammation concerns, the decision was taken to terminate the product [38]. A study on the baseline immune function of patients with XLRS suggested that patients may exhibit a proinflammatory phenotype, which may further increase the risk of post-operative inflammation and hence, transduction efficiency [39], which may indicate an intravitreal delivery route as less suitable for this condition. A lack of efficacy may also be a result of inadequate penetration of the vector through the internal limiting membrane to transduce retinal cells, a problem that is being countered by development of new capsid types (such as with 4D Molecular Therapeutics).
2.9.2 AAV.RS1 (National Eye institute, VegaVect Inc.)
An ongoing, single-centre, phase I/II clinical trial (NCT02317887) sponsored by the National Eye Institute provided AAV8 with a modified human RS1 promoter augmented by an interphotoreceptor retinoid-binding protein enhancer element. Twelve adults with XLRS were enrolled and scAAV8-RS1-hRS1 was delivered at 1 × 109, 1 × 1010, 1 × 1011 or 3 × 1011 vg/eye. Although preliminary data at the 18-month follow-up suggested safety of the vector, no significant improvement in visual acuity was seen in treated versus untreated eyes in all but one patient (injected with the high dose 1 × 1011 vg), who showed a transient closure of the schitic cavities in the study eye, which occurred at 2 weeks post-injection [40].
2.9.3 ATSN-201 (Atsena Therapeutics)
The limitations associated with intravitreal injections may be overcome by the more recent clinical programme announced by Atsena Therapeutics. This will use a novel AAV capsid, AAV-SPR, which is thought to spread laterally beyond the injection site after a subretinal injection and should enable better targeting of photoreceptors [41].
2.10 Achromatoposia
Achromatopsia is caused by loss-of-function mutations in the cyclic nucleotide-gated channels involved in the phototransduction cascade. Being inherited in an autosomal recessive manner, it is an attractive target for gene supplementation therapy.
2.10.1 AAV8.CNGA3 (STZ eyetrial)
The clinical trial sponsored by the STZ eyetrial evaluated the effect of three escalating doses (1 × 1010–1 × 1011 total vector genomes per eye) of AAV8.CNGA3 under the control of the cone-specific arrestin-3 promoter (ARR3) in nine patients with CNGA3-associated achromatopsia and 1-year and 3-year data have been reported [42, 43].
The treatment was well tolerated and resulted in two adverse events that resolved with corticosteroid treatment and without sequelae. Unlike the progressive chorioretinal atrophy observed in patients who received voretigene neparvovec, retinal changes observed after treatment with AAV8.CNGA3 consisted of hypopigmentation around the retinotomy site and a hyperpigmentation delineating the lower border of the retinal bleb raised during the subretinal injection. These changes were visible in the early post-operative period and did not progress with time. Provision of AAV8.CNGA3 resulted in consistent improvements in visual acuity and contrast sensitivity against baseline in all nine treated patients [42], for at least 3 years after treatment [43]. However, as the fellow eye demonstrated simultaneous improvement in visual acuity, there was no statistical significance difference between the treated and untreated eye. The small sample size limited the statistical power of efficacy analyses, but the trial results offer potential endpoints for future clinical trials.
A phase IIb clinical trial (randomised, observer masked) targeting treatment of the second eye of the first patients and treatment of children aged 6–12 years is ongoing with 14 patients anticipated to be enrolled to receive 1 × 1011 vector genome particles in each eye at different timepoints. The cohort in whom the first eye has received treatment will only receive one injection in the second eye. The study is anticipated to be completed in April 2027, which will be of particular interest to identify whether earlier intervention in younger patients results in greater functional gains by avoiding the development of amblyopia.
2.10.2 AGTC-401 (CNGB3) and AGTC-402 (CNGA3) [AGTC, Acquired by Beacon Therapeutics 2023]
AGTC initiated phase I/II trials for CNGB3-related (AGTC-401, NCT02599922) and CNGA3-related (AGTC-402, NCT02935517) achromatopsia. Both vectors used the triple-mutant AAV2 capsid (AAV2tYF) and a cone-photoreceptor specific promoter (PR1.7) delivered by a macular subretinal injection. Interim safety data were presented at ARVO 2022 from both trials, which combined included 37 adults and 18 children [44]. Three serious adverse events were observed in children at the highest dose of 3.2 × 1012 vg/mL (two from CNGA3 treatment and one from CNGB3 treatment). At lower doses, the drugs were well tolerated and had favourable safety profiles in adults and children. Despite the encouraging safety profile, since acquiring AGTC in 2023, Beacon Therapeutics have stated they are currently not pursuing the development of either of these drugs, but the clinical trials will continue to conclusion.
2.10.3 AAV.CNGB3 and AAV.CNGA3 (MeiraGTx)
MeiraGTx is conducting phase I/II trials with AAV.CNGB3 (NCT03001310) and AAV.CNGA3 (NCT03758404) vectors, of which, data have been published for the CNGB3 trial [45]. The AAV8 vector carries a transgene with the CNGB3 coding sequence driven by a 0.4-kb fragment of the human promoter. Eleven adults and 12 children received the vector at 0.1 × 1012, 0.6 × 1012 or 1 × 1012 vg/mL. Adverse events were as expected for a study of this type and manageable with no improvements in visual function assessments achieved from treated eyes compared to untreated eyes at week 24. This drug received Fast Track designation in 2021 and the medicine has been registered as entacingene turiparvovec and is awaiting approval.
3 Secreted Factors
As evidenced by the therapies discussed so far, single-gene disorders, particularly autosomal recessive and X-linked forms, are achieving encouraging outcomes from clinical trials. However, the leading cause of irreversible sight loss amongst the aging population across the developed world is age-related macular degeneration (AMD). This is a complex disorder resulting from genetic and environmental factors that is most common in the form of dry AMD, which progresses slowly over many years with development of geographic atrophy [46]. In many patients, wet AMD (or neovascular AMD) can develop, when blood vessels grow incorrectly in the retina and lead to further significant damage. Gene therapy strategies have been developed to target both forms of AMD and make use of secreted factors to induce prevention of inflammation and atrophy, thereby providing a general regulation of the condition.
3.1 Dry AMD
Genetic risk factors for dry AMD predominantly occur in genes associated with the complement system, a complex system that forms a critical part of the innate response and plays a key role in AMD pathogenesis [47]. It has been considered that provision of inhibitors of complement activation may offer a viable strategy to prevent geographic atrophy in dry AMD.
3.1.1 GT005/PPY988 (Novartis)
Originally developed by Gyroscope Therapeutics, this was acquired by Novartis in 2022 with a phase I/II FOCUS trial (NCT03846193) and two additional phase II trials (EXPLORE, NCT04437368 and HORIZON, NCT04566445) initiated. GT005 uses AAV2 to deliver complement factor I using the ubiquitous CAG promoter (cytomegalovirus enhancer, CBA promoter plus rabbit beta-globin intron), a protein that downregulates complement activation. Dose escalation and expansion was performed with patients undergoing surgery either by a subretinal transvitreal delivery or using the Orbit™ subretinal delivery system. Data from 31 patients identified no serious adverse events and whilst RPE changes were observed in some patients, these were limited to the bleb area and did not cause significant changes to vision [48]. Immunogenicity data were encouraging with no significant association of immune responses or adverse events, vitreous samples were extracted at baseline then again post-treatment at weeks 4–25 and 29–56 with increasing levels of complement factor I detected at each timepoint. Vitreous samples also showed a reduction in complement system factors Ba, C3b/iC3b and C3. However, despite the preliminary data indicating the vector was well tolerated in patients with geographic atrophy, the FOCUS, EXPLORE and HORIZON trials were terminated in 2023 with Novartis announcing it would discontinue GT005 [49].
3.1.2 HMR59 (Hemera Biosciences)
HMR59 is an AAV2 vector for expression of soluble CD59 under the CAG promoter following an intravitreal injection. CD59 is expressed on cell membranes and responds to the complement cascade by forming the membrane attack complex, the final step in the complement cascade. It is hoped that provision of soluble CD59 circulating throughout the retina will reduce the complement activity that is a hallmark of AMD. The HMR-1001 phase I trial (NCT03144999) was a dose-escalation study to identify the maximum tolerated dose of HMR59 in patients with dry AMD. However, the trial was reported as inactive as of January 2020. Data have been discussed online but not in a formal peer-review publication. Online reports have disclosed that 17 patients with dry AMD with geographic atrophy received HMR59 intravitreally at three doses and that these were well tolerated [50]. No further information seems to be publicly available.
3.2 Wet AMD
Wet AMD is less common than the dry form and can develop suddenly, leading to significant and irreversible damage in the macular region. Current treatments require patients to receive regular intravitreal injections of anti-vascular endothelial growth factor (VEGF) to control the unwanted blood vessel growth that leads to neovascularisation. Patients with wet AMD receive regular injections of anti-VEGF throughout a given year as and when it is necessary to control the neovascularisation. The primary intention of gene therapy strategies for wet AMD is to use the cells of the eye as factories to produce secreted anti-VEGF to enable long-term treatment with just a single injection.
3.2.1 ADVM-022 (Adverum Biotechnologies)
This product is delivered by an intravitreal injection and uses an AAV2.7m8 vector and the immediate-early cytomegalovirus enhancer and promoter to achieve anti-VEGF (aflibercept) expression. Preliminary 3-year results from the phase I OPTIC trial (NCT03748784) were presented at the 2022 Retina Society meeting with the presentation available on the Adverum website [51]. Two serious adverse events were reported but other adverse events were deemed to be mild to moderate. Aqueous aflibercept expression was detected across the 3 years with trial patients further considered for their need of supplemental aflibercept injections. In the low-dose cohort (2 × 1011 vg/eye), there was a 53% reduction in the requirement of supplemental injections 2 years after receiving ADVM-022 and in the high-dose cohort (6 × 1011 vg/eye) the rate of reduction was 80% (n = 15 per cohort). Visual acuity was maintained at both doses. A phase II LUNA trial (NCT05536973) is ongoing, continuing with the 2 × 1011 vg/eye dose and an additional lower dose of 6 × 1010 vg/eye.
3.2.2 ABBV-RGX-314 (Regenxbio and AbbVie)
As with ADVM-022, ABBV-RGX-314 is designed as a single-delivery anti-VEGF therapy. In this case, subretinal and suprachoroidal delivery routes of AAV8 are being assessed, with the vector expressing a monoclonal antibody fragment to neutralise VEGF-A using a CAG promoter. Data from the phase I NCT03066258 study were recently published [52]. Doses of the ABBV-RGX-314 vector below 2.5E+11 vg/eye were well tolerated with no drug-related inflammation or serious adverse events. Visual acuity reduced in nine of 46 study eyes with three of these events potentially linked to RGX-314. Providing ≥6E+10 vg/eye enabled sustained anti-VEGF-A protein expression and it was identified that the subretinal delivery route provided better outcomes and will be used in the ongoing phase II/III ATMOSPHERE and AAVIATE trials (NCT04704921 and NCT04514653) and the phase III ASCENT trial (NCT05407636). Patients are undergoing a long-term follow-up but 4 years post-injection, available cohorts appear to show improved or stable best-corrected visual acuity and as with ADVM-022, annual injection rates for supplemental anti-VEGF reduced by 59–85% in cohorts that received ≥6E+10 vg/eye.
3.2.3 4D-150 (4D Molecular Therapeutics)
4D-150 uses the evolved AAV vector R100 for intravitreal delivery of a transgene that expresses both anti-VEGF (aflibercept) and a VEGF-C targeting microRNA for knockdown of VEGF-C. The safety and efficacy of 4D-150 are being assessed in a phase I/II PRISM trial (NCT05197270) in 50 patients. Preliminary data from 15 patients were presented at ARVO 2023 and released on the company website [53]. Three doses were described as being well tolerated with patients showing a reduction in follow-up anti-VEGF injections. The 3 × 1010 vg/eye showed the most encouraging signs with 80% of patients not requiring supplemental anti-VEGF injections 24 weeks after receiving 4D-150.
3.2.4 rAAV.sFLT1 (Avalanche Biotechnologies) and AAV.sFLT1 (Genzyme/Sanofi)
FLT1 encodes Fms-related receptor tyrosine kinase 1, a VEGF receptor that binds VEGF-A and VEGF-B. An AAV2 vector carrying a transgene to express soluble FLT1 was provided either by an intravitreal (NCT01024998) or subretinal (NCT01494805) injection. Following an intravitreal injection at four doses (2 × 108, 2 × 109, 6 × 109 or 2 × 1010 vg/eye), ocular inflammation was observed in the highest dose group. Levels of sFLT1 aqueous humour levels were variable between patients, which was considered to be related to pre-existing antibodies against AAV2 in some patients [54]. It was observed that six of 11 patients who presented with subretinal intraretinal fluid at baseline showed an improvement in fluid after the intravitreal injection with four of these six patients maintaining this response at 52 weeks. However, the other five patients who presented with fluid at baseline showed no significant changes.
Subretinal delivery of rAAV.sFLT1 (Avalanche) was provided to a cohort of six patients at either 1 × 1010 or 1 × 1011 vg. No drug-related adverse events were reported and in the year following treatment, four patients did not require supplemental anti-VEGF injections and two required one supplemental injection of anti-VEGF [55]. Three-year follow-up data for combined phase I/II cohorts further supported this treatment was safe and well tolerated when provided by a subretinal injection [56, 57]. The small cohort numbers used did not enable efficacy to be evaluated and though the subretinal approach was well tolerated, an intravitreal injection is easier to apply in the clinic and less stressful for patients. These trials were some of the earliest gene therapies for wet AMD and new vectors have since become available that may make intravitreal delivery of these worth attempting again as there are now more potent serotypes available that enable effective transgene expression.
3.2.5 HMR59 (Hemera Biosciences)
Unlike the HMR-1001, which has seen no follow-up to date, the HMR-1002 phase I trial assessed outcomes in patients with wet AMD who received intravitreal delivery of an anti-VEGF medication followed by an intravitreal injection HMR59 7 days later. Long-term safety was scheduled for 24 months with supplemental anti-VEGF injections provided to patients as needed. As with other trials, data have not been formally published or peer reviewed but it has been reported online that 24 patients received either 3.56 × 1011 or 1.07 × 1012 vg/eye with three patients experiencing inflammation that was resolved with a course of topical and/or oral corticosteroids. After 6 months, four of 22 patients did not require supplemental anti-VEGF injections [50].
3.3 Other
3.3.1 SPVN06 (Sparing Vision)
This treatment uses subretinal delivery of an AAV8 vector to induce expression of a proprietary neurotrophic factor known as rod-derived cone viability factor and an enzyme for reducing oxidative stress (rod-derived cone viability factor long form). Combined, these factors are intended to slow retinal degeneration, regardless of the genetic mutation causing the disease. A phase I/II PRODYGY trial (NCT05748873) is ongoing, assessing SPVN06 in patients with advanced rod-cone dystrophy due to mutations in rhodopsin or phosphodiesterase alpha and beta. Press releases in 2023 reported encouraging safety data in the first three patients injected but dose escalation is ongoing with a total 33 patients intended to be recruited [58].
4 Optogenetics
The intent of most treatment strategies is to apply them at a stage when sight loss can be prevented, stalled or improved, but there will always be patients in whom the disease has progressed to such an advanced stage whereby these strategies will be of no benefit. In such cases, optogenetics may offer treatment potential. A variety of optogenetic approaches exist that share a similar strategy: to provide a light-sensitive protein (an opsin) to surviving cells of the retina and induce them to become light sensitive. The opsins used for such an approach vary and whilst pre-clinical studies have used human opsins such as rhodopsin, cone opsins and melanopsin, others have used microbial-derived opsins [59]. These opsins vary in their light sensitivity and in the nature of the downstream cellular response to light activation, for example depolarising forms of channel-rhodopsin or hyperpolarising halorhodopsin. Opsins are typically expressed on cell membrane surfaces for optimal light exposure and microbial opsins carry the risk of inducing immune responses post-transduction, which will need to be carefully monitored. Vertebrate opsins may therefore offer safer alternatives; however, the downstream components that enable active signalling pathways for these opsins may need to be co-delivered to enable light responses. Regardless of the opsin used, an optogenetic treatment strategy requires the visual system to adapt to the new form of signalling with data so far indicating this is a viable approach for patients who have lost the ability to sense light. Current clinical trials are using microbial-derived enhanced opsins and are discussed below.
4.1 RST-001 (AbbVie)
Because of the loss of photoreceptor cells, trials of optogenetic gene therapies use an intravitreal delivery route to target the surviving cells at the ganglion cell and inner cell layers. RST-001 is an AAV2 vector carrying a form of channelrhodopsin (ChR2) that was originally developed by Retrosense Therapeutics, which was then acquired by Allergan (2016), later acquired by AbbVie 2020. Fourteen patients with advanced-stage RP were recruited for a phase I/II trial (NCT02556736) and received a single intravitreal injection of RST-001, no serious adverse events have been reported but other outcomes have yet to be published.
4.2 GS030 (GenSight Biologics)
The GenSight PIONEER trial (NCT03326336) began in 2018 and uses intravitreal delivery of a ChrimsonR opsin delivered in an AAV2 7m8 vector under the control of the CAG promoter. ChrimsonR is algal derived and sensitive to red light (590 nm), so GS030-DP gene therapy is paired with GS030-MD stimulating glasses to enhance ChrimsonR excitation. The vector additionally carries a fluorescent reporter tdTomato, included to increase expression of ChrimsonR at cell membranes. A single study case has been reported [60], involving a 58-year-old patient with visual acuity limited to light perception prior to the trial. When wearing the goggles, the patient was able to use the treated eye to locate objects with corresponding object recognition activity detected by electroencephalographic recordings. No safety concerns have been reported and further patient recruitment is ongoing.
4.3 BS01 (Bionic Sight)
BS01 delivers a more light-sensitive form of channel rhodopsin using the CAG promoter, ChronosFP, and also requires an additional device for opsin stimulation. A phase I/II trial began recruiting in 2020 and is ongoing (NCT04278131). A press release has indicated that no safety concerns have been identified and four patients have been able to detect light and motion 2–3 months post-treatment [61]. As with other trials, peer-reviewed publication of data will be critical to determining the viability of this strategy relative to other approaches.
4.4 Sonpiretigene Isterparvovec (vMCO-010) [Nanoscope Therapeutics]
A multi-characteristic opsin (MCO) was designed using a web-based structure predictor, RaptorX, from which a novel gene was synthesised and fused to an mCherry fluorescent reporter, which was shown to be activated at bright ambient light levels [62]. This optogenetic approach differs from the previous therapies as it uses the mGluR6 promoter enhancer for bipolar cell expression. A phase I/II dose-escalation study involving intravitreal delivery of AAV2 vMCO-1 in patients with advanced RP began in 2019 (NCT04919473). In 2021, a Nanoscope press release claimed safety and efficacy results from 11 patients, including improvements in visual acuity in the high-dose group [63]. A phase II study to evaluate the safety and efficacy of MCO-010 in patients with advanced RP was initiated in 2021 (NCT04945772). A press release in March 2024 indicated MCO-010 was well tolerated with best-corrected visual acuity improving in the treatment groups [64], which were maintained at 52 weeks in the high-dose group (1.2E+11 vg/eye). Patients in these trials were diagnosed with advanced RP of different genetic origins. A further trial has been completed delivering MCO-010 to six patients with Stargardt disease (NCT05417126).
5 Genome Editing
The gene therapies discussed so far reflect the range of treatment potential in retinal gene therapy. However, they lack applicability when mutations occur in large genes exceeding the 4.7kb AAV packaging limit and for dominant gain-of-function mutations. Gene editing offers hope for patients who fall into these groups but permanent off-target effects induced by gene editing technologies are a potential significant drawback that will require careful monitoring and consideration. This is the least developed gene therapy form but is rapidly advancing and likely to overtake other strategies in the coming years.
5.1 EDIT-101 and EDIT-102 (Editas Medicine)
The EDIT-101 phase I/II BRILLIANCE trial (NCT03872479) is for the treatment of LCA type 10 caused by the common CEP290 mutation c.2991+1655A>G in intron 26 that produces an aberrant splice donor site and truncated protein. Both trials directed by Editas use the GRK1 promoter to drive CRISPR/Cas9 expression with the EDIT-101 therapy excising the CEP290 deep intronic mutation [65], whereas EDIT-102 makes two intronic incisions to eliminate exon 13 from the USH2A gene. It has been shown that exon 13 may be dispensable to usherin protein function, attributable to the repetitive structure of the gene [66]. EDIT-101 trial data were recently published, showing an encouraging safety profile with six of 14 patients achieving an improvement in cone-mediated vision [67]. This represents a significant milestone in gene therapy but despite these encouraging results, Editas will not progress the programme further, largely owing to the small patient population that might benefit from the gene therapy. However, it will prove a valuable trial in the advancement of future CRISPR-based treatments for IRDs.
5.2 EDIT-103 (Editas)
A third trial by Editas (EDIT-103) will address the gain-of-function problem caused by rhodopsin mutations by simultaneously knocking down the aberrant rhodopsin with CRISPR/Cas9 in addition to providing a CRISPR-resilient codon-optimised replacement gene [68]. Dominant rhodopsin mutations are considered highly prevalent, affecting ~7500 patients in the USA and ~12,100 patients across the European Union and UK. The vector uses a mini rhodopsin promoter to keep transgene activity within rod photoreceptor cells, thereby limiting risks of unwanted off-target effects in other retinal cell types.
Genome interacting approaches include anti-sense oligonucleotide gene therapy treatments, which are showing highly encouraging data from clinical trials. The mechanism of action means they are ideal for silencing mutant transcripts and blocking unwanted splicing. Given the lack of requirement of a viral vector and the intravitreal delivery route, these are an exciting class of gene therapy.
5.3 Sepofarsen (QR-110) [ProQR Therapeutics]
The RNA antisense oligonucleotide targeting the common CEP290 mutation c.2991+1655A>G was assessed by intravitreal delivery in the phase II/III ILLUMINATE trial (NCT03913143). Trial data have been published, presenting a highly encouraging safety profile with significant improvements in visual acuity and retinal sensitivity observed at 12 months [69].
5.4 QR-1123 and QR-421a (ProQR Therapeutics)
QR-1123 is an RNA antisense oligonucleotide for targeting autosomal dominant rhodopsin caused by the common P23H mutation, assessed in 11 patients in the phase I/II AURORA trial (NCT04123626). QR-421a (Ulterversen) targets USH2A mutations in exon 13. The STELLAR phase I/II trial (NCT03780257) was followed by initiation of phase II/III parallel studies CELESTE (NCT05176717), SIRIUS (NCT05158296) and HELIA (NCT05085964). However, CELESTE and HELIA have since been terminated and for business reasons ProQR have suspended their ophthalmology pipeline. Sepofarsen and Ultevursen have been acquired by Laboratories Théa.
6 Conclusions
Gene therapy development for retinal diseases has expanded rapidly in the last 20 years and continues to accelerate. Trials are ongoing employing an array of therapeutic strategies, which are modernising as new molecular tools develop (Table 1). Whilst there is a general trend of these gene therapies to show good tolerance following ocular delivery, outcomes of efficacy are less often observed. In certain patients, there are significant barriers to achieving persistent transgene expression following viral delivery. Transcript levels can sharply decline according to the antibody library of the individual: production of neutralising antibodies by B cells excludes any patient with pre-existing immunity from treatment [70]. Moreover, T-cell responses can lead to complete rejection of virally transduced cells, limiting the duration of a treatment effect [71]. Patient immunity carries implications where a treatment effect declines over time, and repeated treatments are required. Robust and persistent expression levels have been reported years following injection of AAV gene therapy vectors, likely owing to their stable episomal configuration but it is not known how long this will persist. Future strategies such as DNA editing offer an advantage from this perspective: as the edit is permanent, persistent transgene expression is not required or even desirable.
Genome editing is likely to dominate the future of gene therapy, which will require extensive considerations of the risks associated with such an approach. Even when applied to intronic mutations, the safety of the CRISPR/Cas9 system is not as robust as it could be for therapeutic use; preclinical assessment of the EDIT-101 vector observed off-target events [65, 72]. EDIT-101 and EDIT-103 both use Staphylococcus aureus Cas9 in their constructs, for which up to three mismatches are tolerated between the spacer and target DNA, increasing the likelihood of guide-dependent off-target events [73, 74]. A multitude of algorithms now exists to predict the position of off-target sites. Some of these are extremely comprehensive with deep learning capabilities and account for numerous factors including epigenetic features, mismatch position within the sgRNA and mismatch-PAM site distance [75,76,77]. However, these algorithms are guide dependent and do not account for guide-independent off-target edits. Furthermore, standard amplicon sequencing does not detect more drastic events such as chromosomal truncations and extensive rearrangements termed ‘chromothripsis’ [78, 79]. These events raise concerns over carcinogenicity, and double-stranded breaks can directly activate the p53-mediated DNA damage pathway [80].
The strategies utilised by the Editas trials offer broad applicability, but the financial toll of clinical trials means that rare disorders are not yet an economically viable target for investors. Despite demonstrating efficacy, the EDIT-101 trial has been paused because the therapy has a patient population of only 300 people in the USA. Furthermore, although intronic deletions reduce the potential for interrupting the coding sequence, rigorous testing for each genomic scenario will likely be required to ensure unidentified binding/enhancer sequences are not affected. Additionally, exon excision (applied in EDIT-102) lacks applicability where the gene is placed out of frame or where the skipped exon has a crucial role.
Other exciting developments in the field include delivering circular DNA using electroporation rather than relying on AAV vectors. Eyevensys has an ongoing phase II clinical trial (NCT04207983) for non-infectious uveitis, which will pave the way for future clinical trials in which non-viral circular DNA vectors can be delivered by electroporation for the treatment of IRDs [81].
Optogenetics is a bold approach for recovering sight loss in patients with advanced stages of degeneration and trials show encouraging signs that the visual system can adapt to the introduction of light-sensitive opsins in remaining cell types. The requirement of additional devices for some strategies may become unnecessary in the future as trial data from gene therapies employing ambient-light sensitive opsins become available.
Currently, many trials have only provided press releases of data, which make it difficult to review and consider the outcomes. Whilst press releases offer indications of safety and efficacy, a large number of trials discussed in this review cannot be fully appreciated until more thorough reporting is available.
The retinal gene therapy clinical trials reviewed have highlighted the successes, particularly in AAV safety profiles, but also the difficulties in providing gene therapies for inherited retinal conditions. Combined, they offer lessons learned regarding how to design and deliver a successful vector and how to get the most from a clinical trial. Similar vectors have been designed independently to treat the same condition. In some cases, these are so highly similar (e.g. choroideremia vector options) that trial outcomes may be complementary to each other whereas in other cases, varying vector designs may be more polarising and highlight concerns with particular options, as in the case of the mutated RPGR vector or in the application of intravitreal viral vector delivery in patients with XLRS.
Study design and outcome measures need to be reviewed and further discussed between researchers and governing bodies to ensure trials are not considered unsuccessful for failing to reach primary endpoints. The trials conducted to date have enabled a much greater understanding of the inherited retinal disease they aim to treat and how best to assess them and these data should be used to guide future trial designs. As genome editing continues to develop, it will likely overtake other forms of gene therapy and become the primary mechanism for the treatment of IRDs. In the meantime, pre-clinical development and clinical assessment of gene therapies for inherited retinal disorders continues to expand. Despite the encouraging research outputs, pharmaceutical companies funding such studies are dropping their support for treatment options because of a lack of financial reward. Trials are expensive to run and when only small patient populations are identified as beneficiaries, this has led to companies terminating promising pipelines. This is often not a reflection on the treatment potential and highlights that considerations need to be made for how best to fund future clinical trials and ultimately achieve investment to progress promising gene therapies to treatments.
References
McClements ME, MacLaren RE. Gene therapy for retinal disease. Transl Res. 2013;161(4):241–54. https://doi.org/10.1016/j.trsl.2012.12.007.
Athanasiou D, Aguila M, Aguila J, et al. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res. 2018;62:1–23. https://doi.org/10.1016/j.preteyeres.2017.10.002.
Griciuc A, Aron L, Ueffing M. ER stress in retinal degeneration: a target for rational therapy? Trends Mol Med. 2011;17(8):442–51. https://doi.org/10.1016/j.molmed.2011.04.002.
Orlans DH, Barnard DAR, Patricio DMM, McClements DME, MacLaren PRE. Effect of AAV-mediated rhodopsin gene augmentation on retinal degeneration caused by the dominant P23H rhodopsin mutation in a knock-in murine model. Available from: www.iebertpub.com, p. hum.2020.008, May 2020, https://doi.org/10.1089/hum.2020.008.
Coave Therapeutics. Coave Therapeutics announces positive 12-months data from ongoing Phase I/II Clinical Trial of CTx-PDE6b in Patients with Retinitis Pigmentosa Caused by Bi-allelic Mutations in PDE6b. Available from: https://coavetx.com/coave-therapeutics-announces-positive-12-months-data-from-ongoing-phase-i-ii-clinical-trial-of-ctx-pde6b-in-patients-with-retinitis-pigmentosa-caused-by-bi-allelic-mutations-in-pde6b/. Accessed 6 Feb 2024.
IOVS ARVO Journals. Safety and vision outcomes of subretinal gene supplementation therapy in PDE6A-associated retinitis pigmentosa. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2786162. Accessed 6 Feb 2024.
Ghazi NG, Abboud EB, Nowilaty ER, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135(3):1–17. https://doi.org/10.1007/s00439-016-1637-y.
Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. The Am J Hum Genet. 2003;73(5):1131–4. https://doi.org/10.1086/379379.
Cehajic-Kapetanovic J, Martinez-Fernandez de la Camara C, Birtel J, et al. Impaired glutamylation of RPGRORF15 underlies the cone-dominated phenotype associated with truncating distal ORF15 variants. P Natl Acad Sci U S A. 2022;119(49): e2208707119. https://doi.org/10.1073/pnas.2208707119.
US10314924B2: RPGR gene therapy for retinitis pigmentosa: Google Patents. Available from: https://patents.google.com/patent/US10314924B2/en. Accessed 21 Feb 2024.
Sladen PE, Naeem A, Adefila-Ideozu T, et al. AAV-RPGR gene therapy rescues Opsin mislocalisation in a human retinal organoid model of RPGR-associated X-linked retinitis pigmentosa. Int J Mol Sci. 2024;25(3):1839. https://doi.org/10.3390/ijms25031839.
Sun X, Park JH, Gumerson J, et al. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc Natl Acad Sci U S A. 2016;113(21):E2925–34. https://doi.org/10.1073/pnas.1523201113.
American Academy of Ophthalmology. Retinitis pigmentosa responds to gene therapy. Available from: https://www.aao.org/eyenet/academy-live/detail/retinitis-pigmentosa-responds-to-gene-therapy. Accessed 6 Feb 2024.
Michaelides M, Jialin Xu, Dai Wang, et al. AAV5-RPGR (botaretigene sparoparvovec) gene therapy for X-linked retinitis pigmentosa (XLRP) demonstrates localized improvements in static perimetry. IOVS | ARVO Journals. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2781372. Accessed: 21 May 2024.
MacLaren RE. Subretinal gene therapy drug AGTC-501 for X-linked retinitis pigmentosa (XLRP) phase 1/2 multicenter study (Horizon): 24-month interim safety results. EURETINA Congress 23 Amsterdam Abstracts. Available from: https://euretina.softr.app/amsterdam-abstract?recordId=recj7HMgjFI08WXwH. Accessed 6 Feb 2024.
Fischer MD, McClements ME, Martinez-Fernandez de la Camara C, et al. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol Ther. 2017;25(8):1854–65. https://doi.org/10.1016/j.ymthe.2017.05.005.
Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de al Camara C, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020;26(3):354–9. https://doi.org/10.1038/s41591-020-0763-1.
Biogen. Biogen Announces topline results from phase 2/3 gene therapy study for XLRP. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-phase-23-gene-therapy-study#:~:text=CAMBRIDGE%2C%20Mass.%2C%20May%2014,linked%20retinitis%20pigmentosa%20(XLRP). Accessed 6 Feb 2024.
von Krusenstiern L, Liu J, Liao E, et al. Changes in retinal sensitivity associated with cotoretigene toliparvovec in X-linked retinitis pigmentosa with RPGR gene variations. JAMA Ophthalmol. 2023;141(3):275–83. https://doi.org/10.1001/jamaophthalmol.2022.6254.
Kotterman M, Beliakoff G, Croze R, et al. Directed evolution of AAV targeting primate retina by intravitreal injection identifies R100, a variant demonstrating robust gene delivery and therapeutic efficacy in non-human primates. bioRxiv. 2021. https://doi.org/10.1101/2021.06.24.449775.
Besirili CG, Birch D, Mathias M, et al. Phase 1/2 clinical trial of intravitreal 4D-125 AAV gene therapy in patients with advanced XLRP: interim safety & preliminary activity. ASRS 2021 abstracts. Available from: https://www.asrs.org/content/documents/in-the-pipeline.pdf. Accessed 12 Feb 2024.
Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–60. https://doi.org/10.1016/s0140-6736(17)31868-8.
Chung DC, McCague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin Exp Ophthalmol. 2018;46(3):247–59. https://doi.org/10.1111/ceo.13022.
Maguire AM, Russell S, Wellman JA, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy. Ophthalmology. 2019;126(9):1273–85. https://doi.org/10.1016/j.ophtha.2019.06.017.
Maguire AM, Russell S, Chung DC, et al. Durability of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease. Ophthalmology. 2021;128(10):1460–8. https://doi.org/10.1016/j.ophtha.2021.03.031.
Gange WS, Sisk RA, Besirli CG, et al. Perifoveal chorioretinal atrophy after subretinal voretigene neparvovec-rzyl for RPE65-mediated Leber congenital amaurosis. Ophthalmol Retin. 2022;6(1):58–64. https://doi.org/10.1016/j.oret.2021.03.016.
den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79(3):556–61. https://doi.org/10.1086/507318.
Dulla K, Aguila M, Lane A, et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 Models. Mol Ther Nucleic Acids. 2018;12:730–40. https://doi.org/10.1016/j.omtn.2018.07.010.
Leon LM, Zhang W, Sahu B, Gao G, Khanna H. A MiniCEP290 gene replacement therapy to treat CEP290-Leber congenital amaurosis (LCA10), IOVS, ARVO Journals 2022. Available: https://iovs.arvojournals.org/article.aspx?articleid=2781375. Accessed 6 Feb 2024.
MacLaren RE, Fischer MD, Gow JA, et al. Subretinal timrepigene emparvovec in adult men with choroideremia: a randomized phase 3 trial. Nat Med. 2023;29(10):2464–72. https://doi.org/10.1038/s41591-023-02520-3.
Aleman TS, Huckfeldt RM, Serrano LW, et al. Adeno-associated virus serotype 2-hCHM subretinal delivery to the macula in choroideremia two-year interim results of an ongoing phase I/II gene therapy trial. Ophthalmology. 2022;129(10):1177–91. https://doi.org/10.1016/j.ophtha.2022.06.006.
Zhai Y, Xu M, Radziwon A, et al. AAV2-mediated gene therapy for choroideremia: 5-year results and alternate anti-sense oligonucleotide therapy. Am J Ophthalmol. 2023;248:145–56. https://doi.org/10.1016/j.ajo.2022.12.022.
4D Molecular Therapeutics. 4D molecular therapeutics reports interim results from the 4D-310 phase 1/2 clinical trial in patients with Fabry disease and provides clinical data update from the 4D-110 phase 1/2 clinical trial in patients with choroideremia. Available from: https://4dmt.gcs-web.com/news-releases/news-release-details/4d-molecular-therapeutics-reports-interim-results-4d-310-phase. Accessed 7 Feb 2024.
Molday RS, Kellner U, Weber BHF. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31(3):195–212. https://doi.org/10.1016/j.preteyeres.2011.12.002.
Ou J, Vijayasarathy C, Ziccardi L, et al. Synaptic pathology and therapeutic repair in adult retinoschisis mouse by AAV-RS1 transfer. J Clin Investig. 2015;125(7):2891–903. https://doi.org/10.1172/jci81380.
Tolun G, Vijayasarathy C, Huang R, et al. Paired octamer rings of retinoschisin suggest a junctional model for cell–cell adhesion in the retina. Proc Natl Acad Sci U S A. 2016;113(19):5287–92. https://doi.org/10.1073/pnas.1519048113.
Pennesi ME, Yang P, Birch DG, et al. Intravitreal delivery of rAAV2tYF-CB-hRS1 vector for gene augmentation therapy in patients with X-linked retinoschisis 1-year clinical results. Ophthalmol Retin. 2022;6(12):1130–44. https://doi.org/10.1016/j.oret.2022.06.013.
Foundation Fighting Blindness. AGTC announces topline interim six-month data of XLRS gene therapy from ongoing phase I/II clinical trial. Available from: https://www.fightingblindness.org/research/agtc-announces-topline-interim-six-month-data-of-xlrs-gene-therapy-from-ongoing-phase-i-ii-clinical-trial-5666. Accessed 7 Feb 2024.
Mishra A, Sieving PA. X-linked retinoschisis and gene therapy. Int Ophthalmol Clin. 2021;61(4):173–84. https://doi.org/10.1097/iio.0000000000000373.
Cukras C, Wiley HE, Jeffrey BG, et al. Retinal AAV8-RS1 gene therapy for X-linked retinoschisis: initial findings from a phase I/IIa trial by intravitreal delivery. Mol Ther. 2018;26(9):2282–94. https://doi.org/10.1016/j.ymthe.2018.05.025.
Atsena Therapeutics. Atsena Therapeutics receives FDA clearance of IND application for ATSN-201, an investigational gene therapy for the treatment of X-linked retinoschisis. Available from: https://atsenatx.com/press-release/atsena-therapeutics-receives-fda-clearance-of-ind-application-for-atsn-201-an-investigational-gene-therapy-for-the-treatment-of-x-linked-retinoschisis/. Accessed 7 Feb 2024.
Fischer MD, Michalakis S, Wilhelm B, et al. Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia. JAMA Ophthalmol. 2020;138(6):643–51. https://doi.org/10.1001/jamaophthalmol.2020.1032.
Reichel FF, Michalakis S, Wilhelm B, et al. Three-year results of phase I retinal gene therapy trial for CNGA3-mutated achromatopsia: results of a non randomised controlled trial. Br J Ophthalmol. 2022;106(11):1567–72. https://doi.org/10.1136/bjophthalmol-2021-319067.
Huckfeldt RM, Comander J, Pennesi ME, et al. Findings on visual photosensitivity in two phase 1/2 clinical trials of subretinal gene therapy with AGTC-401 and AGTC-402 for CNGB3 and CNGA3 achromatopsia. IOVS, ARVO Journals 2022. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2778982. Accessed 7 Feb 2024.
Michaelides M, Hirji N, Wong SC, et al. First-in-human gene therapy trial of AAV8-hCARp.hCNGB3 in adults and children with CNGB3-associated achromatopsia. Am J Ophthalmol. 2023;253:243–51. https://doi.org/10.1016/j.ajo.2023.05.009.
Rajanala K, Dotiwala F, Upadhyay A. Geographic atrophy: pathophysiology and current therapeutic strategies. Front Ophthalmol. 2023;3:1327883. https://doi.org/10.3389/fopht.2023.1327883.
Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cell Mol Life Sci. 2021;78(10):4487–505. https://doi.org/10.1007/s00018-021-03796-9.
Nielsen J, MacLaren RE, Heier JS, et al. Preliminary results from a first-in-human phase I/II gene therapy study (FOCUS) of subretinally delivered GT005, an investigational AAV2 vector, in patients with geographic atrophy secondary to age-related macular degeneration. IOVS, ARVO Journals 2022. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2780316. Accessed 7 Feb 2024.
Novartis. GT005 (PPY988): development program in geographic atrophy. Available from: https://www.novartis.com/news/gt005-ppy988-development-program-geographic-atrophy. Accessed 7 Feb 2024.
PentaVision. Retinal physician. Available from: https://retinalphysician.com/issues/2020/april/clinical-trial-download-data-on-a-gene-therapy-for-dry-and-wet-amd/. Accessed 7 Feb 2024.
Regillo C. ADVM-022 (ixoberogene soroparvovec) intravitreal gene therapy for neovascular age-related macular degeneration: end of study results from the 2-year OPTIC trial. Available from: https://adverum.com/wp-content/uploads/2022/11/ADVM-Retina-Society-OPTIC-2022.pdf. Accessed 7 Feb 2024.
Campochiaro PA, Avery R, Brown DM, et al. Gene therapy for neovascular age-related macular degeneration by subretinal delivery of RGX-314: a phase 1/2a dose-escalation study. Lancet. 2024;403(10436):1563–73. https://doi.org/10.1016/s0140-6736(24)00310-6.
4D Molecular Therapeutics. 4DMT presents positive interim data from intravitreal 4D-150 phase 1/2 PRISM clinical trial in patients with wet AMD at ARVO 2023. Available from: https://4dmt.gcs-web.com/news-releases/news-release-details/4dmt-presents-positive-interim-data-intravitreal-4d-150-phase-12. Accessed 7 Feb 2024.
Heier J, Kherani S, Desai S, et al. Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: a phase 1, open-label trial. Lancet. 2017;390(10089):50–61. https://doi.org/10.1016/s0140-6736(17)30979-0.
Rakoczy EP, Lai CM, Magno AL, et al. Gene therapy with recombinant adeno-associated vectors for neovascular age-related macular degeneration: 1 year follow-up of a phase 1 randomised clinical trial. Lancet. 2015;386(10011):2395–403. https://doi.org/10.1016/s0140-6736(15)00345-1.
Constable IJ, Lai CM, Magno AL, et al. Gene therapy in neovascular age-related macular degeneration: three-year follow-up of a phase 1 randomized dose escalation trial. Am J Ophthalmol. 2017;177:150–8. https://doi.org/10.1016/j.ajo.2017.02.018.
Rakoczy EP, Magno AL, Lai CM, et al. Three-year follow-up of phase 1 and 2a rAAV.sFLT-1 subretinal gene therapy trials for exudative age-related macular degeneration. Am J Ophthalmol. 2019;204:113–23. https://doi.org/10.1016/j.ajo.2019.03.006.
SparingVision. SparingVision reports positive initial safety data from the first cohort treated in its PRODYGY phase I/II gene therapy trial. Available from: https://sparingvision.com/sparingvision-reports-positive-initial-safety-data-from-the-first-cohort-treated-in-its-prodygy-phase-i-ii-gene-therapy-trial/. Accessed 7 Feb 2024.
McClements ME, Staurenghi F, MacLaren RE, Cehajic-Kapetanovic J. Optogenetic gene therapy for the degenerate retina: recent advances. Front Neurosci. 2020;14: 570909. https://doi.org/10.3389/fnins.2020.570909.
Sahel J-A, Boulanger-Scemama E, Pagot C, et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat Med. 2021;27(7):1223–9. https://doi.org/10.1038/s41591-021-01351-4.
First four patients in Bionic Sight’s optogenetic gene. Available from: https://www.globenewswire.com/news-release/2021/03/30/2201412/0/en/First-Four-Patients-In-Bionic-Sight-s-Optogenetic-Gene-Therapy-Trial-Are-Able-To-Detect-Light-And-Motion.html. Accessed 7 Feb 2024.
Wright W, Gajjeraman S, Batabyal S, et al. Restoring vision in mice with retinal degeneration using multicharacteristic opsin. Neurophotonics. 2017;4(4): 041412. https://doi.org/10.1117/1.nph.4.4.041412.
Nanoscope Therapeutics. Nanoscope’s optogenetic gene therapy restores clinically meaningful vision in 11 patients blinded by retinitis pigmentosa. Available from: https://nanostherapeutics.com/2021/06/03/nanoscopes-optogenetic-gene-therapy-restores-clinically-meaningful-vision/. Accessed 12 Feb 2024.
Nanoscope Therapeutics. Nanoscope Therapeutics announces positive top-line results from randomized controlled trial of MCO-010 for retinitis pigmentosa. Available from: https://nanostherapeutics.com/2024/03/26/nanoscope-therapeutics-announces-top-line-results-from-ph2-trial-of-mco-010-for-retinitis-pigmentosa/. Accessed 29 May 2024.
Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229–33. https://doi.org/10.1038/s41591-018-0327-9.
Dulla K, Slijkerman R, van Diepen HC, et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol Ther. 2021;29(8):2441–55. https://doi.org/10.1016/j.ymthe.2021.04.024.
Pierce EA, Aleman TS, Jayasundera KT, et al. Gene editing for CEP290-associated retinal degeneration. N Engl J Med. 2024. https://doi.org/10.1056/nejmoa2309915.
Liu C-H, Wolf P, Dong R, et al. A mutation-independent CRISPR/Cas9-based ‘knockout and replace’ strategy to treat rhodopsin-associated autosomal dominant retinitis pigmentosa. IOVS, ARVO Journals 2022. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2780948. Accessed 12 Feb 2024.
Russell SR, Drack AV, Cideciyan AV, et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat Med. 2022;28(5):1014–21. https://doi.org/10.1038/s41591-022-01755-w.
Jeune VL, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods. 2013;24(2):59–67. https://doi.org/10.1089/hgtb.2012.243.
Mingozzi F, Maus MV, Hui DJ, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13(4):419–22. https://doi.org/10.1038/nm1549.
Preclinical EDIT-101 poster. Available from: https://www.editasmedicine.com/wp-content/uploads/2019/10/16.pdf. Accessed 12 Feb 2024.
Eid A, Alshareef S, Mahfouz MM. CRISPR base editors: genome editing without double-stranded breaks. Biochem J. 2018;475(11):1955–64. https://doi.org/10.1042/bcj20170793.
Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–42. https://doi.org/10.1038/nbt.3190.
Chuai G, Ma H, Yan J, et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018;19(1):80. https://doi.org/10.1186/s13059-018-1459-4.
Haeussler M, Schönig K, Eckert H, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. https://doi.org/10.1186/s13059-016-1012-2.
Guo C, Ma X, Gao F, Guo Y. Off-target effects in CRISPR/Cas9 gene editing. Front Bioeng Biotechnol. 2023;11:1143157. https://doi.org/10.3389/fbioe.2023.1143157.
Leibowitz ML, Papathanasiou S, Doerfler PA, et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet. 2021;53(6):895–905. https://doi.org/10.1038/s41588-021-00838-7.
Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–71. https://doi.org/10.1038/nbt.4192.
Enache OM, Rendo V, Abdusamad M, et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet. 2020;52(7):662–8. https://doi.org/10.1038/s41588-020-0623-4.
Staurenghi F, McClements ME, Salman A, MacLaren RE. Minicircle delivery to the neural retina as a gene therapy approach. Int J Mol Sci. 2022;23(19):11673. https://doi.org/10.3390/ijms231911673.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
All authors are supported by funding from the National Institute for Health Research. The views expressed are those of the authors and not necessarily those of the National Institute for Health Research or the Department of Health and Social Care.
Conflicts of Interest/Competing Interests
Robert E. MacLaren and Michelle E. McClements are co-founders of Beacon Therapeutics. Robert E. MacLaren and Michelle E. McClements hold consultancy agreements with Beacon Therapeutics. Robert E. MacLaren is a consultant to Scribe Therapeutics, Novartis Pharmaceuticals and Biogen Inc. Robert E. MacLaren also sits on advisory boards for the US Food and Drug Administration and the UK National Institute for Health and Care Excellence. Robert E. MacLaren and Michelle E. McClements are listed as inventors on retinal gene therapy patents owned by the University of Oxford. Maram E.A. Abdalla Elsayed, Lauren Major and Cristina Martinez-Fernandez de la Camara have no conflicts of interest that are directly relevant to the content of this article.
Code Availability
Not applicable.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Authors’ Contributions
All authors provided content and performed manuscript editing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
McClements, M.E., Elsayed, M.E.A.A., Major, L. et al. Gene Therapies in Clinical Development to Treat Retinal Disorders. Mol Diagn Ther (2024). https://doi.org/10.1007/s40291-024-00722-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s40291-024-00722-0