
Abstract
Immunotherapy for colorectal cancer (CRC) is limited to patients with advanced disease who have already undergone first-line chemotherapy and whose tumors exhibit microsatellite instability. Novel technical strategies are required to enhance therapeutic options and achieve a more robust immunological response. Therefore, exploring gene analysis and manipulation at the molecular level can further accelerate the development of advanced technologies to address these challenges. The emergence of advanced genome editing technology, particularly of clustered, regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein (Cas) 9, holds promise in expanding the boundaries of cancer immunotherapy. In this manuscript, we provide a comprehensive review of the applications and perspectives of CRISPR technology in improving the design, generation, and efficiency of current immunotherapies, focusing on solid tumors such as colorectal cancer, where these approaches have not been as successful as in hematological conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Immunotherapy for colorectal cancer (CRC) is currently limited to patients with advanced disease who have undergone first-line chemotherapy and have tumors displaying microsatellite instability. |
Novel technical strategies are needed to enhance therapeutic options and achieve a stronger immunological response in CRC treatment. |
CRISPR can be utilized to genetically modify immune cells, such as chimeric antigen receptor (CAR)-T cells, tumor infiltrating lymphocytes (TILs), and natural killer (NK) cells, enhancing their capacity to recognize and attack cancer cells while maintaining activity within the immunosuppressive tumor microenvironment. |
1 Introduction
Colorectal cancer (CRC) is one of the most frequently diagnosed tumors and a significant cause of death among males and females. In 2021, approximately 140,500 new cases of CRC were diagnosed in the USA, with an estimated 52,980 deaths [1]. CRC is characterized by the accumulation of specific mutations in a single cell, providing tumor cells with advantages in proliferation and metastasis [2]. This sequential accumulation of oncogenic mutations follows the adenoma–carcinoma sequence [3]. While treatment options such as surgery, chemotherapy, and radiation exist for CRC, most cases are diagnosed at later stages, resulting in lower survival rates [4].
There is compelling evidence that acquired mutations allow tumor cells to evade the immune system and adapt to their environment, promoting their survival and preventing elimination [5]. As CRC tumors develop, tissue damage can activate the immune system, releasing chemokines and cytokines that attract various immune cells. These cells can either impact tumor development or have no effect [4]. Furthermore, CRC tumors express aberrant proteins with distinct amino acid sequences, influencing the expression of other proteins in critical signaling pathways and serving as tumor-associated antigens presented to T cells by the major histocompatibility complex (MHC). Cellular and antibody-mediated immune responses against these antigens have been observed in CRC patients [6]. These interactions between the tumor and the immune system suggest that immunotherapy holds promise in delaying or eradicating cancer progression.
Immunotherapy faces challenges in solid tumors due to difficulties in immune cells effectively infiltrating the tumor core. Currently, immunotherapy for CRC is limited to patients with advanced CRC who have undergone first-line chemotherapy and whose tumors have demonstrated microsatellite instability [7]. Novel technical strategies are required to enhance therapeutic options and achieve a more robust immunological response. The discovery and application of clustered, regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas)-based technologies have revolutionized cancer research. CRISPR–Cas is currently employed as a complementary tool in the experimental design of more efficient immunotherapies. This review aims to provide an up-to-date analysis of the role of immunotherapy in CRC and a comprehensive overview of the current understanding and potential applications of the CRISPR–Cas9 method to enhance immunotherapy in CRC.
2 Immunogenicity of Colorectal Cancer
CRC develops through distinct stages, starting from colonic crypt lesions, and progressing to adenomas and ultimately cancer. As colorectal cancer progresses from adenomas to carcinomas, there are shifts in the participation of immune cells. Genetic and epigenetic alterations in genes such as adenomatous polyposis coli, Kirsten rat sarcoma viral oncogene homolog (KRAS), phosphoinositide 3-kinase (PI3K), transforming growth factor β (TGF β), the β-catenin pathway, and others, coupled with interactions with stromal and immune cells, create a unique tumor microenvironment (TME) [8].
During the initial stages of carcinogenesis, nascent tumor cells produce antigens that trigger the recruitment of innate immune cells, such as dendritic cells (DCs), neutrophils, natural killer (NK) cells, and macrophages. These cells secrete cytokines and chemokines, facilitating the recruitment of T and B lymphocytes. Colorectal tumors exhibit inflammatory infiltration and increased expression of proinflammatory cytokines such as tumor necrosis factor alpha (TNFα) and soluble mediators such as prostaglandin E2 (PGE2). These molecules are overexpressed during inflammation and can activate signaling pathways such as ERK, nuclear factor (NF)-κB, signal transducer and activator of transcription 3 (STAT3), and PI3K-AKT in epithelial cells, thereby augmenting β-catenin signaling [4, 9].
During tumor initiation, genotoxic compounds produced by inflammatory cells can induce DNA damage in epithelial cells, contributing to increased mutagenesis required for tumor initiation. Therefore, inflammatory conditions in colon epithelium advantage tumor cells, and it is a key point in tumor progression.
2.1 Antigen Presenting Cells and Tumor-Infiltrating Lymphocytes
Antigen-specific activation of T cells is initiated through the interaction between the T cell receptor (TCR) and tumor antigens presented by antigen presenting cells (APCs) on major histocompatibility complex molecules. The optimal signaling for T cell activation is regulated by inhibitory and co-stimulatory molecules that modulate signal amplification. DCs play a critical role as APCs in recognizing tumor cells, processing neoantigens, and presenting them to T cells, thereby eliciting a reactive response against tumor cells and promoting their elimination. However, in advanced tumors, antigen presentation and T cell activation are inhibited or are inefficient [9].
Late-stage CRC tumors employ various escape mechanisms to evade T cell-mediated responses, including altered expression of human leukocyte antigen class I (HLA-I). Downregulation of the phosphatidylserine receptor TIM4 in DCs by tumors inhibits the phagocytosis of tumor antigens, impairing the activation of tumor-specific CD8+ T cells [10, 11]. Mice with reduced expression of HLA-I molecules exhibit heightened tumor development and increased expression of immunoinhibitory molecules such as PD-L1 [12, 13]. In the context of liver metastasis, enhanced infiltration of DCs into tumors sensitized CRC tumors to immune checkpoint blockade therapy, leading to improved survival outcomes [14]. These studies suggest that optimal HLA expression and functioning of DCs can enhance the efficacy of immunotherapies.
CD8 T cells play a crucial role in the specific destruction of tumor cells by releasing their lytic components [15]. Primary tumors exhibit dense infiltration of tumor-infiltrating lymphocytes (TILs) [16], and a high density of CD8 T cell infiltration is associated with improved disease-free and overall survival, as well as a favorable response to immunotherapy [17]. Furthermore, patients with high densities of memory CD45RO and CD8 T cells experience better outcomes [17]. The expression of specific chemokines and adhesion molecules is critical for achieving high densities of CD8 T cells within the tumor. Tumors with elevated expression levels of CX3CL1, CXCL9, and CXCL10 demonstrate significantly higher densities of CD8 T cells, and the chemokine stromal cell-derived factor 1 (SDF-1) serves as a favorable prognostic marker for overall survival in CRC due to its lymphocyte chemotactic activity [18]. However, tumor cells often evade the intrinsic anti-tumor activity by downregulating T cell activation and recruitment. The migration of T cells toward the tumor is limited due to reduced expression of chemoattractant chemokines such as CXCL10 and CXCL9 [19]. Oncoproteins such as Tribbles homolog 3 (TRIB3) have been identified as inhibitors of CD8+ T cell infiltration in CRC [20]. The ectopic expression of the oncoprotein TRIB3 inhibits the STAT1-CXCL10 signaling axis, resulting in reduced tumor-infiltrating T cells in various CRC mouse models [20]. Genetic ablation of Trib3 or pharmacological acceleration of TRIB3 degradation increases T cell recruitment and sensitizes CRCs to immune checkpoint blockade therapy [20].
2.2 Regulatory T Cell and Tumor-Associated Macrophages
Within the TME, the interaction among tumor cells, nonmalignant stromal cells, and immune cells creates an immunosuppressive environment that recruits myeloid cells and regulatory T cells (Tregs). While these cells are essential for preventing excessive immune responses, they also have pro-tumorigenic roles.
TILs consist of various cell types, predominantly CD8+ and CD4+ T cells, as well as Tregs. The balance between these populations is crucial for anti-tumor immunity. In CRC, increased infiltration of Tregs has been associated with both improved and worsened prognosis and overall survival, indicating their complex role [21]. Further research has identified heterogeneous subpopulations of Tregs in CRC patients, including suppressive and non-suppressive Foxp3+ cells, which have implications for prognosis [22]. Therefore, the infiltration of Tregs may vary during the progression from adenoma to carcinoma.
