
Abstract
Oncology indispensably leads us to personalized medicine, which allows an individual approach to be taken with each patient. Personalized oncology is based on pharmacogenomics and the effect of genetic differences in individuals (germline and somatic) on the way cancer patients respond to chemotherapeutics. Biomarkers detected using molecular biology tools allow the molecular characterization of cancer signatures and provide information relevant for personalized treatment. Biomarkers can be divided into two main subgroups: prognostic and predictive. The aim of the application of prognostic biomarkers, which provide information on the overall cancer outcome in patients, is to facilitate cancer diagnosis, usually with no need for putting invasive methods into use. Predictive biomarkers help to optimize therapy decisions, as they provide information on the likelihood of response to a given chemotherapeutic. Among the prognostic factors that identify patients with different outcome risks (e.g., recurrence of the disease), the following factors can be distinguished: somatic and germline mutations, changes in DNA methylation that lead to the enhancement or suppression of gene expression, the occurrence of elevated levels of microRNA (miRNA) capable of binding specific messenger RNA (mRNA) molecules, which affects gene expression, as well as the presence of circulating tumor cells (CTCs) in blood, which leads to a poor prognosis for the patient. Biomarkers for personalized oncology are used mainly in molecular diagnostics of chronic myeloid leukemia, colon, breast and lung cancer, and recently in melanoma. They are successfully used in the evaluation of the benefits that can be achieved through targeted therapy or in the evaluation of toxic effects of the chemotherapeutic used in the therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Conventional clinical procedures in patients with tumors are based on the proper selection of chemotherapy depending on the histopathological evaluation of the tumor and its organs of origin. Molecular mechanisms of neoplasia involve a great number of genetic aberrations and abnormalities [1], but a small part of them are found in all tumor types, which may be of key importance for oncogenesis and tumor progression [2].
In our paper, we investigate markers and separate them into two subgroups: predictive and prognostic factors. It is possible that the same biomarker might represent a good prognostic and predictive factor, which makes this division more difficult and sometimes can be practically confusing (e.g., MGMT promoter methylation [3], circulating tumor cells [CTCs] [4]). Furthermore, we focus on DNA markers which are less known in diagnostics than serum biomarkers. Samples for DNA biomarkers are mainly collected from tissue after biopsy or blood cells containing nuclei. Genetic variability and the occurrence of specific polymorphisms may participate in susceptibility to diseases, including tumors, and in the type of response to the therapy used. The advances in molecular biology allow the identification of the polymorphisms, which may soon permit us to enter the age of personalized medicine that will facilitate the choice of appropriate clinical procedures [5]. The choice of targeted therapy is based on the previous genetic analysis, which is becoming a complement for conventional diagnostic methods. In order to make these strategies successful, it is necessary to identify those populations of patients carrying genetic abnormalities, in whom the given therapy may be used with a positive outcome [6].
This review presents primarily markers for breast cancer, leukemia, colorectal cancer (CRC), lung cancer, melanoma, and glioblastoma. However, many studies on biomarkers have also been conducted for renal cancer [7, 8], thyroid cancer [9, 10], hepatocellular cancer [11, 12] as well as ovarian cancer [13, 14].
Therefore, it is extremely important to search for novel molecular biomarkers, since their correct selection permits the assessment of the level of malignancy and disease remission, monitoring of therapy advances, as well as the prediction of the response to the therapy used (which facilitates therapy selection from the available options).
2 Prognostic Biomarkers
Prognostic biomarkers enable the monitoring of the advances of anticancer therapy, the assessment of the stage of the tumor and its potential malignancy, as well as the prognosis of disease remission in every case individually [15, 16].
A good biomarker is characterized by the specificity for a given type of tumor and the appropriate level of sensitivity, while the concentration of the biomarker should reflect the stage of the disease and the response to the therapy used [17].
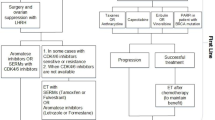
Prognostic biomarkers are assigned to a specific tumor type by determining the occurring polymorphism, mutation or the change in DNA methylation or gene expression, or by detecting the presence of specific microRNA (miRNA) molecules or CTCs in the peripheral blood (Fig. 1).

Examples of prognostic and predictive markers according to the subject of analysis. CTCs circulating tumor cells, miRNA microRNA. * Studies in progress
2.1 Prognostic Biomarkers: Analysis of Mutations and Polymorphisms
Mutations within the genes whose products participate in DNA repair, such as BRCA1, BRCA2, ATM and P53, predispose the patients to an increased risk of developing breast cancer. Mutations of those genes in germline cells may be inherited and a mutation in only one allele of the BRCA2 gene contributes to the inactivation of the BRCA2 protein and impairs the repair system [18]. Therefore, patients with mutations identified within the aforementioned genes should undergo regular screening examinations [19], while mutations within the genes encoding glutathione S-transferase (GSTM1, GSTT1, GSTP1) and a polymorphism within the gene encoding prostate-specific antigen (PSA), G158A single nucleotide polymorphism (SNP), may increase the risk of developing prostate cancer [20, 21]. Constitutive mutations in the APC gene predispose the patients to familial adenomatous polyposis (FAP), a genetic tumor disease of the autosomal dominant type of inheritance, characterized by an increased probability of occurrence of polyps and tumors in the gut [22]. The genome-wide association studies (GWAS) permit a fast detection of genetic variants contributing to the pathogenesis of complex genetic disorders, but also the detection of numerous pharmacogenetic markers [5]. GWAS indicate the clinical potential of the information on the polymorphisms predisposing the patients to aggressive forms of prostate cancer as potential early markers of risk assessment [23].
2.2 Prognostic Biomarkers: Analysis of Gene and MicroRNA (miRNA) Expression
Prostate cancer is one of the most common tumors in men [24]; therefore, it is essential to perform screening examinations. An example of a prognostic biomarker implemented in clinical diagnostics is PSA, whose upper limit of normal serum concentration is 4.0 ng/ml. However, PSA is not an ideal biomarker; elevated PSA levels in blood serum may also be related to nonpathological benign prostatic hyperplasia, prostatitis or inflammations of the genitourinary tract. There have also been reports of patients with diagnosed prostate cancer in whom PSA levels were below 4.0 ng/ml [25–27]. Therefore, an extensive search for markers more specific than PSA is ongoing. By using microarrays and the quantitative real-time PCR (qRT-PCR) technique, Liong et al. [28] looked for those blood-based markers of prostate cancer that would allow the identification of men with aggressive forms of the disease. Detailed analysis of 85 genes allowed them to identify six genes, CRTAM, CXCR3, FCRL3, KIAA1143, KLF12 and TMEM204, with potential prognostic significance. Determining the level of expression permits the stratification of patients with more or less aggressive forms of prostate cancer, which cannot be achieved with PSA [28].
Another example of the implementation of gene expression analysis, this time in female patients with diagnosed breast cancer, is the MammaPrint Symphony analysis, in which the diagnostic test is based on the microarray technique and four different bioinformatics analyses. The first analysis, consisting of a panel of 70 genes, allows the assessment of tumor dynamics and the direction of the neoplastic process, which, in consequence, permits the stratification of patients into the groups of high and low risk of relapse [29, 30] (regardless of the status of receptors of estrogens, progesterone and human epidermal growth factor 2). Separate analyses of these transcripts are conducted as part of the TargetPrint test [31]. The information obtained through these analyses, along with that from the additional analysis of 80 transcripts in the BluePrint test, providing molecular distinction of breast cancers into the basal, luminal and ERBB2 types, helps oncologists to choose the appropriate chemo- and hormonotherapy. The last test in the MammaPrint Symphony test set is TheraPrint, a predictive test providing information on the expression of the selected 56–125 genes identified as predictive biomarkers, which are of key importance for therapy personalization and the assessment of potential benefits from the chemotherapy used. For example, a significant difference in the level of expression of the BCL2, CDH3, GRB7, KRT6B, and KRT17 genes was observed between the groups of patients responding and not responding to treatment [31].
Expression analysis aimed at prognostic assessment may also be conducted using real-time PCR. For example, the diagnostic test Oncotype DX consists of a panel of 21 genes and assesses the probability of relapse of breast cancer within 10 years. The 21 genes include five genes related to proliferation (KI-67, STK 15, SURVIVIN, CYCLIB B1, MYBL2), four genes related to invasiveness (STROMELYSIN 3, CATHEPSIN L2, HER2, GRB7), seven hormonal genes (ER, PR, BCL2, SCUBE2, GTSM1, CD68, BAG 1) and five reference genes (β-ACTIN, GAPDH, RPLPO, GUS, TFRC). Unfortunately, the test based on real-time PCR (Oncotype DX) reports a higher percentage of false negatives with respect to the expression of HER2 [32].
miRNAs are small, noncoding RNA molecules that regulate gene expression by binding specific messenger RNAs (mRNAs). The presence of some miRNA molecules may be related to hepatocellular carcinoma (HCC) [33], multiple myeloma [34] and renal cell carcinoma [35]. HCC is characterized by the overexpression of miRNA-255, which increases the activity of the Wnt signaling path [36]. The presence of miRNA-155 levels in HCC patients suggests a high level of malignancy, potential for metastasis and poor prognosis [36]. In CRC, a prognostic role is played by miRNA-362-3p, whose overexpression leads to cell cycle arrest and inhibits the growth and migration of tumor cells. A high level of miRNA expression correlates with better prognosis for the patient [37].
2.3 Prognostic Markers: Analysis of Methylation
Recent studies indicate that the observation of the changes in DNA methylation may also be a good prognostic marker. Association studies of the entire epigenome, as well as studies of the methylation of single suppressor genes or oncogenes, indicate that these genes may undergo hypermethylation in the tumor tissue, which blocks their promoter sites and thus leads to the loss of gene expression [38]. In their studies, Xu et al. [39] demonstrated that estrogen receptor (ER) levels are correlated with the methylation of RASSF1A, while progesterone receptor (PR) levels are correlated with the methylation of RIL and CDH13. In the RASSF1A gene, the altered methylation pattern alone has been used to determine the time of relapse of the tumor and the time of survival [39]. Vasiljevic et al. [40] concluded that the methylation pattern of the HSBP1 gene encoding heat-shock protein 27 (HSP-27) is a good prognostic biomarker in prostate cancer. In tumor cells, the level of methylation increases toward the 3′ terminus, while that in benign prostatic hyperplasia remains below 5 %, which is considered normal in the human genome [40]. Another promising prognostic marker, analyzed in the peripheral blood instead of the tumor tissue, is the methylation analysis of the SEPT9 gene, whose product is involved in embryo- and carcinogenesis. Not every patient agrees to undergo a prophylactic colonoscopy. Therefore, an interesting alternative to this examination is the assessment of the methylation status of the SEPT9 gene, in which the level of methylation is correlated with the probability of developing CRC [41]. Although the assessment of the methylation status of the SEPT9 gene has no therapeutic benefits, it appears to be a good screening test because of its low invasiveness.
2.4 Prognostic Biomarkers: Circulating Tumor Cells (CTCs)
The presence of CTCs in the peripheral blood is a significant prognostic factor. Some cells that form the tumor acquire the ability to detach from the tumor mass and migrate across the blood vessel wall to the peripheral blood [42]. Chang et al. estimated that approximately 106 separating tumor cells per gram of tumor mass come into contact with blood [43]. It is thought that the presence of tumor cells in the peripheral blood is strongly correlated with metastasis and the formation of secondary tumor foci. Therefore, even a very small quantity of CTCs, such as 1 cell in a 10-ml blood sample, has poor prognosis [44–46]. One of the techniques allowing the determination of the quantity of CTCs is the CellSearch system, capable of detecting as little as 1 cell in 7.5 ml of blood [47]. After their analysis of literature data, Wang et al. [48] estimated that the presence of five or more CTCs correlates with a short overall survival in patients with prostate cancer.
3 Predictive Biomarkers
Predictive biomarkers provide information on the probability of obtaining a response to treatment [49] and support the process of therapeutic decisions [50]. The most important predictive biomarkers with a recognized clinical value in the molecular diagnostics in oncology are somatic mutations, of both the point mutation and chromosomal aberration types, in the following genes: EGFR, KRAS, BRAF, PDGFRA, KIT, HER2, BCR-ABL, and EML4-ALK. These key mutations are considered as predictors for certain therapies, as shown in Fig. 1. The analysis of gene expression or methylation is more common in research studies than in routine diagnostics, with minor exceptions, such as the TheraPrint microarray analysis that allows the assessment of the expression of selected breast cancer genes for the prediction of the response to treatment.
3.1 Predictive Biomarkers: Analysis of Mutations and Polymorphisms
Somatic mutations in the KRAS gene are some of the earliest incidents in the process of colorectal carcinogenesis. The first reports indicating a correlation between the KRAS mutations and the development of this tumor type appeared as early as in 1988 [51]. The most common mutations occur in codons 12 and 13; those less common occur in codons 61 and 146 [52]. The assessment of the mutation status in codons 12 and 13 of the KRAS gene is a standard predictive biomarker in the evaluation of the eligibility of patients with advanced CRC for targeted therapy using monoclonal antibodies such as cetuximab or panitumumab, directed against the extracellular domain of epidermal growth factor receptor (EGFR) [49, 53]. EGFR stimulates the growth and survival of CRC cells (via signaling involving the MAPK, PIK3 and JAK/STAT pathways) [50]. As a result of treatment, the proliferation of tumor cells is inhibited, while the rate of their apoptosis is increased. Therefore, the lack of mutations in codons 12 and 13 of the KRAS gene has a positive predictive value [49, 50]. On the other hand, one has to remember that the patients carrying no mutations in codons 12 and 13 of the KRAS gene but exhibiting a high level of EGFR expression may present a worse response to treatment if the presence of somatic mutations in codons 61 or 146 of the KRAS gene, or the V600E somatic mutation in the BRAF gene, has been determined in their tumor cells [52]. Analyses of mutations in CRC indicate the incidence rate of both the V600E mutation in the BRAF gene and mutations in codon 61 of the KRAS gene as 10 % [54]. Therefore, quick diagnostic tests based on molecular biology techniques have been implemented in the assessment of mutations in both the KRAS gene (codons 12, 13 and 61) and the BRAF gene (V600E), while the use of the very sensitive real-time PCR method seems proper, especially when DNA has to be extracted from tissues with a low-density carcinoma pattern, in which the limit of detection of Sanger sequencing or the traditional single strand conformation polymorphism (SSCP) methods return false negative results [54]. Regardless of the mutations in the KRAS gene, the response to the monoclonal antibody therapy of metastatic CRC may also be decreased by mutations in the PIK3CA gene and deletions in the PTEN gene [55]. Perhaps future recommendations regarding targeted therapy will also include the assessment of mutations in the BRAF, PIK3 and PTEN genes.
Thymidylate synthase (TS) is a critical enzyme for DNA synthesis, so it is an important molecular target for many chemotherapeutics, including 5-fluorouracil (5-FU). TS overexpression is related to resistance to 5-FU. As has been demonstrated, the presence of three copies of tandem repeats (TSER*3) in the TS promoter region induces increased TS expression, as compared with the presence of only two copies of tandem repeats (TSER*2). Moreover, a better response to 5-FU therapy has been observed in patients with the TSER*2/TSER*2 or TSER*2/TSER*3 genotype. Such patients also exhibited increased overall survival, as compared with those with the TSER*3 homozygotes. One should remember that the overexpression of TS (related to a high number of copies of the gene) results in a failure in 5-FU therapy and shorter overall survival rates in patients with metastatic colorectal cancer (mCRC) [56].
The catabolism of 5-FU is mediated by dihydropyrimidine dehydrogenase (DPD), which is approved by the FDA as a pharmacogenetic biomarker. In patients with DPD deficit, a high toxicity of 5-FU occurs, which in some cases leads to death [57, 58]. The most significant mutation leading to the loss of enzymatic activity by the DPD protein is the G>A transition at a donor splice site (IVS14+1G>A), which, in consequence, causes the skipping of exon 14 [59]. Genetic analyses of the presence of the DPD polymorphism (IVS14+1G>A) allow patient qualification to groups of high or low risk of grade 3 or 4 toxicity during 5-FU therapy. Other studies conducted in patients with grade 4 neutropenia demonstrated that 50 % of patients are homozygous or heterozygous with respect to IVS14+1G>A [60].
Microsatellite instability (MSI) analysis of the tumor can provide prognostic and predictive information as well. The results encompassing grade 2 and 3 CRC patients in a single-factor analysis revealed that in patients with stable microsatellites (MSS) or with low-frequency MSI, the fluorouracil therapy had more benefits than in CRC patients with high-frequency MSI (MSI+) (in the latter case, fluorouracil did not result in disease improvement or even cause the disease to worsen) [61].
An opposite tendency is observed with the use of camptothecin-11 (CPT-11), a topoisomerase I inhibitor that eliminates those MSI+ tumor cells, that have damaged genes involved in mismatch system repair (MMR) [62]. However, the predictive value of the MSI status is under discussion, and currently it is not commonly used in oncological practice [50].
In patients with the non-small-cell lung carcinoma (NSCLC), the assessment of mutations in the EGFR kinase domain may be used as a predictive biomarker. It has been proved that somatic mutations in exons 19 or 21 are related to tumor sensitivity to therapies with tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib. A higher median survival rate was observed in the case of exon deletion in exon 19 than in the case of the point mutation L858R in exon 21 [63].
In the Polish population, activating mutations (those in exon 19 and substitution L858R in exon 21) of the EGFR gene were found in 12 % of patients with diagnosed adenocarcinoma during the analysis of deletions in exon 19 and the substitution L858R [64], as well as in 13 % of patients with the same diagnosis but during the analysis of 29 mutations in exons 18, 19, 20 and 21 of the EGFR gene [53]. Unfortunately, NSCLC cells susceptible to TKIs may acquire resistance over time. It has been demonstrated that in nearly half of the cases of lung adenocarcinoma, apart from the mutations sensitizing the tumor to TKIs, the somatic mutation T790M occurs in the EGFR kinase domain of exon 20 [65]. The substitution of threonine for methionine at position 790 increases the affinity of adenosine triphosphate (ATP), which is the main mechanism of formation of drug resistance [66]. Moreover, in approximately 20 % of cases, the MET gene amplification, independent of the mutation T790M in the EGFR gene, may be a potential cause of drug resistance. This may generate acquired resistance via the activation of the ERBB signaling pathway. Studies employing the XL880 molecules (inhibitors of MET kinase) and small interfering RNA (siRNA) preventing MET expression demonstrated that in the H820 cell line, ERBB signaling is highly dependent on the activity of MET. The results of the analysis confirm the promising potential of low molecular weight inhibitors in patients suffering from lung adenocarcinoma with acquired resistance to EGFR inhibitors but carrying multiple copies of the MET gene [67]. In 2007, Soda et al. [68], for the first time in NSCLC patients, identified a small inversion in the p region of chromosome 2, whose consequence was the formation of the fusion gene EML4-ALK. The study revealed the presence of the inversion in 6.7 % of patients; the study group included 75 Japanese patients [68]. In studies involving larger groups (n = 266), a lower incidence of 4.9 % is indicated [69].
One has to remember that the aberration EML4-AKL occurs in different histological types of NSCLC in nonsmoking patients and most often does not occur together with the mutations in the EGFR and KRAS genes [69]. The translocation plays the leading role in tumor development in this group of patients. In the studies involving transgenic mice carrying the overexpressed EML4-ALK fusion gene as the model organism, hundreds of adenocarcinoma tumors developed in both lungs within a few weeks after birth. This led the authors to conduct further studies of the dominating role of this oncogene in lung carcinogenesis [70]. Subsequent studies of ALK and MET inhibitors led to the quick introduction of crizotinib into the market (2011), while the predictive assessment of the fusion gene EML4-ALK, using fluorescence in situ hybridization (FISH) for paraffin-embedded material or reverse transcriptase PCR (RT-PCR) for cytological material [71], is of key importance for the evaluation of the patients’ eligibility for treatment with this selective kinase inhibitor.
Another example of a fusion gene as a predictive biomarker is the presence of BCR-ABL, the key fusion gene for targeted therapy, in patients with chronic myeloid leukemia (CML). The expression of the newly formed chimeric protein, the tyrosine kinase BCR-ABL, is possible thanks to a translocation between chromosomes 9 and 22 [72], whereas the predictive assessment of the presence of this protein has allowed the evaluation of the patients’ eligibility for treatment with imatinib for more than 10 years.
Another interesting predictive biomarker is the assessment of somatic mutations in codon 600 of the BRAF gene in patients with advanced melanoma. For many years, the only medicine approved by the FDA in this indication was dacarbazine (DTIC), and the response to this therapy was very poor, approximately 10 % [73]. The identification of the most common mutation in codon 600 of the BRAF gene (V600E) and the introduction of its strong inhibitor, vemurafenib, into therapy marked the breakthrough in the oncological approach to this tumor type. The BRAF gene encodes a protein of the raf/mil family of serine-threonine kinases. It is involved in the regulation of the MAP kinase and the ERK signaling path, which affects cell division, differentiation and secretion. Missense somatic mutations in the BRAF gene (V600E/K/D/R/M) occur in many tumor types. However, the highest incidence rate has been reported in malignant melanoma (66 %), most of which is represented by the mutation V600E (c. 1799 T>A) [74].
The results of phase III clinical trials have proven that vemurafenib decreased the risk of death by 63 % and the risk of tumor progression by 74 % in untreated inoperable patients with stage III or IV melanoma carrying the mutation V600E, as compared with dacarbazine. The response to the therapy was observed in 48 % of the vemurafenib-treated patients, as compared with 5 % of patients treated with dacarbazine. Moreover, the study group included patients with the mutations V600K and V600D. As many as 40 % of patients with the mutation V600K also responded to the treatment with vemurafenib [73, 75]; therefore, it seems important to determine not only the most common mutation, V600E, but also other known activating mutations, either using the pyrosequencing technique [76] or real-time PCR (Entrogen, Inc).
Unfortunately, as in the case of NSCLC and CRC, the problem of the resistance of melanomas to the aforementioned inhibitors is becoming noticeable. Studies of drug-resistant clones derived from the cell lines of the BRAF V600E M14 melanoma treated with the inhibitor AZ628 revealed an increased activity of the CRAF protein. In light of these studies, the CRAF protein may gain a high predictive value in the assessment of efficacy of BRAF inhibitor therapy. The potential predictive value has also been assessed for geldanamycin (tumor cells with a high level of CRAF expression were highly susceptible to this medicine) [77]. Further molecular studies related to personalized medicine revealed somatic point mutations in the MEK1 gene (in a cell line of the BRAF V600E A375 melanoma), whose presence contributes to the resistance to the MEK inhibitor (AZD6244) [78], as well as a somatic mutation in the KIT oncogene, characteristic of mucosal melanomas (21 %), acral melanomas (11 %) and melanomas related to chronic sun-induced damage (16.7 %). Moreover, the mutations in the KIT gene are observed in melanomas susceptible to imatinib [79], which makes the gene a potential therapeutic target, studied for almost 10 years.
Breast cancer is the most common malignant tumor in women, and the studies aimed at identifying its predictive biomarkers have been conducted for over 10 years. The predictive biomarkers listed below include both polymorphisms and somatic mutations.
The main medicine used to treat breast cancer is tamoxifen. The polymorphism CYP2D6*10/*10 (and CYP2D6*5/*10) is related to a lower concentration of endoxifen (active metabolite of tamoxifen) and a high concentration of N-desmethyltamoxifen (NDM). This means that NDM accumulation in blood plasma is a direct consequence of the damage to the metabolic conversion of NDM into endoxifen [80]. The correlation between CYP2D6*10/*10 and the low concentrations of main active metabolites of tamoxifen (like endoxifen and 4-hydroxytamoxifen), as well as the suggestion of a correlation between these results and the weaker response to tamoxifen in the treated female patients, have been mentioned before [81]. Some studies appear to confirm this hypothesis. Xu et al. [82], for the first time, studied this correlation in the female population in China. In patients who were homozygous with respect to the CYP2D6*10 variant T/T, a considerably lower concentration of 4-hydroxytamoxifen was reported, as compared with the control group of wt C/C homozygotes. Subsequently, the effect of this polymorphism on the therapy was investigated. As expected, the CYP2D6*10 T/T patients had worse clinical results than those with the C/C and C/T genotypes. Importantly, among the nontreated women, no effect of this genetic alteration on the survival was observed [82]. On the other hand, in a study investigating the correlation between the alleles CYP2D6*1, *4, *5, *9, *10, *41, and *UM and overall survival or breast cancer-specific survival (BCSS) in 3,155 patients treated with tamoxifen and 3,485 nontreated patients during the period of 7 years, no statistically significant correlations were found [83]. A similar lack of correlation was observed in the Japanese population of female breast cancer patients receiving tamoxifen as adjuvant therapy and carrying the genotype CYP2D6*10 [84].
Similarly, the analysis of the HER2 gene amplification is the basic genetic test used in cancer diagnostics for the evaluation of the eligibility of breast cancer patients for treatment with trastuzumab or lapatinib. Unfortunately, trastuzumab therapy does not always induce the desired response in patients. Studies conducted with the NIH3T3 and MCF-7 tumor cell lines indicate that the aforementioned drug resistance may be related to the presence of the isoform HERΔ16, which, once coupled with Src kinase, increases its metastatic and oncogenic properties. In such circumstances, one of the proposed strategies involves the use of appropriate inhibitors to separate the HER and kinase pathways. In the described experiment, the use of dasatinib TKI resulted in the inhibition of Src kinase [85].
3.2 Predictive Biomarkers: Analysis of Gene and miRNA Expression
An example of RNA expression analysis as a predictive biomarker is the analysis of the transcript of the ERCC1 gene encoding the key enzyme for DNA repair. Due to the poor prognosis for patients with HCC, prognostic and predictive markers are highly desired. Increased ERCC1 expression accompanies hepatic fibrinogenesis and neoplasia. Similarly to NSCLC, it has been demonstrated that in patients with HCC, higher ERCC1 expression is related to resistance to cisplatin. Apart from ERCC1, the analysis of mTOR and c-erb-B2 expression may as well provide information on the aggressiveness of the tumor due to the involvement of these genes in hepatocarcinogenesis [86].
In light of the small number of predictive markers practically applicable in the analysis of RNA expression, the use of miRNA diagnostics is particularly promising (Fig. 1). Studies involving the MCF-7 breast cancer cell lines exhibiting the expression of the aforementioned isoform HER2Δ16 suggest a relationship between miRNA-342 and the response to tamoxifen. It has been demonstrated that miRNA-324 is present in the tumor cells susceptible to this chemotherapeutic, and its suppression correlates with resistance. Since clinical data indicate a correlation between lower miRNA-342 expression and resistance to the medicine, restoring the expression might constitute a new and efficacious therapeutic approach [87].
Deregulation of the miRNA levels plays an equally important role in bladder cancer, in which it is correlated with dysplasia and may become both a prognostic and a predictive biomarker. An analysis of over 600 miRNAs from tumor tissues derived from patients with bladder cancer treated with cisplatin allowed the narrowing of the signature to 15 miRNAs that correlate with the response to chemotherapy, five miRNAs that correlate with survival time, and three miRNAs (886-3p, 923, 944) that correlate with both factors [88].
3.3 Predictive Biomarkers: Analysis of Methylation
One of the most recognized predictive factors is the methylation of the MGMT promoter (Fig. 1). The current standard treatment of patients with glioblastoma includes surgeries followed by radiation therapy and chemotherapy involving the use of DNA alkylating agents capable of an efficient penetration of the blood–brain barrier (temozolomide [TMZ]) [89]. It has been proven that such a combination is much more efficacious than the radiation therapy alone [90]. Moreover, this treatment option is strongly predicted by the methylation of the MGMT promoter [90–92] (Fig. 1), which is beneficial because of its relatively common use.
Methylations are observed in nearly 40 % [93] to 44 % [94] of all glioblastoma cases, but there are subgroups characterized by a higher frequency of methylation (more common in women than in men: 53 vs. 39 %); moreover, the MGMT promoter methylation occurs much more frequently in secondary tumors than in primary tumors (73 vs. 43 %) [94].
MGMT is an enzyme responsible for removing methylated adducts from the O-6 position of guanine, which is one of the main biological targets for alkylating agents [91]. The high repair activity of MGMT ‘protects’ cancer cells from chemotherapeutics such as carmustine or the above mentioned TMZ [91, 92]. A comparative study revealed a positive correlation between MGMT methylation and the response to carmustine (including overall survival and disease-free survival, as compared with the response to the drug in the patients with no methylation).
Earlier in vitro studies demonstrated that the methylation of the CpG islands in the MGMT promoter region is responsible for the silencing of the gene [95–97]. Further studies revealed a correlation between the loss of expression and a high frequency of primary tumors. In an analysis of over 500 primary tumors, it was determined that the highest percentage of methylation occurs in gliomas and CRC (40 % each), as well as in NSCLC, leukemias and head and neck tumors (25 % each) [98]. Thus the inactivation of the MGMT gene is a common epigenetic mechanism in malignant tumor progression [91]. Therefore, when the MGMT promoter undergoes methylation resulting in the inhibition of MGMT expression, the effect of the medicine (TMZ) is more pronounced as the results of its activity are not repaired by O-6-methylguanine-DNA methyltransferase [89, 99].
The determination of the methylation status of the MGMT promoter instead of the analysis of gene or protein expression has the technical advantage of not being susceptible to contamination by the lymphocytes infiltrating the tumor or normal tissue. Since the MGMT promoter in normal tissue is not methylated, the analysis returns simple results, which enables its inclusion in routine diagnostics [91].
3.4 Predictive Biomarkers: Circulating Tumor Cells (CTCs)
Presence or absence of CTCs brings prognostic value. Assessment of CTC count before, during and after therapies at different times also enables prediction of treatment outcome [4, 100]. Although the amount of isolated CTCs from patient samples are very low, some researchers have evaluated new methods for highly efficient enrichment of CTCs in order to perform gene expression profiling [101]. Molecular characterization of CTCs would be helpful to predict response to therapy. For example, Reinholz et al. [102] demonstrated that decreased mammaglobin 1 (MGB1) mRNA level in CTCs collected from metastatic breast cancer patients may help predict response to therapy.
4 Conclusions
Molecular diagnostics is one of the key tools allowing the personalization of oncology [103]. However, it is important that the clinical tests allowing the assessment of the information provided concurrently by prognostic and predictive markers permit quicker diagnoses and accurate choices of therapeutic procedures. Predictive biomarkers can provide information helpful in avoiding the toxicity of standard (systemic) therapies and in evaluating the eligibility of patients for targeted therapy [49]. The greatest number of biomarkers has been found for breast cancer, prostate cancer and lung cancer, as they are the most common tumor types in general population. However, to date, this has not led to a significant extension in patient survival, as these are malignant metastatic tumors that have a poor prognosis overall. The development of high-throughput technologies, such as microarrays, new generation sequencing methods and mass spectrometry focused on nucleic acids, allows the use of a growing range of DNA biomarker analyses (e.g., Onco DEEP, Onco TRACE) [104]. Last but not least, The Cancer Genome Atlas (TCGA) project creates the possibility to compare whole genomes of the same cancer from different patients and can be used to identify novel biomarkers, for example, prognostic presence of IDH1 p.R132H mutation in glioma [105, 106].
References
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75. doi:10.1038/nature07423.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8. doi:10.1038/nature05610.
Wick W, Meisner C, Hentschel B, Platten M, Schilling A, Wiestler B, Sabel MC, Koeppen S, Ketter R, Weiler M, Tabatabai G, von Deimling A, Gramatzki D, Westphal M, Schackert G, Loeffler M, Simon M, Reifenberger G, Weller M. Prognostic or predictive value of MGMT promoter methylation in gliomas depends on IDH1 mutation. Neurology. 2013;81(17):1515–22. doi:10.1212/WNL.0b013e3182a95680.
de Albuquerque A, Kubisch I, Stolzel U, Ernst D, Boese-Landgraf J, Breier G, Stamminger G, Fersis N, Kaul S. Prognostic and predictive value of circulating tumor cell analysis in colorectal cancer patients. J Transl Med. 2012;10:222. doi:10.1186/1479-5876-10-222.
Ziegler A, Koch A, Krockenberger K, Grosshennig A. Personalized medicine using DNA biomarkers: a review. Hum Genet. 2012;131(10):1627–38. doi:10.1007/s00439-012-1188-9.
Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, Stubbs H, McDermott U, Settleman J, Kwak EL, Clark JW, Isakoff SJ, Sequist LV, Engelman JA, Lynch TJ, Haber DA, Louis DN, Ellisen LW, Borger DR, Iafrate AJ. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–58. doi:10.1002/emmm.201000070.
Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, Maher ER, Stanley AJ, Harnden P, Joyce A, Knowles M, Selby PJ. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006;66(4):2000–11. doi:10.1158/0008-5472.CAN-05-3074.
Young AC, Craven RA, Cohen D, Taylor C, Booth C, Harnden P, Cairns DA, Astuti D, Gregory W, Maher ER, Knowles MA, Joyce A, Selby PJ, Banks RE. Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res. 2009;15(24):7582–92. doi:10.1158/1078-0432.CCR-09-2131.
Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol. 2011;7(10):569–80. doi:10.1038/nrendo.2011.142.
Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, Herrero A, Fusco A, Cameselle-Teijeiro J, Santoro M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65(22):10199–207. doi:10.1158/0008-5472.CAN-04-4259.
Chaiteerakij R, Addissie BD, Roberts LR. Update on biomarkers of hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2013. doi:10.1016/j.cgh.2013.10.038.
Lin KT, Shann YJ, Chau GY, Hsu CN, Huang CY. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene. 2013. doi:10.1038/onc.2013.424.
Yang N, Kaur S, Volinia S, Greshock J, Lassus H, Hasegawa K, Liang S, Leminen A, Deng S, Smith L, Johnstone CN, Chen XM, Liu CG, Huang Q, Katsaros D, Calin GA, Weber BL, Butzow R, Croce CM, Coukos G, Zhang L. MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008;68(24):10307–14. doi:10.1158/0008-5472.CAN-08-1954.
Yang H, Kong W, He L, Zhao JJ, O’Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68(2):425–33. doi:10.1158/0008-5472.CAN-07-2488.
Panczyk MMM. Farmakogenetyka—znaczenie w chemioterapii raka jelita grubego. Nowotwory. 2008;58(1):62–9.
Paduch RKJ. Markery Nowotworowe. Onkologia Polska. 2003;6(2):77–82.
Bates SE. Clinical applications of serum tumor markers. Ann Intern Med. 1991;115(8):623–38.
Cui J, Antoniou AC, Dite GS, Southey MC, Venter DJ, Easton DF, Giles GG, McCredie MR, Hopper JL. After BRCA1 and BRCA2-what next? Multifactorial segregation analyses of three-generation, population-based Australian families affected by female breast cancer. Am J Human Genet. 2001;68(2):420–31. doi:10.1086/318187.
Synowiec EMA, Krupa R, Woźniak K. Gene’s polymorphism of DNA double-stranded repair in breast cancer. PostBiolKom. 2010;37(2):433–8.
Gong M, Dong W, Shi Z, Xu Y, Ni W, An R. Genetic polymorphisms of GSTM1, GSTT1, and GSTP1 with prostate cancer risk: a meta-analysis of 57 studies. PLoS ONE. 2012;7(11):e50587. doi:10.1371/journal.pone.0050587.
Samzadeh M, Hasanzad M, Jamaldini SH, Haghdoost AA, Afshari M, Ziaee SA. Association of G/A polymorphism, rs266882, in AREI region of the prostate-specific antigen gene with prostate cancer risk and clinicopathological features. Urol J. 2012;9(4):691–9.
Plawski A, Slomski R. APC gene mutations causing familial adenomatous polyposis in Polish patients. J Appl Genet. 2008;49(4):407–14. doi:10.1007/BF03195640.
Xu J, Zheng SL, Isaacs SD, Wiley KE, Wiklund F, Sun J, Kader AK, Li G, Purcell LD, Kim ST, Hsu FC, Stattin P, Hugosson J, Adolfsson J, Walsh PC, Trent JM, Duggan D, Carpten J, Gronberg H, Isaacs WB. Inherited genetic variant predisposes to aggressive but not indolent prostate cancer. P Natl Acad Sci USA. 2010;107(5):2136–40. doi:10.1073/pnas.0914061107.
Ferlay J, Shin H, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v2.0, Cancer incidence and mortality worldwide: IARC CancerBase No. 10 [Internet]. International Agency for Research on Cancer, 2010; 2008.
Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, Minasian LM, Ford LG, Lippman SM, Crawford ED, Crowley JJ, Coltman CA Jr. Prevalence of prostate cancer among men with a prostate-specific antigen level < or =4.0 ng per milliliter. N Engl J Med. 2004;350(22):2239–46. doi:10.1056/NEJMoa031918.
Barry MJ. Clinical practice. Prostate-specific-antigen testing for early diagnosis of prostate cancer. N Engl J Med. 2001;344(18):1373–7. doi:10.1056/NEJM200105033441806.
Strittmatter F, Stieber P, Nagel D, Fullhase C, Walther S, Stief CG, Waidelich R. Detection of prostate cancer with complexed PSA and complexed/total PSA ratio—is there any advantage? Eur J Med Res. 2011;16(10):445–50.
Liong ML, Lim CR, Yang H, Chao S, Bong CW, Leong WS, Das PK, Loh CS, Lau BE, Yu CG, Ooi EJ, Nam RK, Allen PD, Steele GS, Wassmann K, Richie JP, Liew CC. Blood-based biomarkers of aggressive prostate cancer. PLoS ONE. 2012;7(9):e45802. doi:10.1371/journal.pone.0045802.
van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–36. doi:10.1038/415530a.
Bueno-de-Mesquita JM, Linn SC, Keijzer R, Wesseling J, Nuyten DS, van Krimpen C, Meijers C, de Graaf PW, Bos MM, Hart AA, Rutgers EJ, Peterse JL, Halfwerk H, de Groot R, Pronk A, Floore AN, Glas AM, Van’t Veer LJ, van de Vijver MJ. Validation of 70-gene prognosis signature in node-negative breast cancer. Breast Cancer Res Treat. 2009;117(3):483–95. doi:10.1007/s10549-008-0191-2.
Nguyen B, Cusumano PG, Deck K, Kerlin D, Garcia AA, Barone JL, Rivera E, Yao K, de Snoo FA, van den Akker J, Stork-Sloots L, Generali D. Comparison of molecular subtyping with BluePrint, MammaPrint, and TargetPrint to local clinical subtyping in breast cancer patients. Ann Surg Oncol. 2012;19(10):3257–63. doi:10.1245/s10434-012-2561-6.
Dabbs DJ, Klein ME, Mohsin SK, Tubbs RR, Shuai Y, Bhargava R. High false-negative rate of HER2 quantitative reverse transcription polymerase chain reaction of the Oncotype DX test: an independent quality assurance study. J Clin Oncol. 2011;29(32):4279–85. doi:10.1200/JCO.2011.34.7963.
Qi J, Wang J, Katayama H, Sen S, Liu SM. Circulating microRNAs (cmiRNAs) as novel potential biomarkers for hepatocellular carcinoma. Neoplasma. 2013;60(2):135–42.
Jones CI, Zabolotskaya MV, King AJ, Stewart HJ, Horne GA, Chevassut TJ, Newbury SF. Identification of circulating microRNAs as diagnostic biomarkers for use in multiple myeloma. Br J Cancer. 2012;107(12):1987–96. doi:10.1038/bjc.2012.525.
Wotschofsky Z, Busch J, Jung M, Kempkensteffen C, Weikert S, Schaser KD, Melcher I, Kilic E, Miller K, Kristiansen G, Erbersdobler A, Jung K. Diagnostic and prognostic potential of differentially expressed miRNAs between metastatic and non-metastatic renal cell carcinoma at the time of nephrectomy. Clin Chim Acta. 2013;416:5–10. doi:10.1016/j.cca.2012.11.010.
Han ZB, Chen HY, Fan JW, Wu JY, Tang HM, Peng ZH. Up-regulation of microRNA-155 promotes cancer cell invasion and predicts poor survival of hepatocellular carcinoma following liver transplantation. J Cancer Res Clin Oncol. 2012;138(1):153–61. doi:10.1007/s00432-011-1076-z.
Lotte Christensen L, Tobiasen H, Holm A, Schepeler T, Ostenfeld MS, Thorsen K, Rasmussen MH, Birkenkamp-Demtroeder K, Sieber OM, Gibbs P, Lubinski J, Lamy P, Laurberg S, Oster B, Hansen KQ, