
Abstract
Background
Childhood epilepsies are caused by heterogeneous underlying disorders where approximately 40% of the origins of epilepsy can be attributed to genetic factors. The application of next-generation sequencing (NGS) has revolutionized molecular diagnostics and has enabled the identification of disease-causing genes and variants in childhood epilepsies. The objective of this study was to use NGS to identify variants in patients with childhood epilepsy, to expand the variant spectrum and discover potential therapeutic targets.
Methods
In our study, 55 children with epilepsy of unknown etiology were analyzed by combining clinical-exome and whole-exome sequencing. Novel variants were characterized using various in silico algorithms for pathogenicity and structure prediction.
Results
The molecular genetic cause of epilepsy was identified in 28 patients and the overall diagnostic success rate was 50.9%. We identified variants in 22 different genes associated with epilepsy that correlate well with the described phenotype. SCN1A gene variants were found in five unrelated patients, while ALDH7A1 and KCNQ2 gene variants were found twice. In the other 19 genes, variants were found only in a single patient. This includes genes such as ASH1L, CSNK2B, RHOBTB2, and SLC13A5, which have only recently been associated with epilepsy. Almost half of diagnosed patients (46.4%) carried novel variants. Interestingly, we identified variants in ALDH7A1, KCNQ2, PNPO, SCN1A, and SCN2A resulting in gene-directed therapy decisions for 11 children from our study, including four children who all carried novel SCN1A genetic variants.
Conclusions
Described novel variants will contribute to a better understanding of the European genetic landscape, while insights into the genotype-phenotype correlation will contribute to a better understanding of childhood epilepsies worldwide. Given the expansion of molecular-based approaches, each newly identified genetic variant could become a potential therapeutic target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A cohort of 55 patients with childhood epilepsy of unknown etiology was analyzed using next-generation sequencing and a 50.9% detection rate was reached. |
This cohort holds particular significance, given that almost half of the successfully resolved cases (46.4%) harbor novel genetic variants. |
Identification of variants in ALDH7A1, KCNQ2, PNPO, SCN1A and SCN2A resulted in gene-directed therapy decisions for 11 children from our study, including four children who all carried novel SCN1A genetic variants. |
1 Introduction
Childhood epilepsies are caused by heterogeneous underlying disorders (structural, genetic, infectious, metabolic, immune, and unknown) and it is estimated that approximately 40% of the etiological causes of epilepsy are now known to be due to genetic factors [1, 2]. Over the past three decades, the process of identifying the genetic cause of epilepsy has transitioned from Sanger sequencing of candidate genes to epilepsy-specific gene panels, clinical-exome sequencing (CES), whole-exome sequencing (WES), and ultimately whole-genome sequencing (WGS) [3]. Thus, the application of next-generation sequencing (NGS) has completely revolutionized the diagnostics of patients with epilepsy and has enabled the identification of disease-causing genes and variants in childhood epilepsies [4]. Prompt establishment of a specific diagnosis is essential to determine prognosis, and enable counseling on the recurrence risk in future pregnancies [5]. Moreover, in the case of monogenic epilepsy, it could lead to a specific treatment that targets the underlying pathophysiology and successfully alleviates at least some phenotypic features [6].
Childhood monogenic epilepsies associated with a developmental delay comprise a wide spectrum of different disorders where the disease-causing variants affect an extensive range of targets, from ion channels to various genes regulating cell metabolism and signaling. A large percentage of monogenic epilepsies are comprised of channelopathies, where the impaired function of voltage-gated ion channels (whether they be sodium, calcium, potassium, and chloride channels) or ligand-gated ion channels (as with acetylcholine, glutamate, and γ-aminobutyric acid type A (GABAA) receptors) stand as the hallmark of the disease [7].
The SCN1A gene codes for voltage-gated sodium channel alpha-1 subunit Nav1.1, crucial for neuronal excitability, and it is commonly associated with Dravet syndrome, a severe form of early-onset treatment-resistant epilepsy characterized by a developmental delay, various types of seizures including generalized tonic-clonic, myoclonic, and febrile, as well as other neurological abnormalities. It is estimated that more than 80–90% of Dravet syndrome cases result from pathogenic de novo variants in the SCN1A gene, but variants in other genes such as GABRA1, STXBP1, SCN9A, SCN1B, GABRG2, HCN1, and CHD2 have also been described [8]. Nevertheless, not all SCN1A variants lead to Dravet syndrome, as this gene has been associated with other conditions including familial febrile seizures, genetic epilepsy with febrile seizures plus, and familial hemiplegic migraine type 3 [9]. Other voltage-gated sodium channels such as SCN2A and SCN8A are associated with early infantile epileptic encephalopathy, where additional clinical manifestations of the patients include a developmental delay and ataxia [10]. Voltage-gated calcium channels play a critical role in regulating the influx of calcium ions into cells in response to membrane potential changes, where impairment due to pathogenic variants can give rise to abnormal neuronal excitability and lead to the development of seizures. A growing number of genes such as CACNA1A, CACNA1B, CACNB4, CACNA1C, CACNA1D, CACNA1E, CACNA1G, and CACNA1H have been associated with different types of childhood epilepsies. This heterogeneous group of disorders has been characterized by early-onset seizures and a developmental delay, and also implicated in epilepsy that occurs in the context of other neurodevelopmental disorders, such as autism spectrum disorder and intellectual disability [11]. However, pathogenic variants in genes encoding potassium and chloride channels are less common as causes of epilepsy (except for the frequent KCNQ2-related disorders), but they have been implicated in a diverse range of disorders characterized by various types of seizure, with KCNMA1, KCNQ2, KCNA2, CLCN2, and CLCN4 being among the most prevalent [12].
Impaired function of ligand-gated ion channels represents another prevalent cause of childhood epilepsies. Nicotinic acetylcholine receptors mediate fast signal transmission at synapses through permeation of sodium and potassium ions, therefore modulating the neurotransmitter release. Pathogenic variants in the genes CHRNA2, CHRNA4, and CHRNB2, which code for this category of receptors, impair the synaptic function and lead to the phenotype of nocturnal frontal lobe epilepsy [13]. Mediation of major inhibitory functions in the neurons is accomplished via the GABAA receptor, a group of ligand-gated chloride channels. Pathogenic variants in genes encoding various types of receptors (GABRA1, GABRB1, GABRB2, GABRB, GABRD, and GABRG2) are among those most frequently found in patients presenting with this type of developmental and epileptic encephalopathy (DEE) [14]. However, even though ionotropic glutamate receptors facilitate excitatory synaptic transmission and plasticity of the neurons, variants in the genes GRIN1, GRIN2A, GRIN2B, and GRIN2D, which code for several types of receptors, similarly lead to the phenotype of DEE [15].
In addition to synaptic excitability at the level of ion channels, other key players involved in the appropriate connection between the pre-synaptic and post-synaptic neurons (the pre-synaptic and post-synaptic membranes, the synaptic cleft, and the neighboring glia with extracellular matrix) can cause derangements of synaptic transmission and plasticity [16]. Furthermore, disordered neuronal cell signaling in numerous pathways, including the mechanistic target of rapamycin (mTOR), mitogen-activated protein kinase, JAK-STAT, WNT/β-catenin, and cAMP signaling have been suggested to play a pivotal role in pathophysiologic mechanisms that lead to seizures, making proteins involved in these pathways potential therapeutic targets [17]. Moreover, given the substantial energy needs in highly metabolically active neuronal cells, any derangement in cellular or mitochondrial metabolism can result in seizure genesis [18].
Therefore, a growing number of diverse proteins included in cell signaling and metabolism are being associated with childhood epilepsies. Among the prevalent proteins are regulators of the actin cytoskeleton during synaptogenesis, as in the case of proline-rich transmembrane protein 2 (PRRT2), where pathogenic variants in the PRRT2 gene can lead to seizures, but also to movement disorders [19]. A range of enzymes, their cofactors, and other proteins involved in cell metabolic pathways are being recognized as causes of epilepsy. Variants in the gene encoding the enzyme pyridoxine 5′-phosphate oxidase (PNPO) lead to pyridoxine-dependent epilepsy, a condition that typically presents with early-onset seizures with a developmental delay, while another pyridoxine-dependent early-onset epilepsy is caused by deficiency of the aldehyde dehydrogenase 7 family, member A1 (ALDH7A1) enzyme [20]. In the case of glucose transporter type 1 deficiency syndrome due to pathogenic variants in the SLC2A1 gene, impaired glucose transport across the blood–brain barrier results in early-onset treatment-resistant seizures [21]. Finally, various metabolic diseases such as mitochondrial disorders, urea cycle disorders, organic acidurias, maple syrup urine disease, and untreated hyperphenylalaninemia can present with epilepsy as a result of enzyme/cofactor deficiency [22].
When considering the broad nature of possible targets, epilepsy can be a presenting symptom in a myriad of genetic diseases including encephalopathies, movement disorders, and neurodevelopmental disorders, as well as different metabolic diseases. The underlying molecular defect itself determines the disease severity as well as the possible treatment options. Nevertheless, pinpointing not only the particular gene but rather the exact variant allows for strategic treatment decisions, whether that involves prescribing the most suitable medication or circumventing those that could potentially exacerbate the condition [23]. In disorders resulting from enzyme/cofactor deficiency, therapeutic strategies may go toward enzyme replacement therapy and supplementation with the missing cofactor, such as in the case of pyridoxine-dependent epilepsy. In metabolism-based treatments, as with glucose transporter deficiency, the therapeutic intervention circumvents the high energy demands and brings the cell into a homeostasis using alternative energy sources, with the high-fat anti-seizure ketogenic diet [7]. Bearing all this in mind, the first step towards possible therapeutic options for childhood epilepsies should clearly be the identification of the precise underlying genetic etiology.
With the advancement of knowledge of the pathophysiology of seizures, the number of genes that are linked to monogenic epilepsy has been rapidly expanding. Simultaneously, the process of identifying the genetic cause of epilepsy has transitioned from Sanger sequencing of candidate genes to epilepsy-specific gene panels, CES (a commercially available panel containing 4813 OMIM-referenced genes), WES, and ultimately WGS [3, 24].
Recently, a comprehensive Genes4Epilepsy list of all genes associated with monogenic epilepsy was compiled to facilitate establishing the genetic causes of epilepsy [25]. To achieve a high diagnostic success rate and minimize the diagnostic journey of patients with epilepsy, it is crucial to conduct a thorough analysis of all genes that have been linked to the particular phenotype. Therefore, we employed both CES and WES in order to reach a high diagnostic success rate. This is the first comprehensive study on molecular and phenotypic characteristics of patients with childhood epilepsies from Serbia aiming to describe and characterize new genetic variants. The relevance of describing new gene variants lays in expanding the spectrum of variants causing epilepsy. In addition, each new genetic variant could become a potential therapeutic target.
2 Materials and Methods
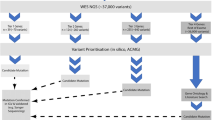
Patients who presented with drug-resistant epilepsy associated with a development delay of unknown etiology in childhood, with suspicion of a genetic cause were included in this study. All patients initially underwent anamnesis, comprehensive clinical examinations, serum biochemical and hematological analyses, along with electroencephalography and neuroimaging tests. The criteria for including patients in this study and proceeding with genetic testing were as follows: (1) the underlying cause of seizures could not be determined through clinical examinations, electroencephalography, or neuroimaging; (2) neuroimaging results were either normal or clinically insignificant, with no connection to the seizures; and (3) biochemical results (electrolytes, transaminases, and other specific tests depending on the suspicion of inborn error of metabolism) indicated a potential genetic disorder. The International League Against Epilepsy’s classification of epilepsy was used to classify the seizures [2].
The data of patients with epilepsy were collected at the Department for Pediatric Neurology and Department for Metabolic Diseases of the Institute for Mother and Child Healthcare of Serbia “Dr Vukan Cupic”, a tertiary-level institution, recognized as the reference center for rare diseases. Subsequently, patients were referred to the Institute of Molecular Genetics and Genetic Engineering for a genetic analysis. This study was approved by the Ethics Committees of both institutions (No. 31/2020 and No. O-EO-017/2020). During the study period, from January 2021 to April 2022, 55 samples from patients from unrelated non-consanguineous families were collected. Parents’ samples were also collected to enable a segregation analysis.
Genomic DNA was isolated from venous blood using the QIAamp DNA-Blood-Mini-Kit (Qiagen, Dusseldorf, Germany) and further analyzed by NGS either using the CES panel (TruSight One Gene Panel, MiSeq; Illumina, San Diego, California, USA) or WES sequencing (BGI WES Library construction kit, DNBSEQ-G400; MGI, Shenzhen, China) with alignment and analysis performed against the GRCh38/hg38 reference genome assembly. Systematic interpretation of variants was performed in the same way regardless of the used sequencing approach [26]. All variants were prioritized using Variant Interpreter (Illumina, San Diego, California, USA) and VarSome (Saphetor, Lausanne, Switzerland) and were classified according to the recommendations of the American College of Medical Genetics and Genomics [27, 28]. For an in silico analysis of all detected variants, computational programs REVEL, BayesDel addAF, BayesDel nodAF, MetaRNN, PROVEAN, CADD, SIFT, PolyPhen-2, and MutPred2 for variant’ pathogenicity prediction were used (Table 1 of the Electronic Supplementary Material [ESM]) [29,30,31,32,33,34]. A segregation analysis was performed using conventional Sanger sequencing when parental DNA was available (13 out of 31 cases). Novel variants were confirmed with Sanger sequencing (Fig. 1 of the ESM).

Predicted three-dimensional molecular models of protein folding resulting from missense substitutions with a close-up view of the regions harboring novel missense variants. Residues affected by variants are depicted in yellow. First inline image represents the wt protein whilst the image next to it represents the variant protein. a ADSL:p.Leu121Phe; b ASH1L:p.Ala2147Val; c DCX:p.Arg102Pro; d HSD17B10:p.Leu227Arg. Novel missense variant GNAS:p.Asp295Val is not shown as the server used for automatic fold recognition was not able to predict the structure of the protein domains where the affected residues are located
For in silico modeling of novel variants, Phyre2 and EzMol for structure prediction (http://www.sbg.bio.ic.ac.uk/phyre2 and http://www.sbg.bio.ic.ac.uk/ezmol) were used [35, 36]. The wild-type protein structures served as references for comparison with structures incorporating missense substitutions. Databases used for the prediction of protein evolutionarily conserved regions and post-translational and other protein modifications were Aminode (http://www.aminode.org [Accessed March 2024]) and MutPred2 (http://mutpred.mutdb.org/ [Accessed March 2024]) [34, 37].
3 Results
3.1 Genetic Analysis and Diagnostic Success
In this study, we analyzed 55 children with epilepsy of unknown etiology. Using CES we analyzed 38 patients, and for 16 of them a diagnosis was established (42.1%). For 17 patients with childhood epilepsy, our primary method of choice was WES, which led to the identification of disease-causing genes in ten of them (58.8%). Then, 11 selected negative CES cases were re-sequenced using WES. With this strategy, nine cases remained unsolved, and two were associated with a recently described epilepsy genes (ASH1L and CSNK2B), which were not present in the CES panel [38]. Therefore, the overall diagnostic success rate of our study was 50.9% (28/55) [16 primarily CES, ten primarily WES, and two WES after re-sequencing].
Genotype and phenotype characteristics of 28 cases with detected variants are presented in Table 1. Pathogenic, likely pathogenic variants or variants of uncertain significance (VUS) [above 50% posterior probability] have been detected in 22 epilepsy genes, which correlated well with the observed phenotype. SCN1A gene variants were found in five unrelated patients, while ALDH7A, and KCNQ2 gene variants were found twice. In the other 19 genes, variants were found only in a single patient (Table 1). Of 22 disease-causing genes in patients from our cohort, several genes (ASH1L, CSNK2B, RHOBTB2, and SLC13A5) were just recently associated with epilepsy [38,39,40,41].
3.2 Pathogenicity Predictions and Molecular Modeling of Novel Genetic Variants
Thirteen novel genetic variants were identified (Table 1). Therefore, 46.4% of patients patients in whom a genetic diagnosis could be confirmed carried novel variants. Two novel variants that were found in the ADSL and PMPCA genes follow an autosomal-recessive inheritance pattern, two in genes DCX and HSH1L were X-linked, and nine follow the autosomal-dominant model of inheritance (Table 1). A segregation analysis of available parents is given in Table 1.
Eight novel variants were truncating variants (five nonsense, three frameshift, and zero splice site) and were found in genes for which loss of function is a known mechanism of disease. Five other novel variants were missense. According to American College of Medical Genetics and Genomics guidelines, one of these missense variants was classified as VUS, this was based on its low frequency/absence in population databases and in silico predicting pathogenicity, while for other variants additional evidence such as location in a hot-spot region and the presence of an alternative variant previously classified as pathogenic was available.
Predicted three-dimensional molecular models of protein folding resulting from missense substitutions in ADSL, ASH1L, DCX, and HSD17B10 proteins are presented in Fig. 1. The affected residues were highlighted in yellow. The structural effect could not be predicted for the novel missense variant in the GNAS protein because of limitations in automatic fold recognition for the relevant protein domain.
The amino acid sequence alignment of proteins ADSL, ASH1L, DCX, GNAS, HSD17B10 across various species was conducted using the Aminode platform. Multiple sequences were aligned for each protein, revealing complete evolutionary conservation of amino acid residues affected by variants p.Leu121Phe (ADSL) and p.Ala2147Val (ASH1L), among 25 orthologs. However, for amino acid changes in DCX, GNAS, and HSD17B10 proteins (p.Arg102Pro, p.Asp295Val, and p.Leu227Arg, respectively), multiple sequence alignments showed high but not complete evolutionary conservation of the affected residues (http://www.aminode.org).
MutPred2 was used to predict the impact of more than 50 different protein properties and, thus, to imply the molecular mechanisms of pathogenicity. It was shown that the p.(Leu121Phe) variant in the ADSL gene causes the loss of the allosteric site at R125 with a probability of 0.20. The DCX protein exhibits an altered ordered interface with a probability of 0.33 and a loss of the allosteric site at R102 with a probability of 0.2 due to the c.305G>C (p.Arg102Pro) variant in the gene. Variant c.680T>G (p.Leu227Arg) in the HSD17B10 gene causes a gain of intrinsic disorder, with a probability of 0.32, alongside the acquisition of an allosteric site and a catalytic site at residue R226, with a probability of 0.22. Because of the c.6440C>T (p.Ala2147Val) variant in the ASH1L gene, the protein undergoes a change in DNA binding, with a probability of 0.24, and exhibits an increase in acetylation at residue K2150, with a probability of 0.21. There is a 0.10 probability of gain of methylation at K300 within the GNAS protein because of the c.884A>T (p.Asp295Val) variant in this gene.
4 Discussion
4.1 Concept of Diagnostic Procedure of Childhood Epilepsy Utilizing Various NGS Approaches
The number of genes associated with epilepsy is constantly growing and a systematic analysis of NGS data based on all genes associated with epilepsy, including recently published and/or candidate epilepsy genes may be the most important factor in achieving high detection rates [42]. Obviously, some genes are more frequent causes of monogenic epilepsy, while other genes are rarely the cause of epilepsy. However, analyzing all genes associated with epilepsy is essential in order to reach high diagnostic rates and provide fast and accurate diagnosis to all patients in the spirit of “leaving no one behind”. Recently, a comprehensive list of 953 epilepsy genes was compiled and displayed to the scientific community for future improvements and editing [25]. The Genes4Epilepsy list included all genes firmly associated with monogenic diseases in which epilepsy is the primary clinical presentation, as well as neurodevelopmental disorder, and malformations, and genes responsible for inborn errors of metabolism that may cause secondary epilepsy in some patients [25].
In order to assess the current utility of CES in diagnostic genetic testing of childhood epilepsies, we confirmed that CES contains 536 out of 953 genes for epilepsy, meaning that currently 56% of all epilepsy genes can be analyzed using the CES panel [25]. With an increase in the number of genes linked to epilepsy, the percentage of epilepsy-specific genes analyzed by CES and thus its diagnostic success rate will decline. Thus, for cases that remained unresolved after CES, an additional WES analysis is recommended. Therefore, the first study reporting the molecular genetic basis of childhood epilepsy in Serbia combined both CES and WES approaches to ensure a high detection rate.
Furthermore, in order to evaluate the difference in their diagnostic success rates, CES and WES were performed in two separate cohorts of patients with childhood epilepsy. The previously reported detection rate for CES with 4813 OMIM genes was 25%, while for WES it can reach 70% in different cohorts of patients with epilepsy [42, 43]. In our study, the diagnostic success rate was 42.1% for CES while it was only slightly higher for WES at 58.8%. The relative detection rates for both CES and WES approaches obtained in our study confirm the importance of careful interpretation of each detected variant. In addition to the broadness of the applied strategy for genetic testing, a diagnostic success rate is a consequence of comprehensive interpretation of identified genetic variants and active consultations with physicians to align genetic variants with the observed phenotype. Etiology of epilepsy in two cases were based on recent publications in which ASH1L and CSNK2B have been associated with epilepsy [38, 39]. Furthermore, a regular re-analysis of sequenced but undiagnosed patients should yield new diagnoses and therefore have a positive impact on the clinical management of patients, reproductive planning of parents and family members, as well as on the healthcare cost savings [44]. In our study, reanalysis after only a 6-month period resulted in the identification of a genetic variant one additional patient.
Although differences between diagnostic rates of CES and WES reported in independent publications can be up to 45% [42, 43], in our study where the same principles for the interpretation of CES and WES results were applied, the diagnostic rate of WES is 16.7% higher than CES. Therefore, we suggest that currently CES is still a valuable strategy for diagnostic genetic testing of childhood epilepsies if comprehensive interpretation and re-analysis of identified genetic variants are performed. In addition, CES is superior to any individual epilepsy-specific gene panel when it comes to containing genes for epilepsy. In the settings where sequencing capacity is not a drawback, as the price of the WES decreases and the number of genes associated with epilepsy increases, it is expected that WES will completely replace CES in diagnostic genetic testing of childhood epilepsies.
4.2 Characterization of 13 Novel Genetic Variants
Thirteen novel variants were detected in ten different genes (Table 1). Interestingly, four novel variants were found in the SCN1A gene, all with a loss-of-function effect. We further detected novel variants leading to truncated protein in the genes CHD2, CSNK2B, HNRNPU, and PMPCA. A patient harboring a novel variant in the CHD2 gene was classified as having Lennox–Gastaut syndrome with childhood onset, a patient with a novel variant in the CSNK2B gene was diagnosed with focal-onset seizures and epileptic spasms, while a patient with a novel variant in the HNRNPU gene was found to have combined focal and myoclonic seizures beginning in infancy. However, a patient with a novel variant in the recessive inheritance gene PMPCA (along with a previously described variant) coding for nuclear-encoded mitochondrial protein presented with childhood-onset spinocerebellar ataxia, generalized epilepsy, and brain abnormalities.
We also detected novel missense variants in the following genes: ADSL, ASH1L, DCX, GNAS, and HSD17B10. These variants were classified as pathogenic (DCX and GNAS genes), likely pathogenic (ADSL and HSD17B10 gene), and VUS (ASH1L). We assessed their effect on protein by various in silico algorithms linked to VarSome (Table 1 of the ESM) and characterized them in a three-dimensional model structure, confirming their pathogenicity (Fig. 1).
The analysis of amino acid sequence alignment across species revealed complete evolutionary conservation of specific variants in ADSL and ASH1L, suggesting their functional importance, while variability was observed in DCX, GNAS, and HSD17B10 proteins. In the majority of aberrant proteins, novel genetic variants introduced structural modifications such as the “gain of intrinsic disorder” in HSD17B10, causing augmentation in regions within a protein that lack a fixed or ordered three-dimensional structure. Other structural changes are predicted to alter proteins’ ordered interface, which can disrupt normal protein–protein interactions, as observed in DCX, or resulting in the loss of an allosteric site, which compromises the regulatory responsiveness of ADSL. Analysis of post-translational modifications showed that the aberrant ASH1L may acquire a new acetylation site at a near K2150 residue, whereas the aberrant GNAS is predicted to develop a new methylation site adjacent to the K300 residue. Functional change, namely the gain of catalytic site, was predicted only for the aberrant HSD17B10 protein.
The predicted structural models offer insight into potential functional consequences of these missense variants, for instance, changes in local conformation and potential disruptions of protein–protein or protein–ligand interactions. However, experimental validation is recommended to precisely establish the impact of these substitutions on protein function and consequently their impact on disease phenotype. Description of 13 novel variants will contribute to a better understanding of the European genetic landscape, while insights on phenotype specificities reported in Table 1 and the genotype-phenotype correlation will contribute to better understanding of childhood epilepsies worldwide.
Furthermore, we describe in detail an interesting genotype-phenotype observation of patients with variants in ADSL, ASH1L, CSNK2B, GRIN1, and RHOBTB2 genes.
The ADSL gene encodes an enzyme that belongs to the lyase 1 family, and it is an essential enzyme involved in purine metabolism. ADSL catalyzes two non-sequential reactions in the de novo purine biosynthetic pathway. Pathogenic variants in this gene are associated with adenylosuccinase deficiency, a rare autosomal recessive disorder characterized by epilepsy, psychomotor retardation, or autistic features (https://www.orpha.net/en/disease/detail/46) [45]. Since the first ADSL variant described (Fon EA), approximately 100 pathogenic/likely pathogenic variants and more than 500 VUS have been reported in the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) [46]. Moreover, 50% of patients with molecularly confirmed ADSL are compound heterozygotes [47]. In this study, compound heterozygous variants c.340T>C and c.363G>T were identified in the ADSL gene of a patient presenting with DEE characterized by focal-onset seizures, epileptic spasms, and apneic episodes as an early sign of acute encephalopathy. At the time of genetic testing, the patient was 1 month old; therefore, there was no available information about autistic features. However, ongoing monitoring of the patient will enable observation of any potential manifestation of autism spectrum disorder as part of the disease spectrum.
The ASH1L gene encodes a histone methyltransferase that catalyzes histone H3K36 methylation and is crucial for chromatin modification and gene transcription [48]. Heterozygous pathogenic variants in ASH1L cause an autosomal dominant neurodevelopmental disorder with a broad range of features, including intellectual disability, autism spectrum disorder, congenital anomalies, and dysmorphic features [49]. Only 11 individuals with heterozygous loss-of-function variants in the ASH1L have been described in the literature, limiting the understanding of the disorder’s full phenotypic spectrum. Missense variants have also been reported, often associated with more severe phenotypes [38, 49]. In addition to developmental delay/intellectual disability and autism spectrum disorder, some patients also present with seizures, feeding difficulties, hypotonia, musculoskeletal abnormalities, ophthalmological abnormalities, hearing impairments, and gastrointestinal disturbances [38, 49]. Our patient, a female, was born from an uneventful pregnancy and delivery. Developmental delay was recognized early, and she never developed speech or walking abilities. At the age of 3 years, she began experiencing epilepsy with focal-onset seizures of long duration, leading to status epilepticus and postictal hemiparesis. As the disease progressed, she exhibited cognitive and neurological regression, with seizures becoming highly resistant to antiseizure medications and leading to severe life-threatening episodes of super-refractory status epilepticus. Genetic analysis revealed that she is a heterozygous carrier of the VUS, c.6440C>T (p.Ala2147Val), in the ASH1L gene. Her clinical phenotype, which includes developmental delay, absence of speech and walking, and severe seizures, is strikingly similar to those observed in previously reported patients with missense variants in the ASH1L gene as summarized in the publication by Cordova et al. [49]. This case adds to the growing evidence that missense variants in ASH1L contribute to severe neurodevelopmental disorders.
A novel neurodevelopmental disorder was recently described by (and subsequently named after) Poirier et al. [39]. Poirier–Bienvenu neurodevelopmental syndrome is caused by pathogenic variants in the CSNK2B gene, coding for the beta regulatory subunit of the casein kinase 2 enzyme. This enzyme is ubiquitously expressed and constitutively active, with high levels found in the brain, consequently confirming the major role it plays in the neurodevelopmental processes [39]. Furthermore, pathogenic variants in genes coding for other subunits of this enzyme were previously found to cause neurodevelopmental abnormalities [50]. Poirier–Bienvenu neurodevelopmental syndrome is characterized by early-onset seizures, hypotonia, and developmental delay. The severity of the intellectual development impairment seems to vary greatly among cases, where some patients are severely affected, nonverbal, and with seizures refractory to treatment, whereas others attain normal psychomotor development with manageable seizures [51, 52]. Our patient presented with pharmacoresistant epilepsy in the first months of life along with generalized hypotonia. Interestingly, the patient’s father was also diagnosed with early-onset epilepsy, and his seizures are now controlled by lamotrigine. A genetic analysis revealed heterozygosity for a novel stop gained variant, c.35G>A (p.Trp12Ter) in the CSNK2B gene, adding another truncating variant in this gene to the spectrum of Poirier–Bienvenu neurodevelopmental syndrome causes. This variant was also detected in the father, reflecting the variability of the symptoms even inside the family, pointing out to the additional genetic and environmental factors that contribute to the fine tuning of the phenotype development in this disease.
The GRIN1 gene codes for subunit 1 of the N-methyl-d-aspartate receptor, a glutamate-gated calcium ion channel with a crucial role in synaptic plasticity, whereas pathogenic variants in the GRIN1 gene lead to neurodevelopmental disorder with or without hyperkinetic movements and seizures [15]. Our patient presented with generalized epilepsy with onset at 14 months of age. His differential diagnosis was juvenile myoclonic epilepsy, thus a genetic analysis was performed in order to exclude Unverricht–Lundborg disease and Lafora disease, and it revealed heterozygosity for c.734A>T (p.Tyr245Phe), a variant classified as a VUS in ClinVar. While seizures are a common and significant symptom of the condition associated with the GRIN1 gene variants, in some cases, given the variability in the clinical presentation, seizures might be the only symptom that manifests. Particularly, some individuals may only have seizures and no other symptoms such as hyperkinetic movements, which highlights the spectrum of clinical manifestations that can be associated with the disorder.
A recent discovery of a novel type of DEE has shed new light on a gene previously associated with cancer. The RHOBTB2 gene (also known as DBC2), coding for RhoBTB2 (Rho-related BTB domain-containing) protein 2, a small atypical Rho GTPase from the BTB domain-containing protein family, has been reported to cause type 64 DEE, with a distinctive phenotype [41]. This protein plays vital roles in many cellular processes, including cell polarity and migration, as well as regulation of the cell cycle and cytoskeleton dynamics [53]. It has been shown that most of the cases were caused by de novo missense variants affecting the BTB region of the RHOBTB2 protein, which led to a neurodevelopmental phenotype with seizures, severe to profound intellectual disability, and other neurological abnormalities [41, 54]. Later studies added alternating hemiplegia of childhood and other movement disorders as indicative symptoms of RHOBTB2-DEE [55], and recent studies continue to widen the phenotypic spectrum of this extremely rare disorder, as only around 70 patients have been described worldwide to date [56]. Our patient presented with seizures at a later infantile age, along with a developmental delay and aphasia, and she developed alternating childhood hemiplegia a year later. She was found to be a heterozygous carrier of the p.Arg511Gln de novo variant, which was shown to be recurrent and a frequent cause of RHOBTB2-DEE [57]. Her phenotype was in line with previously described cases and she showed good seizure control in response to levetiracetam and carbamazepine.
This study provided the first comprehensive genotype and phenotype characterization of patients with childhood epilepsies from Serbia, thus setting the base for molecular genetic diagnostics and genetic counseling of various epilepsy-related diseases. The limitation of our study is the modest sample size of 55 children with epilepsy, and although CES and WES methods are powerful tools for the analysis, variants in non-coding regions cannot be captured by the sequencing methods employed, potentially leading to false-negative results. While in silico algorithms were used to predict the impact of novel variants, experimental (in vitro) validation of the identified variants is recommended to confirm the pathogenicity of novel variants.
4.3 Implications for Targeted Therapeutic Strategies
Developmental and epileptic encephalopathy was the most frequent disorder in our cohort as it was documented in 14 patients (50%), with disease-causing variants found in the CACNA1A, CACNA1E, CHD2, HNRNPU, KCNQ2, SCN1A, SCN2A, SLC13A5, and RHOBTB2 genes. This outcome aligns with findings from other studies [58], including the fact that DEE was the most common indication for a genetic analysis. Among DEE, Dravet syndrome was observed in five patients, all harboring disease-causing variants in the SCN1A gene.
Pyridoxine-dependent epilepsy was diagnosed in three patients, two of whom had biallelic pathogenic variants identified in the ALDH7A1 gene, and the third patient had pathogenic variants in the PNPO gene. This type of epilepsy is characterized by a very early onset, typically in the neonatal period. For all infants with early-onset drug-resistant epilepsy and status epilepticus unresponsive to first-line medications, therapeutic doses of vitamin B6 (pyridoxine and pyridoxal 5′-phosphate [for PNPO deficiency]) are introduced [59]. All three of our patients presented with seizures under 2 months of age, and their seizure control in response to therapy was sufficient to continue with the high-dose pyridoxine in their daily treatment regimen. However, given the potential risks associated with the prolonged and high-dose administration of pyridoxine, its use would be discontinued if genetic testing did not confirm variants associated with pyridoxine-dependent epilepsy. Because of a timely diagnosis, their treatment continued with high-dose pyridoxine and the introduction of a low-lysine diet. Thus, the outcome of the molecular genetic analysis directly influenced the subsequent treatment of these three children.
In addition to patients with pyridoxine-dependent epilepsy, four more children were found to have an inborn errors of metabolism (ADSL, ALDH7A1, AMT, HSD17B10, MMAA, and PNPO). For inborn errors of metabolism, it has been noted that epilepsies are more likely to present in the neonatal period, with infantile spasms or myoclonic seizures [60], as was the case with our patients (Table 1). Although epilepsy is one of the most common symptoms in inborn errors of metabolism, they are underrepresented within our cohort (25%) as their diagnosis predominantly relies on other symptomatic manifestations. A patient homozygous for the frameshift variant in the MMAA gene presented with symptoms suggestive of metabolic disorder, and therefore the diagnosis of methylmalonic aciduria caused by the deficiency of the cobalamin cofactor rather than the apoenzyme mutase enabled the therapeutic choice of vitamin B12. This variant has already been described in a Serbian population [61]. Glycine encephalopathy resulting from defects in the glycine cleavage system due to AMT gene deficiency is the second most common disorder of amino acid metabolism after phenylketonuria. The more severe neonatal form is characterized by encephalopathy with seizures and often a fatal outcome, while atypical forms have a more heterogeneous disease course [62]. Our patient presented with seizures prenatally as he was homozygous for a truncating variant. HSD10 mitochondrial disease is a rare, it is a recently described X‐linked metabolic disorder caused by pathogenic variants in the HSD17B10 gene. This neurodegenerative disorder exhibits a highly variable onset age (from neonatal to childhood) and severity, with a myriad of multisystem clinical manifestations, reflecting mitochondrial impairment [63]. Our patient had a childhood onset of seizures accompanied by movement disorders, as he was shown to be hemizygous for a novel missense variant in the HSD17B10 gene.
Interestingly, 35.7% of patients from our cohort were found to harbor disease-causing variants in genes encoding ion channels. The majority of the patients had variants in sodium channels (SCN1A and SCN2A), which was expected, given that SCN1A is one of the most common and well researched of the epilepsy genes, with an incidence varying from 1:15,400 to 1:40,900 for Dravet syndrome, the most severe end of SCN1A-related disorders [64]. All of our patients with pathogenic variants in the SCN1A gene presented with a typical form of Dravet syndrome, with infantile onset and several types of seizures (focal, myoclonic, generalized), while three patients also presented with febrile seizures.
As previously mentioned, we identified four novel variants in the SCN1A gene, all of which are truncating mutations (either nonsense or frameshift) that lead to the loss of protein function. The fifth patient carried a missense variant, p.Tyr1770His in the SCN1A gene, which was confirmed to have arisen de novo. This variant was shown to be recurrent and already described in patients with Dravet syndrome [65]. For SCN1A loss-of-function variants, therapeutic intervention should avoid sodium channel blockers, as this approach could aggravate the pathogenic defect even further [7, 60]. Therefore, for our patients with Dravet syndrome, with prolonged unilateral febrile seizures and atypical absences, early genetic diagnosis and early introduction of therapy included stiripentol and fenfluramine, while carbamazepine and phenytoin were avoided. However, patients with gain-of-function variants in the SCN1A gene are associated with early-onset DEEs and have different clinical manifestations in comparison with Dravet syndrome due to SCN1A loss-of-function variants, making them likely to respond to sodium channel-blocking therapies [66]. Thus, for each gene, characterization of a specific variant is essential as a prerequisite for determining appropriate therapeutic options. The same principle is applicable for the SCN2A gene, where gain-of-function versus loss-of-function variants in SCN2A govern the selection of therapy, making sodium channel blockers an eligible therapeutic choice only for some patients [67]. Our patient presented with focal-onset seizures and had a missense variant.
From a wide and heterogeneous spectrum of potassium channels widespread across almost every cell type, voltage-gated potassium channels remain the main focus in epilepsy research, given that pathogenic variants in the KCNQ2 gene stand as one of the most prevalent causes of infantile-onset epilepsy, with an incidence of 1 per 17,000 live births [68]. Most of the disease-causing variants are loss of function, leading to DEE type 7, characterized by early-onset seizures (which respond well to sodium channel blockers) and intellectual disability, whereas only several heterozygous KCNQ2 missense variants increase the channel function, leading to a different phenotype with developmental delay and a characteristic nonepileptic myoclonus. Two of our patients were found to have variants in KCNQ2, of which one had a gain-of-function p.Arg201His with a phenotype corresponding to one previously described in the literature [69], while the other had a loss of function previously associated with early-onset epileptic encephalopathy. In these cases, the genetic diagnosis directed the therapy regimen towards carbamazepine.
In our cohort of patients, two had pathogenic variants in genes coding for calcium channels, CACNA1A and CACNA1E. Similar to other voltage-gated channels, the type of variant denotes the corresponding phenotype as well as therapy choice, where the phenotypic spectrum in CACNA1A-related disorders encompasses movement disorders along with epilepsies [11]. One of our patients presented with childhood onset DEE and was found to have a heterozygous missense variant in CACNA1A. This variant was previously reported in a patient with early-onset encephalopathy with myoclonic epilepsy [70]. The other patient presented with a similar phenotype and was found to have a heterozygous missense variant in CACNA1E.
The identification of a monogenic etiology underlying epilepsy instills optimism that a targeted therapeutic approach that corrects or improves at least some phenotypic features may be applied. A recent review reported current and emerging treatments for 42 forms of monogenic epilepsy [60]. They include inborn errors of metabolism, channelopathies, and diseases caused by variants in the mechanistic target of rapamycin pathway genes [19, 60]. Furthermore, a recent systematic review of genetic testing in epilepsy, encompassing 24 studies with heterogeneous patient cohorts, revealed that once genetic diagnostics were established, they influenced the treatment strategies in 12–80% of the patients [6]. Taken all together, we identified variants in ALDH7A1, KCNQ2, PNPO, SCN1A, and SCN2A gene-directed therapy decisions of 11 children from our study, including four children who carry entirely novel genetic variants.
Although therapeutic options are currently available for a minority of patients, drug discovery for monogenic rare disease is skyrocketing. More than ever before, RNA therapeutics based on antisense oligonucleotides, RNA interference, messenger RNA or gene therapy based on adeno-associated virus vectors, or the CRISPR/Cas gene editing system are being tested in clinical studies [71]. Consequently, every genetic variant should be perceived as a potential therapeutic target, now when the field of hyper-personalized medicine is gaining momentum.
5 Conclusions
Our study emphasizes the importance of NGS in diagnosing childhood epilepsy. With an increasing number of genes associated with epilepsy, a comprehensive analysis using CES and WES is crucial for high rates of diagnostic success. Characterization of novel variants improves genotype-phenotype understanding, and enables personalized therapy. Advancements in drug discovery offer promising targeted treatments, highlighting the potential of each genetic variant as a therapeutic target in personalized medicine. Integrating NGS technology, variant characterization, and personalized therapies will improve epilepsy management.
References
Zhang D, Liu X, Deng X. Genetic basis of pediatric epilepsy syndromes. Exp Ther Med. 2017;13:2129–33.
Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:522–30.
Krey I, Platzer K, Esterhuizen A, Berkovic SF, Helbig I, Hildebrand MS, et al. Current practice in diagnostic genetic testing of the epilepsies. Epileptic Disord. 2022;24:765–86.
Kothur K, Holman K, Farnsworth E, Ho G, Lorentzos M, Troedson C, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure. 2018;59:132–40.
Graifman JL, Lippa NC, Mulhern MS, Bergner AL, Sands TT. Clinical utility of exome sequencing in a pediatric epilepsy cohort. Epilepsia. 2023;64:986–97.
Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, et al. Genetic testing for the epilepsies: a systematic review. Epilepsia. 2022;63:375–87.
Perucca P, Perucca E. Identifying mutations in epilepsy genes: impact on treatment selection. Epilepsy Res. 2019;152:18–30.
Gao C, Pielas M, Jiao F, Mei D, Wang X, Kotulska K, et al. Epilepsy in Dravet syndrome: current and future therapeutic opportunities. J Clin Med. 2023;12:2532.
Ding J, Li X, Tian H, Li W, Wang F, Sun T. SCN1A mutation: beyond Dravet syndrome: a systematic review and narrative synthesis. Front Neurol. 2021;12: 743726.
Mangano GD, Fontana A, Antona V, Salpietro V, Mangano GR, Giuffrè M, et al. Commonalities and distinctions between two neurodevelopmental disorder subtypes associated with SCN2A and SCN8A variants and literature review. Mol Genet Genomic Med. 2022;10: e1911.
Lauerer RJ, Lerche H. Voltage-gated calcium channels in genetic epilepsies. J Neurochem. 2023. https://doi.org/10.1111/jnc.15983.
Wei F, Yan L-M, Su T, He N, Lin Z-J, Wang J, et al. Ion channel genes and epilepsy: functional alteration, pathogenic potential, and mechanism of epilepsy. Neurosci Bull. 2017;33:455–77.
Becchetti A, Aracri P, Amadeo A. The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front Physiol. 2015;6: 125966.
Absalom NL, Lin SXN, Liao VWY, Chua HC, Møller RS, Chebib M, et al. GABAA receptors in epilepsy: elucidating phenotypic divergence through functional analysis of genetic variants. J Neurochem. 2024. https://doi.org/10.1111/jnc.15932.
Amin JB, Moody GR, Wollmuth LP. From bedside-to-bench: what disease-associated variants are teaching us about the NMDA receptor. J Physiol. 2021;599:397–416.
Spoto G, Valentini G, Saia MC, Butera A, Amore G, Salpietro V, et al. Synaptopathies in developmental and epileptic encephalopathies: a focus on pre-synaptic dysfunction. Front Neurol. 2022;13: 826211.
Gautam V, Rawat K, Sandhu A, Kumari P, Singh N, Saha L. An insight into crosstalk among multiple signaling pathways contributing to epileptogenesis. Eur J Pharmacol. 2021;910: 174469.
Rho JM, Boison D. The metabolic basis of epilepsy. Nat Rev Neurol. 2022;18:333–47.
Savino E, Cervigni RI, Povolo M, Stefanetti A, Ferrante D, Valente P, et al. Proline-rich transmembrane protein 2 (PRRT2) regulates the actin cytoskeleton during synaptogenesis. Cell Death Dis. 2020;11:856.
Mastrangelo M, Gasparri V, Bernardi K, Foglietta S, Ramantani G, Pisani F. Epilepsy phenotypes of vitamin B6-dependent diseases: an updated systematic review. Children. 2023;10:553.
Gabriel M, Loos MA, Armeno M, Alonso CN, Roberto H. Glucose transporter type 1 deficiency syndrome: clinical aspects, diagnosis, and treatment. Arch Argent Pediatr. 2023;121: e202202677.
Boyer SW, Barclay LJ, Burrage LC. Inherited metabolic disorders: aspects of chronic nutrition management. Nutr Clin Pract. 2015;30:502–10.
Mei D, Parrini E, Marini C, Guerrini R. The impact of next-generation sequencing on the diagnosis and treatment of epilepsy in paediatric patients. Mol Diagn Ther. 2017;21:357–73.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Oliver KL, Scheffer IE, Bennett MF, Grinton BE, Bahlo M, Berkovic SF. Genes4Epilepsy: an epilepsy gene resource. Epilepsia. 2023;64:1368–75.
Skakic A, Djordjevic M, Sarajlija A, Klaassen K, Tosic N, Kecman B, et al. Genetic characterization of GSD I in Serbian population revealed unexpectedly high incidence of GSD Ib and 3 novel SLC37A4 variants. Clin Genet. 2018;93:350–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C, et al. ACGS best practice guidelines for variant classification in rare disease. 2020. Available from: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classificationv4-01-2020.pdf. [Accessed 6 Apr 2024].
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–94.
Li C, Zhi D, Wang K, Liu X. MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Med. 2022;14:115.
Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020;11:5918.
Phyre2. Protein Homology/analogY Recognition Engine V 2.0. Available from: http://www.sbg.bio.ic.ac.uk/phyre2. [Accessed 6 Apr 2024].
EzMol. Available from: http://www.sbg.bio.ic.ac.uk/ezmol. [Accessed 6 Apr 2024].
Aminode. Available from: http://www.aminode.org. [Accessed 6 Apr 2024].
Liu H, Liu D-T, Lan S, Yang Y, Huang J, Huang J, et al. ASH1L mutation caused seizures and intellectual disability in twin sisters. J Clin Neurosci. 2021;91:69–74.
Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat. 2017;38(8):932–41.
Thévenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Jugé C, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. 2014;95:113–20.
Straub J, Konrad EDH, Grüner J, Toutain A, Bok LA, Cho MT, et al. Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in Drosophila. Am J Hum Genet. 2018;102:44–57.
Rochtus A, Olson HE, Smith L, Keith LG, El Achkar C, Taylor A, et al. Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia. 2020;61:249–58.
Riza AL, Streață I, Roza E, Budișteanu M, Iliescu C, Burloiu C, et al. Phenotypic and genotypic spectrum of early-onset developmental and epileptic encephalopathies: data from a Romanian cohort. Genes (Basel). 2022;13(7):1253.
Epilepsy Genetics Initiative. The Epilepsy Genetics Initiative: systematic reanalysis of diagnostic exomes increases yield. Epilepsia. 2019;60(5):797–806.
Orphanet. Knowledge on rare diseases and orphan drugs. Available from: https://www.orpha.net/en/disease/detail/46. [Accessed 6 Apr 2024].
ClinVar. ClinVar National Library of Medicine. Available from: https://www.ncbi.nlm.nih.gov/clinvar. [Accessed 6 Apr 2024].
Mastrogiorgio G, Macchiaiolo M, Buonuomo PS, Bellacchio E, Bordi M, Vecchio D, et al. Clinical and molecular characterization of patients with adenylosuccinate lyase deficiency. Orphanet J Rare Dis. 2021;16:1–10.
Gregory GD, Vakoc CR, Rozovskaia T, Zheng X, Patel S, Nakamura T, et al. Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol. 2007;27(24):8466–79.
Cordova I, Blesson A, Savatt JM, Sveden A, Mahida S, Hazlett H, et al. Expansion of the genotypic and phenotypic spectrum of ASH1L-related syndromic neurodevelopmental disorder. Genes (Basel). 2024;15(4):423.
Okur V, Cho MT, Henderson L, Retterer K, Schneider M, Sattler S, et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum Genet. 2016;135(7):699–705.
Li J, Gao K, Cai S, Liu Y, Wang Y, Huang S, et al. Germline de novo variants in CSNK2B in Chinese patients with epilepsy. Sci Rep. 2019;9(1):17909.
Nakashima M, Tohyama J, Nakagawa E, Watanabe Y, Siew CG, Kwong CS, et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet. 2019;64(4):313–22.
Siripurapu V, Meth J, Kobayashi N, Hamaguchi M. DBC2 significantly influences cell-cycle, apoptosis, cytoskeleton and membrane-trafficking pathways. J Mol Biol. 2005;346:83–9.
Belal H, Nakashima M, Matsumoto H, Yokochi K, Taniguchi-Ikeda M, Aoto K, et al. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat. 2018;39:1070–5.
Zagaglia S, Steel D, Krithika S, Hernandez-Hernandez L, Custodio HM, Gorman KM, et al. RHOBTB2 mutations expand the phenotypic spectrum of alternating hemiplegia of childhood. Neurology. 2021;96:e1539–50.
Langhammer F, Maroofian R, Badar R, Gregor A, Rochman M, Ratliff JB, et al. Genotype-phenotype correlations in RHOBTB2-associated neurodevelopmental disorders. Genet Med. 2023;25: 100885.
Fonseca J, Melo C, Ferreira C, Sampaio M, Sousa R, Leão M. RHOBTB2 p. Arg511Trp mutation in early infantile epileptic encephalopathy-64: review and case report. J Pediatr Genet. 2023;12:155–8.
Myers KA, Scheffer IE. Precision medicine approaches for infantile-onset developmental and epileptic encephalopathies. Annu Rev Pharmacol Toxicol. 2022;62:641–62.
Coughlin CR, Gospe SM Jr. Pyridoxine-dependent epilepsy: current perspectives and questions for future research. Ann Child Neurol Soc. 2023;1:24–37.
Balestrini S, Mei D, Sisodiya SM, Guerrini R. Steps to improve precision medicine in epilepsy. Mol Diagn Ther. 2023;27:661–72.
Stojiljkovic M, Klaassen K, Djordjevic M, Sarajlija A, Brasil S, Kecman B, et al. Molecular and phenotypic characteristics of seven novel mutations causing branched-chain organic acidurias. Clin Genet. 2016;90:252–7.
Poothrikovil RP, Al Thihli K, Al Futaisi A, Al MF. Nonketotic hyperglycinemia: two case reports and review. Neurodiagn J. 2019;59:142–51.
Falk MJ, Gai X, Shigematsu M, Vilardo E, Takase R, McCormick E, et al. A novel HSD17B10 mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol. 2016;13:477–85.
Strzelczyk A, Lagae L, Wilmshurst JM, Brunklaus A, Striano P, Rosenow F, et al. Dravet syndrome: a systematic literature review of the illness burden. Epilepsia Open. 2023;8:1256–70.
Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46:183–91.
Brunklaus A, Brünger T, Feng T, Fons C, Lehikoinen A, Panagiotakaki E, et al. The gain of function SCN1A disorder spectrum: novel epilepsy phenotypes and therapeutic implications. Brain. 2022;145:3816–31.
Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–36.
Grayson C, Harden C, Butterfield N, Aycardi E. Evaluating the epidemiological burden of KCNQ2 epilepsy. Neurology. 2021. https://doi.org/10.1212/WNL.96.15_supplement.5169.
Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grønborg S, Jiang Y, et al. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ 2 gain-of-function variants R201C and R201H. Epilepsia. 2017;58:436–45.
Hayashida T, Saito Y, Ishii A, Yamada H, Itakura A, Minato T, et al. CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. 2018;40:130–3.
Arabi F, Mansouri V, Ahmadbeigi N. Gene therapy clinical trials, where do we go? An overview. Biomed Pharmacother. 2022;153: 113324.
Acknowledgements
Genetic analyses and interpretations were performed by the Rare Disease Research and Therapeutics Development research group as part of activities of the Center for Genetic Diagnosis of Rare Diseases. The authors express their gratitude to Sara Stankovic and Djordje Pavlovic for their invaluable assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Ministry of Science, Technological Development and Innovations Republic of Serbia (No.: 451-03-66/2024-03/200042).
Conflicts of Interest/Competing Interests
Marina Andjelkovic, Kristel Klaassen, Anita Skakic, Irena Marjanovic, Ruzica Kravljanac, Maja Djordjevic, Biljana Vucetic Tadic, Bozica Kecman, Sonja Pavlovic, and Maja Stojiljkovic have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
This study has been approved by the Ethics Committee of the Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic” in Belgrade, Serbia (No. 31/2020 dated 25 September, 2020.) and the Research Ethics Committee of the Institute of Molecular Genetics and Genetic Engineering, University of Belgrade (O-EO-017/1/2020 dated 3/9/2020) and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
Not applicable.
Availability of Data and Material
Raw genetic data are generated and stored at the Institute of Molecular Genetics and Genetic Engineering, University of Belgrade, and are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Authors’ Contributions
MA, KK, AS, and IM performed the genetic analysis and data interpretation, MA and KK wrote the paper, RK, MD, BVT, and BK collected the patients’ phenotype data and reviewed the paper, SP provided funding, and MS conceived and designed the study and wrote the paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Andjelkovic, M., Klaassen, K., Skakic, A. et al. Characterization of 13 Novel Genetic Variants in Genes Associated with Epilepsy: Implications for Targeted Therapeutic Strategies. Mol Diagn Ther (2024). https://doi.org/10.1007/s40291-024-00720-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s40291-024-00720-2