In colitis-associated colorectal cancer (CAC), inflammation plays a driving role in neoplasm development, making Tregs crucial in the early stages to limit immune infiltration. Several studies have highlighted the therapeutic potential of Tregs in this context [23,24,25]. In a study by Delgado-Ramírez et al. [26], it was found that Tregs could help prevent inflammation and tumor progression. The study demonstrated that STAT6-deficient (STAT6−/−) mice exhibited less tumor progression during the early stages of the disease due to increased Tregs infiltration, which helped limit inflammation. Thus, STAT6 deficiency favored Tregs function and limited CAC in this model compared with wild-type mice. In another study, Tianzhen He et al. [27] used two-pore channel (TPC) inhibitors to enhance transmembrane TNF expression, inducing proliferative expansion of Tregs. In a colitis mouse model, the use of this inhibitor increased the number of Tregs and consequently attenuated colon inflammation. The role of Tregs in the context of CRC is bimodal and particularly crucial in the first steps of tumorigenesis; therefore, studying the role of Tregs in CRC is essential to developing new therapeutics.
Tumor-associated macrophages (TAMs) are highly prevalent in the TME. TAMs secrete chemokines and cytokines that promote tumor development, immunosuppression, angiogenesis, metastasis, and resistance to therapy [28]. TAMs produce arginase I, which alters T cell function [29], and recruit Tregs by secreting the chemokine CCL2 [30]. Additionally, TAMs promote angiogenesis and metastasis by increasing vascular endothelial growth factor (VEGF) expression [31]. Junhui Yu et al. [32] found that TAMs produce matrix metalloproteinases 1 (MMP1), which significantly facilitate colon cancer cell proliferation by accelerating the cell cycle transition. Consequently, the increased presence of TAMs in the TME is associated with a poor prognosis [33].
In solid tumors, the extracellular matrix (ECM) reduces the efficacy of treatments. This occurs through the formation of a drug diffusion barrier and limited nutrient and oxygen influx, which promote stress- and hypoxia-induced pathways, thereby reducing the efficacy of radiotherapy and stimulating drug-resistance pathways. Additionally, in the case of cellular immunotherapy, the tumor ECM forms a barrier that chemoattracts tumor-reactive cells and traps them, making them unable to reach the tumor [34]. Furthermore, the TME supports inappropriate metabolic reprogramming, which dampens T cell function and impacts the antitumor immune response and tumor progression. Due to the high nutrient consumption by tumor cells, immune cells that manage to infiltrate the tumor do not obtain the necessary nutrients to carry out their cytotoxic activities [35].
2.3 Natural Killer Cells
Natural killer (NK) cells, an integral part of the innate immune system, play a vital role in promoting cytotoxicity against tumor cells or infected cells, regardless of MHC recognition. The activity of NK cells is regulated by both activator and inhibitory receptors. Activating receptors, such as natural killer group 2 member D (NKG2D) (NKG2D), DNM-1, natural cytotoxicity receptors (NCRs), and the type 2 receptor (KIR) family, are responsible for triggering NK cell responses [36]. Upon activation, NK cells release inflammatory cytokines and lytic granules, leading to the destruction of tumor cells. In the context of CRC, circulating NK cells serve as a prognostic marker. Immune cell therapy for CRC aims to enhance tumor cell recognition by NK cells [37]. Improvements in genetics and cancer immunology have led to a better understanding of the drivers of immunogenicity in cancer and potential mechanisms for treatment, especially in cancers that do not respond to current therapies. Therefore, the role of immunotherapy in treating CRC is becoming more prominent and needs the development of new adjuvants and tools.
3 Current Immunotherapy Approaches in Colorectal Cancer
Chimeric antigen receptor (CAR)-T cells consist of genetically modified receptors that recognize surface antigens expressed for tumor cells; this recognition is independent of the MHC receptor, and consequently T cells have a powerful anti-tumoral activity [38].
CARs have four structure components that confer different functions of recognition and specificity to T cells. These components are: (1) the antigen binding domain, which confers recognition specificity for the target antigen; (2) the hinge region, an extracellular structural region that extends the binding units from the transmembrane domain and provides flexibility and contribute to the length of the binding domain; (3) the transmembrane domain, which anchors the CARs to the T cell membrane and, in addition, influences the stability and expression level of CARs, can be active in signaling or synapse formation, and dimerizes with endogenous signaling molecules (most transmembrane domains derive from natural proteins such as CD3ζ, CD4, CD8α, or CD28 [38, 39]); and (4) intracellular signaling domain(s)—the durability and persistence of CAR-T cells depends on their co-antitumor stimulatory domains; for this reason, it is necessary to associate intracellular signaling domains to the transmembrane domain, allowing for their co-stimulation. CD28 and 4-1BB (CD137) are the most used co-stimulatory domains due to their high patient response rate [40, 41].
CAR-T cells are used for the treatment of hematologic cancers, such as B-cell lymphomas or non-Hodgkin lymphoma [39,40,41]. However, this therapy is still limited in solid tumors due to the immunosuppressive microenvironment that can limit proliferation and persistence in tumor sites [42]. Solid tumors present unique challenges for CAR-T cell therapy, requiring the T cells to navigate through stromal barriers and reach the tumor site to exert their tumor-specific cytotoxic effects. However, even if successful infiltration occurs, T cells can face dysfunction within the toxic TME. This hostile environment is characterized by metabolic disruptions, the presence of soluble inhibitory factors and cytokines, increased numbers of suppressive immune cells, and tumor cells that secrete these mediators and express high levels of ligands for negative immune checkpoint receptors such as PD-L1.
The use of CAR-T cell therapy in human patients has been limited in terms of both efficacy and safety. Adverse effects such as cytokine release syndrome (CRS) and the ineffectiveness of CAR-T cells upon reaching the tumor site have been observed. As a result, preclinical trials have focused on improving the signaling of CAR receptors, preventing the inactivation of CAR-T cells, and evaluating cytotoxicity through in vitro and in vivo studies. To improve the efficacy of CAR-T cells, the addition of the co-stimulatory molecule CD27 improved CAR-T cell proliferation and anti-tumor activity in a xenografts model of CRC [43]. CRC antigen GUCY2C expressed in CAR-T cells promoted antigen-dependent T-cell activation. GUCY2C-CAR-T cells are effective against metastatic tumors in mouse CRC models, as well as xenograft models of human CRC [44]. Other target antigens have been explored; CD6 receptors CD166 and CD318 are highly expressed in CRC. CD166-CAR-T cells have cytotoxic effect in target-positive human CRC cell lines and cancer stem cells [45], highlighting the need for identification and characterization of novel cancer-related ligand receptors for CAR design and evaluation.
Another challenge arises from the absence of appropriate target antigens for CAR-T cells in solid tumors. Numerous research teams have concentrated on pre-clinical investigations centered on CAR-T cell biology. Their goal is to create secure therapeutic approaches and confirm their applicability in treating CRC. Table 1 provides a summary of the distinct antigens that CAR-T cell therapies in CRC are aimed at, as well as those identified in preclinical research. Nevertheless, none of the studies have yielded conclusive results to date.
For instance, new strategies are necessary for improving CAR-T cell function in solid tumors by prolonging their persistence, trafficking, tumor infiltration, and tumor elimination. CRISPR–Cas9 systems are emerging genetic editors that enable the introduction of desired genetic modifications into the genome, with or without the need for creating double-stranded breaks (DSBs). These techniques and approaches can target genes that inhibit T-cell function, precisely insert therapeutic genes into specific genomic positions, and produce consistently secure and effective allogeneic universal CAR-T cell products for customized cancer immunotherapy.
CRC can evade detection by the immune system by controlling certain aspects of the immune response through immune checkpoints (ICP). These control points can either stimulate or inhibit immune cell activity, which contributes to self-tolerance or immune reactivity. When T cells are activated, they express a protein called PD-1, which can bind to its ligand PDL-1/PDL-2. Tumor cells express these ligands, which can inhibit tumor cell apoptosis, leading to the exhaustion of peripheral T effector cells, and convert T effector cells into Treg cells. This process allows the tumor cells to evade the normal immune system. [60]. The CTLA-4 (CD 152) molecule belongs to the B7/CD28 family and plays a role in immune suppression by indirectly inhibiting signals through CD28. CD28 is a co-stimulatory receptor expressed on the surface of T cells. When CD28 interacts with CD80/CD86, T cells produce IL-12 and cytotoxic enzymes, which are essential for immune function. However, CTLA-4 has a higher affinity for CD80/CD86 and can outcompete CD28, leading to a decrease in the effector activity of T cells. Tregs cells recruited by the tumor also contribute to the inhibition of cytotoxic T cells by blocking their immune checkpoints, which results in the T cells’ exhaustion [60, 61].
Cancer cells depend on immune checkpoints to evade the immune response and persist. Targeted therapies such as immune checkpoint inhibitors (ICIs) have been developed to inhibit these pathways. The first immune checkpoint antibody to be approved by the Food and Drug Administration (FDA) was anti-CTLA-4 ipilimumab [62]. Later, anti-PD-1 inhibitors such as nivolumab, pembrolizumab, and cemiplimab, and anti-PD-L1 inhibitors such as atezolizumab, avelumab, and durvalumab were developed [60, 63,64,65]. Currently, new immune checkpoints are being investigated as possible therapeutic targets, including LAG-3, TIM-3, A2aR, CD73, NKG2A, and PVRIG/PVRL2 [60, 61].
There is a wide interest in combining CAR-T cell adoptive transfer with checkpoint blockade alternatives, such as the administration of monoclonal antibodies against PD-1 and CTLA-4, especially in solid tumors, where CAR-T cells have not been as effective as in the treatment of hematological malignancies. Particularly, the lack of neoantigens, a deficient cell infiltration and the immunosuppressive microenvironment are obstacles to overcome in CRC [66]. It has been shown that PD-1 blockade can rescue CAR-T cells from exhaustion [67, 68]. However, the success of this approach will be influenced by the Treg/Teff balance, as it has been shown that administration of PD-1 antibodies also stimulate suppression by PD1+ Tregs counteracting CD8+ cytotoxic T-cells in a murine colon cancer model [69, 70].
CRC is divided into two groups on the basis of the mutation pattern: deficient MMR/microsatellite instability-high (dMMR-MSI-H) and mismatch repair-proficient/microsatellite stable tumors (pMMR-MSI-L) [62]. Many clinical trials have confirmed that ICIs, particularly PD-1/PD-L1 inhibitors, may be more beneficial for treating dMMR-MSI-H CRC. Consequently, the FDA has authorized immune checkpoint therapy as a treatment for dMMR-MSI-H CRC mainly in advanced stages to improve patient survival.
Numerous studies have indicated the potential of ICIs in treating patients with dMMR/MSI-H [71]. In particular, a meta-analysis of 14 studies comprising 1129 subjects examining the use of PD-1/PD-L1 inhibitors in patients with metastatic colorectal cancer (mCRC) showed encouraging clinical benefits, including improved progression-free survival [72]. This may be due to the fact that MSI tumors are more immunogenic, characterized by a high infiltration of cytotoxic T lymphocytes and T helper 1 (Th1) cells, which produce interferon (IFN)-γ. Given that these tumors express high levels of PD-1/PD-L1 and/or CD80/CTLA-4, blocking these immune checkpoints using ICIs can elicit a robust immune response [73].
Despite the potential of ICIs in treating CRC, some patients may not respond well to the treatment, and others may develop resistance to it. Additionally, ICIs have been shown to cause inflammatory side effects in CRC treatment, and they do not significantly improve overall survival rates. Furthermore, there are still patients who do not benefit from targeted PD-1/PD-L1 antibodies [74]. Fortunately, the CRISPR–Cas9 system offers a potent and efficient method for editing PD-1 genes in various immune cells to turn off immune checkpoints.
4 CRISPR–Cas System as a Gene Editing Tool and Expanded Alternatives
CRISPR–Cas based technologies have had a profound impact on genome editing in eukaryotic organisms due to their simplicity and programmability [75]. CRISPR is the only known adaptive immune system that prokaryotes have developed against foreign genetic elements such as viruses and noxious plasmids and consists of basically two elements:
-
Effector protein(s): These are known as CRISPR-associated (Cas) nucleases and are activated only upon binding to a guide RNA and hybridization to the target DNA. A specific protospacer adjacent motif (PAM), which is a sequence located 3′ to the guide RNA-binding site, is also required for Cas nucleases to bind and cut the target DNA at a specific position, usually at two base pairs (bp) upstream the PAM, within the guide RNA binding site. Each Cas protein has a specific PAM requirement. So far, six types of CRISPR systems have been discovered depending on their architecture and number of effector proteins. Type II systems, such as CRISPR–Cas9, are the most widely used as gene editors, as they consist of a single nuclease effector, facilitating the experimental setup.
-
Guide RNA: It is a duplex RNA molecule that combines a CRISPR RNA (crRNA ) or spacer, which is a short sequence complementary to the target DNA, and a trans-activating CRISPR RNA (tracrRNA), which is responsible to transactivate the nuclease domains of the Cas protein. Currently, both short molecules are combined into a single molecule known as single guide RNA (sgRNA) without losing any activity for genome editing.
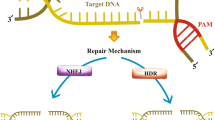
In the case of CRISPR–Cas9 [from Streptococcus pyogenes (SpCas9)], to program the system, only a short 21-necleotide (nt) guide RNA and the presence of a PAM sequence (NGG for SpCas9, occurring once every 42 bp in the human genome adjacent to its 3′ end are necessary [75]. Genome editing is a powerful tool for investigating the genetic foundations of physiological and metabolic processes in any organism. It is important to note, that the CRISPR–Cas9 system generates double-strand breaks at the target gene, and only when the DNA repair mechanisms are recruited, random insertions or deletions (indels) leading to gene inactivation are introduced due to the error-prone nature of the non-homologous end-joining (NHEJ) pathway.
CRISPR–Cas-based technologies for genome and epigenome editing are shown in Fig. 1. The CRISPR–Cas9 gene editing mechanism enables the introduction of specific mutations by incorporating a DNA repair template to leverage the homologous-directed repair (HDR) pathway, which may vary in efficiency (Fig. 1A). CRISPR-associated Prevotella and Francisella 1 (Cpf1) is an endonuclease of the class 2 CRISPR family. Cpf1 (also known as Cas12a) serves a dual role as a nuclease, operating both as an endoribonuclease to process crRNA and as an endodeoxyribonuclease for cleaving target sequences and creating double-stranded breaks with sticky ends, facilitating HDR (Fig. 1B). CRISPR–Cas9-based genome editing allows the introduction of point mutations in the DNA without generating double-strand breaks (DSBs), e.g., cytidine (C→T) and adenine (A→G) deaminases for precise single-base mutations (Fig. 1C) [76]. More complex edition can also be accomplished via prime editing, where a Cas9 nickase is combined with a specialized guide RNA that contains the desired edit in its sequence, which will be inserted at the target gene location by a coupled reverse transcriptase (Fig. 1D). Epigenome editing by associating dead Cas9 (dCas9; a mutated version without nuclease activity, but with intact DNA recognition and binding ability) with DNA methlyltransferase [e.g., DNA methyltransferase 3a (DNMT3)] and demethylase domains [e.g., Tet methylcytosine dioxygenase 1 (TET1)] has been used recently in lymphocytes and other cells (Fig. 1E) [77,78,79]. Cas13, an alternative RNA-targeting CRISPR-associated nuclease, has been harnessed to liberate a fluorophore from an RNA-bound quencher upon activation triggered by the specific binding of crRNA to tumor-associated microRNAs. Consequently, it proves to be a valuable diagnostic tool (Fig. 1F) [80]. These techniques and approaches can be readily employed to focus on inhibitory controllers of T-cell activity, guide therapeutic genetic material to precise genomic positions, and produce consistently secure and effective allogeneic universal CAR T-cell treatments for customized cancer immunotherapy as needed.

CRISPR–Cas based technologies for genome and epigenome editing. A Genome editors: “Classic” Cas9/sgRNA gene knock-outs are initiated by a double-strand break at the binding region of the sgRNA. B Cpf1 is a double-strand nickase, making intercalated DNA breaks, which can be used for more efficient gene knock-ins through homologous DNA repair. C Base editors combine a Cas9 nickase with a cytosine or adenine deaminase to catalyze C to T or A to G conversions respectively, at the opposing strand to the sgRNA binding region. D In prime-editing, nCas9 is combined with a reverse transcriptase that uses a derived single guide RNA (sgRNA) [prime editing guide RNA (pegRNA)] encoding a desired edit to directly perform genome editing at the target DNA site. E Epigenome editors: DNA sequence is not altered by these tools; instead, gene transcription is modulated. dCas9 can be combined with Tet1 or DNA methyltransferase 3a (DNMT3a) to induce DNA demethylation or methylation, leading to the activation or inhibition of gene transcription, respectively. F Molecular diagnostic tools: Cas13 exhibit sequence-independent RNA degradation upon activation with crRNA-specific RNA binding. This activity can be harnessed to split and activate a fluorochrome-RNA-quencher fusion, to detect tumor-secreted miRNAs. Cas9 CRISPR-associated protein 9, dCas9 “dead” Cas9, nCas9 Cas9 nickase, Cas12a CRISPR-associated protein 12a, Cas13 CRISPR-associated protein 13, Cpf1 CRISPR-associated Prevotella and Francisella 1, crRNA CRISPR RNA, DNMT3a DNA methyltransferase 3a, miRNA microRNA, pegRNA prime editing guide RNA, sgRNA single guide RNA, TET1 Tet methylcytosine dioxygenase 1
5 CRISPR–Cas system and Its Applications in Colorectal Cancer Research
CRISPR–Cas9 has emerged as a powerful tool for elucidating the exact functions of mutations that underlie the development of CRC. It is employed to investigate the natural course of CRC progression and to elucidate the sequence of mutations that contribute to tumorigenesis. This versatile system facilitates efficient genome editing, enabling the simultaneous insertion and deletion of multiple genes. Through genome-wide CRISPR–Cas9 knockout screens, certain gene knockouts were found to have either favorable or unfavorable effects when KRAS activation was present, thus revealing particular metabolic vulnerabilities. These findings underscore the potential of targeting cancer metabolism in human mutant KRAS tumors for therapeutic purposes [81, 82]. When the mutation in the β-catenin pathway was corrected using CRISPR–Cas9 in HCT-116 cell line human colon carcinoma, it resulted in the recovery of Wnt phosphorylation, a decrease in β-catenin nuclear translocation, and survivin and c-myc expression. This led to a decrease in cell growth and impaired tumor formation in mouse xenografts [83]. Drost et al. [84] employ CRISPR–Cas9 technology to precisely modify four of the frequently mutated genes associated with CRC (APC, P53 or TP53, KRAS, and SMAD4) within cultured human intestinal stem cells. After systematically introducing mutations for APC, P53, KRAS, and eventually SMAD4 through specific gRNAs, these cells were transplanted into recipient mice, and they were effectively transformed into tumors exhibiting histology resembling adenocarcinoma leading to the identification of essential driver mutations [84]. Similar works have since confirmed the validity of CRISPR–Cas9 technology for in vivo genome editing and organoid transplantation of colon tumors in mice without predisposing mutations [82]. In addition, CRISPR–Cas9 technology has been fruitful for detecting other driver mutations in genes such as Acvr1b, Acvr2a, and Arid2 [85], and the MUC5AC-CD44 axis [86].
Current cancer treatments have not effectively addressed the inadequate therapeutic outcomes, and while immunotherapy has shown promise, the adverse effects limit its clinical implementation. Therefore, there is a need to explore molecular-level gene analysis and manipulation to enhance the advancement of cutting-edge technologies and overcome these limitations. CRISPR–Cas9 can enhance current immunotherapies, such as CAR-T, CAR-NK, and TCR-T cell therapies.
6 The Application of the CRISPR–Cas9 System in the Immune Cell Engineering for Colorectal Cancer
As mentioned before, with limited clinical data, CAR-T cell therapy for CRC is in its initial phase. This therapy faces significant challenges, including high toxicity, a tendency for relapses, and difficulties in effectively infiltrating the tumor microenvironment.
An expected complication of this therapy is T-cell exhaustion. It is characterized by a complex phenotype involving a gradual decline in effector functions and a diminished memory T-cell response, contributing to poor persistence and limiting the durability of CAR-T cells. This exhaustion state is marked by heightened expression of various inhibitory receptors, such as PD-L1 and CTLA-4 [87]. Hence, employing CRISPR–Cas9 gene editing to target inhibitory coreceptors such as PD-1 and CTLA-4 in CAR-T cells could potentially enhance their resistance to suppression by cancer cells and Tregs. Several CRISPR approaches have been developed to target different tumor antigens. Pre-clinical and clinical evidence where CRISPR–Cas9 has been used to enhance adoptive cell transfer, mainly CAR-T and tumor-infiltrating lymphocyte (TILs) therapy are shown in Table 2.
In vitro experiments employing the CRISPR–Cas9 technique to disable the PD-1 gene in CAR-T cells showcased increased cytokine production and heightened cytotoxicity of CAR-T cells when targeting PD-L1-expressing cancer cells [95]. Furthermore, in a breast cancer model based on xenografts, this approach exhibited a superior synergistic response compared with the use of a PD-1 blocking antibody [95]. However, a phase I study was conducted in 2018 utilizing CAR-T cells targeting mesothelin in CRC and other solid tumors. In this study, the patients were administered mesothelin-directed CAR-T cells with a knockdown in PD-1 using CRISPR–Cas9. However, CAR-T cells’ expansion and long-term presence with PD-1 disruption did not show significant improvement beyond 2 weeks [93].
Knocking out CTLA-4 through CRISPR–Cas9 technology has demonstrated its ability to enhance cytotoxic lymphocytes’ anti-tumor activity. By boosting cell activation through the elimination of CTLA-4, which competes with CD28 ligand binding, and by elevating the secretion of TNF-α and IFN-γ, significant outcomes were achieved: reduced tumor size and extended survival of mice in a xenograft colon cancer model [96]. Cytokine-induced SH2 protein (CISH) is an intracellular immune checkpoint and plays a crucial role in negatively regulating T-cell signaling and function. Inhibiting CISH in CD8 T-cells led to a significant enhancement in their capacity to induce tumor regression when administered to mice with tumors [97]. In a current clinical trial, the researchers have developed a CRISPR–Cas9-based strategy that enables precise and efficient genetic engineering in primary human T-cells. In this protocol, the gene responsible for the intracellular checkpoint target CISH is inhibited in lymphocytes obtained from patients with colorectal and gastrointestinal metastatic cancers. These modified lymphocytes, selected for their anti-tumor activity, are then used to evaluate the safety and effectiveness of genetically engineered T-cell therapy for solid tumors [98].
Epithelial cell adhesion molecule glycoprotein (EpCAM) is a cell surface marker that is commonly overexpressed in various types of carcinomas, such as colorectal, gastric, pancreatic, and endometrial cancers [99]. In an effort to enhance the effectiveness of CAR T cells against tumors overexpressing EpCAM, the in vitro performance of CAR T cells specific to both EpCAM and intercellular adhesion molecule-1 (ICAM-1) was validated through CRISPR–Cas9 editing of cell lines [100]. Due to the heterogenous expression of antigens in solid tumors, to replicate the heterogeneity observed in EpCAM expression, EpCAM knockout tumor cell lines were mixed with wild-type cell lines at specific ratios. EpCAM and ICAM-1 CAR T cells facilitate cytotoxicity against heterogenous tumor cell lines in vitro [100]. In addition, Tag-72, an oncofetal mucin overexpressed by colon adenocarcinomas has been used to generate CAR-T cells specific for TAG-72 (CART72) as a treatment for patients with CRC and liver metastases. However, the rapid clearance of subsequent CART72 infusions in most patients limited their efficacy [101]. Using CRISPR–Cas9 technology, a new and improved version of CART72 cells was developed. These modified CAR-T cells lacked diacylglycerol (DAG) kinase α and ζ (DGKαζ), resulting in elevated intracellular levels of DAG. This increase in DAG enhanced the metabolic activity of the cells. When these modified CAR-T cells were administered in a xenograft model, they effectively eliminated the tumors and enhanced the killing of tumor cells by CD8 T cells [102]. Carcinoembryonic antigen (CEA) is a crucial biomarker for diagnosing and predicting the prognosis of CRC, and it is highly expressed in more than 98% of CRC tissue samples. The anti-tumor activity of CAR-T cells targeting CEA (CEA-CAR-T cells) has shown remarkable efficacy both in laboratory studies and animal models [103]. The identification of molecules associated with exhaustion is crucial for enhancing the efficacy of CEA-CAR T cells. CbI-I is a E3-ubiquitin ligase upregulated in exhausted (PD1+Tim3+) CD8+ TILs. CbI-I deletion via CRISPR–Cas9 restores the effector functions of cytotoxic TILs, and also avoids exhaustion of transplanted CAR-T cells, resulting in enhanced tumor inhibition and survival without reported toxicity in the syngeneic MC38-CEA colon cancer model [104].
Another approach consisted of TILs extracted from patients with advanced-stage CRC, and subsequently, CRISPR–Cas9 technology is utilized to genetically modify these TILs by selectively disrupting genes such as PD-1 or hematopoietic progenitor kinase (HPK1). These genes are recognized for their role in suppressing T- and B-cell responses against tumors. Subsequently, these modified TILs are transfused back into the patients. The current evaluation focuses on assessing the safety, tolerance, and initial clinical efficacy of the treatment [105]. TILs can also be collected from biopsies and engineered by CRISPR–Cas9 to knock-down immune checkpoint genes to enhance their anti-tumor activity. Natural killer group 2 member D (NKG2D) is an important activating receptor expressed on the surface of human natural killer (NK) cells. Normal cells typically have minimal or undetectable amounts of these molecules, but they quickly emerge on the surface of cells that are stressed, malignantly transformed, or infected. Ligands for NKG2D are expressed during neoplastic cell transformation; indeed, high expression of NKG2D ligands (NKG2D-L) was correlated with improved disease-free survival in human colorectal carcinomas [106]. Certainly, NKG2D CAR-T cells exhibit precise and effective killing capabilities against human colorectal cancer cells in vitro and in vivo [46]. Sekiba et al [107] demonstrated the potential of CRISPR–Cas9 in vitro by using it to activate NKG2D-L loci and ensure their surface expression in human hepatocellular carcinoma cell lines. Their findings highlight the power of CRISPR–Cas9 as a tool for effectively enhancing NKG2D-L expression on tumor cells and promoting their elimination through the interaction between NKG2D receptors on NK cells and NKG2D ligands on pathogenic cells [107]. In addition, by employing CRISPR–Cas9 gene editing to boost the expression of CXCR2 and IL-2 in NK-92 cells, there was an increase in NK cell infiltration at tumor sites, heightened cell-killing and proliferation activities, ultimately leading to a decreased tumor burden in a human colon cancer xenograft mouse model [108].
7 Expanded Alternatives of the CRISPR–Cas9 System for CRC Therapy
As mentioned earlier, CRISPR–Cas9 holds substantial potential for augmenting immunotherapy in the context of CRC. In this section, we will explore alternative approaches that, while not yet applied to CRC, have been developed in other contexts and may offer potential benefits in the treatment of CRC.
7.1 Cas12a–Cpf1, a Class 2 Type V CRISPR System for Multiplex Gene Editing of CAR-T Cells
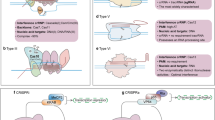
CARs are proteins that allow the T cells to recognize an antigen on targeted tumor cells. The currently approved method for delivering CAR-T cell transgenes involves the random integration of lentiviral and γ-retroviral vectors. This approach comes with the potential risk of causing insertional oncogenesis and translational silencing. Xiaoyun Dai (2019) [109] described a methodology for the use of Cas12a–Cpf1, a class 2 type V CRISPR system for precise gene editing in multiple loci in human primary T cells. By employing this approach in conjunction with adeno-associated virus (AAV), they successfully engineered stable CAR-T cells with high-efficiency homology-directed-repair knock-in and immune-checkpoint knockout in a single step. In the context of solid tumors such as CRC, resistance patterns involving antigen escape are frequently encountered during CAR-T cell therapy. Strategies centered on targeting multiple antigens have the potential to yield more robust anti-tumor responses compared with single-targeted therapies. The Cpf1 platform facilitates the highly efficient development of advanced CAR-T cells with multiple CARs, simplifying the creation of more intricate genetically engineered T cells through modular processing, which can include the deletion of endogenous TCR for avoiding allogeneic rejection [110]. In the case of CRC, this technique could be useful for the generation of universal CAR-T cells more stable in transgene expression levels and with improved ability to recognize and destroy cancer cells (Fig. 2 A).

CRISPR–Cas9 modifications for improving immunotherapy in the tumor microenvironment in CRC. A Cpf1-multiplex CAR-targeting and TCR deletion. TRAC gene inactivation leads to the loss of endogenous TCRα, allowing a more specific immune response against the neoantigen through only the chimeric receptor, and also allows the allogeneic use of CAR-T cells; moreover, the two alleles can be targeted to include two different chimeric receptors to target antigenic variability in CRC. B Prime-editing of oncogene mutations such as naturally occurring KRAS variants in tumor cells allows to reverse CRC progression. C STAT3 signaling activation in CAR-T cells enhances anti-tumor activity by enhancing cytokine-mediated killing. D Inhibition of TGF-β-mediated Treg induction in CAR-T cells. TGFBR2 gene inactivation inhibits TGF-β receptor expression and renders CAR-T cells refractory to Treg induction, increasing their stability within the TME. E Disabling immune checkpoint genes. Tumor cells express ligands of inhibitory coreceptors such as PD-1 and CTLA-4, leading to T-cell exhaustion and curbing anti-tumoral responses. PD1 and CTLA-4 gene inactivation blocks PD1 and CTLA-4 expression with similar effects to immune checkpoint blockade. CISH is another recently characterized immune checkpoint whose inhibition via CRISPR–Cas9 gene inactivation leads to improved CAR-T cell survival, cytokine secretion, and anti-tumor killing. F Cas9/gRNA-mediated release of PD1-targeting DNA aptamer. DNA polyaptamers against PD1 can be injected into the tumor site and be activated when combined with Cas9/gRNA complexes that release the monomeric aptamers. The DNA aptamers bind to and block PD-1 activity with a similar effect to immune checkpoint antibodies, but with reduced systemic complications due to their localized activation within the tumor site
7.2 CRISPR–Cas9 DNA Base-Editing and Prime-Editing
The most frequently encountered side effect of CAR T-cell therapy is known as cytokine release syndrome, abbreviated as CRS. It can impact as many as 9 out of 10 CAR T-cell patients [111]. CRS includes marked increases in IFN-gamma, granulocyte macrophage-colony stimulating factor (GM-CSF), TNF-alpha, interleukin (IL)-6, and IL-10 after administration of retrovirally-transduced autologous CAR-T cells [112]. In certain cases, the presence of cytokine gene polymorphisms associated with elevated IL-6 and IL-10 levels, which are linked to shock and fatality in patients, suggests the importance of recommending a cytokine gene profile assessment before administering CAR-T cell therapy. CRISPR–Cas9 base editors afford programmable enzymatic nucleotide conversion at targeted loci and could be used to knockout gene function in human T cells. CRISPR–Cas9 can also be useful to down-modulate the excessive cytokine responses caused by CAR-T cells (e.g., base-editing of specific cytokine gene polymorphisms). In particular, base editors bring advantages over “traditional” CRISPR–Cas9 techniques, as no double-strand breaks are generated and are useful to edit human T cells [113]. Prime editing also shares these advantages and allows for more complex genome modifications. Recently, prime editing has been deployed to correct oncogene mutations, such as the diverse, naturally occurring KRAS variants in pancreatic cancer and the colon cancer cell line HCT-116, opening novel therapeutic options [114] (Fig. 2B).
7.3 Activation of STAT Pathways in Progenitor Lineages for CAR-T Cells
Other factors that influence a favorable response or resistance to CAR-T cell therapy have also been identified [115]. CAR-T cells exhibiting an activated IL-6/STAT3 pathway demonstrated high functionality in patients with chronic lymphocytic leukemia. Specific phenotype signatures, such as CD27+CD45RO–CD8+ T cells, have been found to be the most effective progenitors for generating active CAR-T cells. In this context, CRISPR–Cas editing could be utilized to activate the STAT3 pathway and engineer specific genotypes that offer greater anti-tumor potential in the progenitor lineages of CAR-T cells (Fig. 2C). It is worth noting that directly editing progenitor cells with lower levels of differentiation has been associated with more significant and prolonged anti-tumor activity than differentiated T cells [105].
7.4 Blocking Tregs Conversion of CAR-T Cells and TILs
CAR-T cell therapy still faces many challenges in solid tumors such as CRC, including the suppressive TGF-β signaling by both tumor and stromal cells. TGF-β can induce Foxp3 activation in CAR-T cells, leading to a Treg signature epigenome and to an exhaustion phenotype with high PD1 expression [116]. Tang et al. (2020) [116] showed that knocking out endogenous TGF-β receptor II (TGFBR2) via CRISPR–Cas9 reduces Treg induction, prevents CAR-T exhaustion, and promotes tumor-killing activity in vivo against patient-derived xenograft solid tumor models (Fig. 2D). Similarly, resistance to immunosuppressive TGF-β signaling has been reported in CRISPR–Cas9-edited TGFBR2-knockout tumor infiltrating lymphocytes against ovarian cancer (Fig. 2D) [117]
7.5 Disabling Immune Checkpoint Genes in Tumor-Infiltrating Lymphocytes
As an alternative to blockade immunotherapy and its associated drawbacks, CRISPR–Cas9 technology can be utilized to directly edit cytotoxic lymphocytes (CTLs) and disable immune checkpoint genes such as PD-1. This approach offers a potential means to enhance immune responses without relying on the use of checkpoint inhibitors and its disadvantages such as Treg activation [118, 119]. Zhang et al. [119] developed a method to generate PD-1-deficient CTLs from primary T-cells of patients by CRISPR–Cas9, and demonstrated improved effector functions, including enhanced degranulation and cytokine production in the presence of PD-L1, suggesting an improved resistance to the immunosuppressive TME (Fig. 2E). Similarly, Zhao et al. [118] translated these results into an in vivo situation where PD-1 edited CTLs inhibited tumor growth in a human multiple-myeloma xenograft murine model by inducing apoptosis and increased TNF-α and IFN-γ secretion. Moreover, CRISPR–Cas9 edited PD1-deficient T-cells have shown superior efficacy against solid tumors when combined with total body irradiation in a melanoma mouse model [120].
7.6 Polyaptamers Targeting PD-1
The CRISPR–Cas tool might help to make the effect of immunotherapy more specific by directly editing the immune checkpoint molecules in cytotoxic lymphocytes, potentially avoiding the administration of ICIs that are commonly used antibodies, with a similar boosting effect on their cytotoxicity [121]. Another alternative approach is to deliver DNA aptamers targeting the PD-1/PD-L1 axis via injection at the tumor site [122]. When combined with Cas9/gRNA, it has been shown that DNA polyaptamers targeting PD-1 are retained only at the site of injection (Fig. 2F). Basically, the DNA aptamers remain in their inactive polymeric form, until the CRISPR/gRNA targets the sequence bonding aptamers, releasing them into their monomeric active form. As the activation occurs locally, this avoids systemic complications of current PD-1 antibody inhibitors. Interestingly, the Cas9/gRNA-mediated release of the PD-1 targeting aptamer also prolongs the retention time and enhances the anti-tumor activity and survival compared with the aptamer or PD-1 antibody treatment alone in a B16F10 melanoma model [123].
8 Concluding Remarks
In recent years, significant advances have been made in the field of cancer research, specifically in the areas of CRISPR gene editing, immunotherapy, and colorectal cancer. CRISPR has emerged as a powerful tool for genetic manipulation, allowing researchers to precisely modify DNA sequences and potentially cure genetic diseases, including cancer. In CRC, CRISPR has been used to identify new targets for therapy and to develop more effective treatment strategies.
Immunotherapy has transformed cancer treatment by harnessing the body’s immune system to target and combat cancer cells. In the case of CRC, immunotherapy has demonstrated promising outcomes, particularly in patients with advanced disease that have not responded to other treatments, specifically those with microsatellite instability. The combination of CRISPR technology with immunotherapy holds the potential to augment the effectiveness of both treatments. CRISPR can be utilized to genetically modify immune cells, such as CAR-T cells, TILs, and NK cells, enhancing their capacity to recognize and attack cancer cells while maintaining activity within the immunosuppressive tumor microenvironment. Simultaneously, immune checkpoint blockade can be employed to activate and bolster the immune response. Figure 2 illustrates the CRISPR–Cas9 modifications for improving immunotherapy in the tumor microenvironment in CRC. While these approaches are still in the early stages of development, they offer great promise for improving outcomes for patients. Ongoing research will continue to refine these techniques and bring them closer to clinical use.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature [Internet]. 2009;458:719–24.
Hanahan D, Weinberg RA. The Hallmarks of cancer review evolve progressively from normalcy via a series of pre. Cell. 2000;100:57–70.
Lichtenstern CR, Ngu RK, Shalapour S, Karin M. Immunotherapy, inflammation and colorectal cancer. Cells [Internet]. 2020;9:618.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov [Internet]. 2022;12:31–46. https://doi.org/10.1158/2159-8290.CD-21-1059.
de Vries NL, Swets M, Vahrmeijer AL, Hokland M, Kuppen PJK. The immunogenicity of colorectal cancer in relation to tumor development and treatment. Int J Mol Sci [Internet]. 2016;17:1030.
Golshani G, Zhang Y. Advances in immunotherapy for colorectal cancer: a review. Therap Adv Gastroenterol [Internet]. 2020;13:175628482091752.
Smit WL, Spaan CN, De Boer RJ, Ramesh P, Garcia TM, Meijer BJ, et al. Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc Natl Acad Sci USA [Internet]. 2020;117:25560–70.
Mortezaee K. Immune escape: a critical hallmark in solid tumors. Life Sci [Internet]. 2020;258:118110.
Anderson P, Aptsiauri N, Ruiz-Cabello F, Garrido F. HLA class I loss in colorectal cancer: implications for immune escape and immunotherapy. Cell Mol Immunol [Internet]. 2021;18:556–65.
Caronni N, Piperno GM, Simoncello F, Romano O, Vodret S, Yanagihashi Y, et al. TIM4 expression by dendritic cells mediates uptake of tumor-associated antigens and anti-tumor responses. Nat Commun [Internet]. 2021;12:2237.
Kriegsman BA, Vangala P, Chen BJ, Meraner P, Brass AL, Garber M, et al. Frequent loss of IRF2 in cancers leads to immune evasion through decreased MHC Class I antigen presentation and increased PD-L1 expression. J Immunol [Internet]. 2019;203:1999–2010.
Romero I, Garrido C, Algarra I, Chamorro V, Collado A, Garrido F, et al. MHC intratumoral heterogeneity may predict cancer progression and response to immunotherapy. Front Immunol [Internet]. 2018;9:102.
Ho WW, Gomes-Santos IL, Aoki S, Datta M, Kawaguchi K, Talele NP, et al. Dendritic cell paucity in mismatch repair-proficient colorectal cancer liver metastases limits immune checkpoint blockade efficacy. Proc Natl Acad Sci USA [Internet]. 2021;118:e2105323118. https://doi.org/10.1073/pnas.2105323118.
Pluhar GE, Pennell CA, Olin MR. CD8+ T cell-independent immune-mediated mechanisms of anti-tumor activity. Crit Rev Immunol [Internet]. 2015;35:153.
Guo G, Wang Y, Zhou Y, Quan Q, Zhang Y, Wang H, et al. Immune cell concentrations among the primary tumor microenvironment in colorectal cancer patients predicted by clinicopathologic characteristics and blood indexes. J Immunother Cancer [Internet]. 2019;7:179.
Wu K, Zheng X, Yao Z, Zheng Z, Huang W, Mu X, et al. Accumulation of CD45RO+CD8+ T cells is a diagnostic and prognostic biomarker for clear cell renal cell carcinoma. Aging (Albany NY) [Internet]. 2021;13:14304.
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138:1429–40.
Karin N. CXCR3 ligands in cancer and autoimmunity, chemoattraction of effector t cells, and beyond. Front Immunol [Internet]. 2020;11:976.
Shang S, Yang YW, Chen F, Yu L, Shen SH, Li K, et al. TRIB3 reduces CD8 + T cell infiltration and induces immune evasion by repressing the STAT1-CXCL10 axis in colorectal cancer. Sci Transl Med [Internet]. 2022;14. https://www.researcher-app.com/paper/10107495
Loddenkemper C, Schernus M, Noutsias M, Stein H, Thiel E, Nagorsen D. In situ analysis of FOXP3+ regulatory T cells in human colorectal cancer. J Transl Med [Internet]. 2006;4:52.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–84. https://www.nature.com/articles/nm.4086
Argon A, Vardar E, Kebat T, Erdinç Ö, Erkan N. The prognostic significance of FoxP3+ T cells and CD8+ T cells in colorectal carcinomas. J Environ Pathol Toxicol Oncol [Internet]. 2016 [cited 2023 Jun 19];35:121–31. https://www.dl.begellhouse.com/journals/0ff459a57a4c08d0,1618a93b7a136570,0753649479f7fa11.html
Kelsen J, Agnholt J, Hoffmann HJ, Rømer JL, Hvas CL, Dahlerup JF. FoxP3+CD4+CD25+ T cells with regulatory properties can be cultured from colonic mucosa of patients with Crohn’s disease. Clin Exp Immunol [Internet]. 2005;141:549–57. https://doi.org/10.1111/j.1365-2249.2005.02876.x.
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol [Internet]. 2008;27:186–92. http://intl.jco.org/cgi/content/full/27/2/186
Delgado-Ramirez Y, Ocaña-Soriano A, Ledesma-Soto Y, Olguín JE, Hernandez-Ruiz J, Terrazas LI, et al. STAT6 is critical for the induction of regulatory T cells in vivo controlling the initial steps of colitis-associated cancer. Int J Mol Sci [Internet]. 2021;22. https://www.mdpi.com/1422-0067/22/8/4049
He T, Yang D, Li XQ, Jiang M, Islam MS, Chen S, et al. Inhibition of two-pore channels in antigen-presenting cells promotes the expansion of TNFR2-expressing CD4+Foxp3+ regulatory T cells. Sci Adv. 2020;6.
Yahaya MAF, Lila MAM, Ismail S, Zainol M, Afizan NARNM. Tumour-associated macrophages (TAMs) in colon cancer and how to reeducate them. J Immunol Res [Internet]. 2019;2019:2368249.
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits t-cell receptor expression and antigen-specific t-cell responses. Cancer Res [Internet]. 2004;64:5839–49. https://doi.org/10.1158/0008-5472.CAN-04-0465.
Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One [Internet]. 2011;6:e19495. https://doi.org/10.1371/journal.pone.0019495.
Feng Y, Ye Z, Song F, He Y, Liu J. The role of TAMs in tumor microenvironment and new research progress. Stem Cells Int [Internet]. 2022;2022:1.
Yu J, Xu Z, Guo J, Yang K, Zheng J, Sun X. Tumor-associated macrophages (TAMs) depend on MMP1 for their cancer-promoting role. Cell Death Discov 2021 7:1 [Internet]. 2021;7:1–10. https://www.nature.com/articles/s41420-021-00730-7
Troiano G, Caponio VCA, Adipietro I, Tepedino M, Santoro R, Laino L, et al. Prognostic significance of CD68+ and CD163+ tumor associated macrophages in head and neck squamous cell carcinoma: a systematic review and meta-analysis. Oral Oncol. 2019;93:66–75.
Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci [Internet]. 2019;6:160.
Kouidhi S, Ayed FB, Elgaaied AB. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol. 2018;9:336488.
Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Cristina M, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197.
Chiesa MD, Setti C, Giordano C, Obino V, Greppi M, Pesce S, et al. NK cell-based immunotherapy in colorectal cancer. Vaccines (Basel). 2022.
Globerson Levin A, Rivière I, Eshhar Z, Sadelain M. CAR T cells: building on the CD19 paradigm. Eur J Immunol [Internet]. 2021;51:2151–63. https://doi.org/10.1002/eji.202049064.
Jackson Z, Roe A, Sharma AA, Lopes FBTP, Talla A, Kleinsorge-Block S, et al. Automated manufacture of autologous CD19 CAR-T cells for treatment of non-hodgkin lymphoma. Front Immunol [Internet]. 2020;11.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med [Internet]. 2017;377:2531–44.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J [Internet]. 2021;11.
Dana H, Chalbatani GM, Jalali SA, Mirzaei HR, Grupp SA, Suarez ER, et al. CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharm Sin B [Internet]. 2021;11:1129–47.
Zhang C, Jia J, Heng G, Li Y, Wang M, Chen J, et al. CD27 agonism coordinates with CD28 and 4-1BB signal to augment the efficacy of CAR-T cells in colorectal tumor. Med Oncol [Internet]. 2023;40.
Magee MS, Abraham TS, Baybutt TR, FlickingerJr JC, Ridge NA, Marszalowicz GP, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res [Internet]. 2018;6:509–16.
He S, Li S, Guo J, Zeng X, Liang D, Zhu Y, et al. CD166-specific CAR-T cells potently target colorectal cancer cells. Transl Oncol [Internet]. 2023;27:101575.
Deng X, Gao F, Li N, Li Q, Zhou Y, Yang T, et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res [Internet]. 2019;9:945–58.
Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G, Hacein-Bey-Abina S. Advances in CAR-T cell genetic engineering strategies to overcome hurdles in solid tumors treatment. Front Immunol [Internet]. 2022;13.
Wang W, Bandara V, Lokman N, Napoli S, Gundsambuu B, Oehler M, et al. Abstract 5183: LGR5 CAR-T cells: a novel potential treatment against high grade serous ovarian cancer. Cancer Res [Internet]. 2022;82:5183–5183. https://doi.org/10.1158/1538-7445.AM2022-5183.
Cha SE, Kujawski M, Yazaki PJ, Brown C, Shively JE. Tumor regression and immunity in combination therapy with anti-CEA chimeric antigen receptor T cells and anti-CEA-IL2 immunocytokine. Oncoimmunology [Internet]. 2021;10:1899469. https://doi.org/10.1080/2162402X.2021.1899469.
Mei Z, Zhang K, Lam AK-Y, Huang J, Qiu F, Qiao B, et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carcinoma. Cancer Med [Internet]. 2020;9:640–52. https://doi.org/10.1002/cam4.2733
Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR t cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity [Internet]. 2016;44:1444–54.
Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology [Internet]. 2017;6:e1284722. https://doi.org/10.1080/2162402X.2017.1284722.
Ahmad R, Alam M, Hasegawa M, Uchida Y, Al-Obaid O, Kharbanda S, et al. Targeting MUC1-C inhibits the AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal cancer. Mol Cancer [Internet]. 2017;16:33. https://doi.org/10.1186/s12943-017-0608-9.
Zhang B-L, Li D, Gong Y-L, Huang Y, Qin D-Y, Jiang L, et al. Preclinical evaluation of chimeric antigen receptor–modified T cells specific to epithelial cell adhesion molecule for treating colorectal cancer. Hum Gene Ther [Internet]. 2019;30:402–12. https://doi.org/10.1089/hum.2018.229.
Lee SJ, Lee J, Park SH, Park JO, Lim HY, Kang WK, et al. c-MET overexpression in colorectal cancer: a poor prognostic factor for survival. Clin Colorectal Cancer [Internet]. 2018;17:165–9. http://www.clinical-colorectal-cancer.com/article/S1533002818300045/fulltext
Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Benamouzig R, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol [Internet]. 2005;16:102–8. http://www.annalsofoncology.org/article/S0923753419416293/fulltext
Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res [Internet]. 2015;75:3596–607. https://doi.org/10.1158/0008-5472.CAN-15-0159.
Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology [Internet]. 2018. https://doi.org/10.1080/2162402X.2018.1440169
Park YY, An CH, Oh ST, Chang ED, Lee J, Kok VC. Expression of CD133 is associated with poor prognosis in stage II colorectal carcinoma. Medicine (United States) [Internet]. 2019 [cited 2023 Jun 20];98. https://journals.lww.com/md-journal/Fulltext/2019/08090/Expression_of_CD133_is_associated_with_poor.37.aspx
Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel) [Internet]. 2020;12. https://www.mdpi.com/2072-6694/12/3/738
Marin-Acevedo JA, Kimbrough EO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol [Internet]. 2021;14:45. https://doi.org/10.1186/s13045-021-01056-8.
McDermott D, Haanen J, Chen T-T, Lorigan P, O’Day S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann Oncol [Internet]. 2013;24:2694–8. https://doi.org/10.1093/annonc/mdt291.
Necchi A, Lo Vullo S, Giannatempo P, Raggi D, Perrone F, Nicolai N, et al. Association of androgen receptor expression on tumor cells and PD-L1 expression in muscle-invasive and metastatic urothelial carcinoma: insights for clinical research. Clin Genitourin Cancer [Internet]. 2018;16:e403–10. https://doi.org/10.1016/j.clgc.2017.09.016.
Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol [Internet]. 2016;17:1374–85. https://doi.org/10.1016/S1470-2045(16)30364-3.
Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and efficacy of durvalumab (MEDI4736), an anti–programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol [Internet]. 2016;34:3119–25. https://doi.org/10.1200/JCO.2016.67.9761.
Aparicio C, Belver M, Enríquez L, Espeso F, Núñez L, Sánchez A, et al. Cell therapy for colorectal cancer: the promise of chimeric antigen receptor (CAR)-T cells. Int J Mol Sci [Internet]. 2021;22:11781. https://www.mdpi.com/1422-0067/22/21/11781/htm.
Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood [Internet]. 2017;129:1039.
Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (1979) [Internet]. 2017;355:1423–7. https://doi.org/10.1126/science.aaf0683.
Aksoylar HI, Boussiotis VA. PD-1+ Treg cells: a foe in cancer immunotherapy? Nat Immunol 2020 21:11 [Internet]. 2020;21:1311–2. https://www.nature.com/articles/s41590-020-0801-7.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Sahin IH, Akce M, Alese O, Shaib W, Lesinski GB, El-Rayes B, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer [Internet]. 2019;121:809–18. https://doi.org/10.1038/s41416-019-0599-y.
Zhang X, Yang Z, An Y, Liu Y, Wei Q, Xu F, et al. Clinical benefits of PD-1/PD-L1 inhibitors in patients with metastatic colorectal cancer: a systematic review and meta-analysis. World J Surg Oncol [Internet]. 2022;20:93. https://doi.org/10.1186/s12957-022-02549-7.
Boukouris AE, Theochari M, Stefanou D, Papalambros A, Felekouras E, Gogas H, et al. Latest evidence on immune checkpoint inhibitors in metastatic colorectal cancer: a 2022 update. Crit Rev Oncol Hematol [Internet]. 2022;173:103663.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol [Internet]. 2019;16:361–75. https://doi.org/10.1038/s41575-019-0126-x.
Arroyo-Olarte RD, Bravo Rodríguez R, Morales-Ríos E. Genome editing in bacteria: CRISPR-Cas and beyond. Microorganisms. 2021;9:844.
Arroyo-Olarte RD, Rodríguez-Hernández KD, Morales-Ríos E. Genome engineering in bacteria: current and prospective applications. Methods Microbiol. 2023;52:35–76.
Kressler C, Gasparoni G, Nordström K, Hamo D, Salhab A, Dimitropoulos C, et al. Targeted de-methylation of the FOXP3-TSDR is sufficient to induce physiological FOXP3 expression but not a functional treg phenotype. Front Immunol [Internet]. 2021;11.
Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucl Acids Res [Internet]. 2016;44:5615–28.
Wilk C, Effenberg L, Abberger H, Steenpass L, Hansen W, Zeschnigk M, et al. CRISPR/Cas9-mediated demethylation of FOXP3-TSDR toward Treg-characteristic programming of Jurkat T cells. Cell Immunol. 2022;371:104471.
Palaz F, Kalkan AK, Can Ö, Demir AN, Tozluyurt A, Özcan A, et al. CRISPR-Cas13 system as a promising and versatile tool for cancer diagnosis, therapy, and research. ACS Synth Biol [Internet]. 2021;10:1245–67. https://doi.org/10.1021/acssynbio.1c00107.
Yau EH, Kummetha IR, Lichinchi G, Tang R, Zhang Y, Rana TM. Genome-wide crispr screen for essential cell growth mediators in mutant KRAS colorectal cancers. Cancer Res [Internet]. 2017;77:6330–9. https://doi.org/10.1158/0008-5472.CAN-17-2043.
Roper J, Tammela T, Cetinbas NM, Akkad A, Roghanian A, Rickelt S, et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat Biotechnol [Internet]. 2017;35:569–76. https://doi.org/10.1038/nbt.3836.
Li Y, Li X, Qu J, Luo D, Hu Z. Cas9 mediated correction of β-catenin mutation and restoring the expression of protein phosphorylation in colon cancer HCT-116 cells decrease cell proliferation in vitro and hamper tumor growth in mice in vivo. Onco Targets Ther [Internet]. 2020;13:17–29. https://www.dovepress.com/cas9-mediated-correction-of-beta-catenin-mutation-and-restoring-the-ex-peer-reviewed-article-OTT
Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature [Internet]. 2015;521:43–7. https://doi.org/10.1038/nature14415.
Takeda H, Kataoka S, Nakayama M, Ali MAE, Oshima H, Yamamoto D, et al. CRISPR-Cas9–mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc Natl Acad Sci [Internet]. 2019;116:15635–44. https://doi.org/10.1073/pnas.1904714116.
Pothuraju R, Rachagani S, Krishn SR, Chaudhary S, Nimmakayala RK, Siddiqui JA, et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol Cancer [Internet]. 2020;19:37. https://doi.org/10.1186/s12943-020-01156-y.
Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion.’ Nat Rev Immunol [Internet]. 2019;19:665–74. https://doi.org/10.1038/s41577-019-0221-9.
Bernard P-L, Delconte R, Pastor S, Laletin V, Silva CC Da, Goubard A, et al. Targeting CISH enhances natural cytotoxicity receptor signaling and reduces NK cell exhaustion to improve solid tumor immunity. J Immunother Cancer [Internet]. 2022;10. https://jitc.bmj.com/content/10/5/e004244
Lv J, Qin L, Zhao R, Wu D, Wu Z, Zheng D, et al. Disruption of CISH promotes the antitumor activity of human T cells and decreases PD-1 expression levels. Mol Ther Oncolyt. 2023;28:46.
Chamberlain CA, Bennett EP, Kverneland AH, Svane IM, Donia M, Met Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol Ther Oncolyt. 2022;24:417.
Su S, Zou Z, Chen F, Ding N, Du J, Shao J, et al. CRISPR-cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. 2017;6:e1249558.
Zhang N, Si J, Li G, Wang Y, Long F, Wang T, et al. Decreasing HPK1 expression in CD19 CAR-T cells: a novel strategy to overcome challenges of cell therapy for adult (r/r) B-ALL. J Clin Oncol. 2022;40:7041.
Wang Z, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, et al. Phase I study of CRISPR-engineered CAR-T cells with PD-1 inactivation in treating mesothelin-positive solid tumors. J Clin Oncol [Internet]. 2020;38:3038. https://doi.org/10.1200/JCO.2020.38.15_suppl.3038.
Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18:2188.
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother [Internet]. 2019;68:365–77. https://doi.org/10.1007/s00262-018-2281-2.
Shi L, Meng T, Zhao Z, Han J, Zhang W, Gao F, et al. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene [Internet]. 2017;636:36–41.
Palmer DC, Guittard GC, Franco Z, Crompton JG, Eil RL, Patel SJ, et al. CISH actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J Exp Med [Internet]. 2015;212:2095–113. https://doi.org/10.1084/jem.20150304.
A study of metastatic gastrointestinal cancers treated with tumor infiltrating lymphocytes in which the gene encoding the intracellular immune checkpoint CISH is inhibited using CRISPR genetic engineering—Full Text View—ClinicalTrials.gov [Internet]. [cited 2023 Jun 20]. https://classic.clinicaltrials.gov/ct2/show/NCT04426669
Keller L, Werner S, Pantel K. Biology and clinical relevance of EpCAM. Cell Stress. Shared Science Publishers OG; 2019. p. 165–80.
Yang Y, McCloskey JE, Yang H, Puc J, Alcaina Y, Vedvyas Y, et al. Bispecific CAR T cells against EpCAM and inducible ICAM-1 overcome antigen heterogeneity and generate superior antitumor responses. Cancer Immunol Res. 2021;9:1158.
Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5.
Evtimov V, Hammett M, Nhu-Y N, Zhuang J, Nisbet I, Trounson A, et al. 12P CRISPR/Cas9-induced knock-out of DGKαζ in TAG-72 CAR-T cells improves function and persistence in ovarian cancer. ESMO Open. 2023;8:100978.
Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019;8:4753.
Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of Cbl-b inhibits CD8 + T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer. 2021;9:e001688.
Zhao Y, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L, et al. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via notch signaling. Cancer Res. 2007;67:2425.
Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55.
Sekiba K, Yamagami M, Otsuka M, Suzuki T, Kishikawa T, Ishibashi R, et al. Transcriptional activation of the MICA gene with an engineered CRISPR-Cas9 system. Biochem Biophys Res Commun. 2017;486:521.
Gao L, Yang L, Zhang S, Ge Z, Su M, Shi Y, et al. Engineering NK-92 cell by upregulating CXCR2 and IL-2 via CRISPR-Cas9 improves its antitumor effects as cellular immunotherapy for human colon cancer. J Interferon Cytokine Res. 2021;41:450.
Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat Methods. 2019;16:247.
Wei F, Cheng XX, Xue JZ, Xue SA. Emerging strategies in TCR-engineered T cells. Front Immunol. 2022;13.
Yan Z, Zhang H, Cao J, Zhang C, Liu H, Huang H, et al. Characteristics and risk factors of cytokine release syndrome in chimeric antigen receptor T cell treatment. Front Immunol. 2021;12.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther [Internet]. 2010;18:843–51.
Webber BR, Lonetree C lin, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10.
Jang G, Kweon J, Kim Y. CRISPR prime editing for unconstrained correction of oncogenic KRAS variants. Commun Biol. 2023;6.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24.
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5.
Fix SM, Forget MA, Sakellariou-Thompson D, Wang Y, Griffiths TM, Lee M, et al. CRISPR-mediated TGFBR2 knockout renders human ovarian cancer tumor-infiltrating lymphocytes resistant to TGF-β signaling. J Immunother Cancer. 2022;10:e003750.
Zhao Z, Shi L, Zhang W, Han J, Zhang S, Fu Z, et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget. 2018;9:5208.
Zhang C, Peng Y, Hublitz P, Zhang H, Dong T. Genetic abrogation of immune checkpoints in antigen-specific cytotoxic T-lymphocyte as a potential alternative to blockade immunotherapy. Sci Rep. 2018;8.
Yang Z, Wu H, Lin Q, Wang X, Kang S. Lymphopenic condition enhanced the antitumor immunity of PD-1-knockout T cells mediated by CRISPR/Cas9 system in malignant melanoma. Immunol Lett. 2022;250:15.
Khalaf K, Janowicz K, Dyszkiewicz-Konwińska M, Hutchings G, Dompe C, Moncrieff L, et al. CRISPR/Cas9 in cancer immunotherapy: animal models and human clinical trials. Genes (Basel). 2020;11:921.
Prodeus A, Abdul-Wahid A, Fischer NW, Huang EHB, Cydzik M, Gariépy J. Targeting the PD-1/PD-L1 immune evasion axis with DNA aptamers as a novel therapeutic strategy for the treatment of disseminated cancers. Mol Ther Nucl Acids. 2015;4:e237.
Lee J, Le QV, Yang G, Oh YK. Cas9-edited immune checkpoint blockade PD-1 DNA polyaptamer hydrogel for cancer immunotherapy. Biomaterials. 2019;218:119359.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grants from Universidad Nacional Autónoma de México (UNAM)-Dirección General de Asuntos del Personal Académico (DGAPA)-Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT; grant number IN215421) and from the Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT; grant A1S23944). A. Mejía-Muñoz is a student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and the receiver of a fellowship (1269973) from CONAHCYT. R. Arroyo-Olarte received a post-doctoral fellowship from CONAHCYT (application number 877758). The Universidad Nacional Autónoma de México is the sponsor of the open-access fee.
Conflicts of interest
Rubén Arroyo-Olarte, Aranza Mejía-Muñoz, and Sonia León-Cabrera declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval
Not applicable.
Consent (participation and publication)
Not applicable.
Author contributions
Literature review: R.A.O, A.M.M, and S.L.C. Initial draft of manuscript: R.A.O, A.M.M, and S.L.C. Critical review and editing of manuscript: R.A.O, A.M.M, and S.L.C.
Data availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Code availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Arroyo-Olarte, R., Mejía-Muñoz, A. & León-Cabrera, S. Expanded Alternatives of CRISPR–Cas9 Applications in Immunotherapy of Colorectal Cancer. Mol Diagn Ther 28, 69–86 (2024). https://doi.org/10.1007/s40291-023-00680-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00680-z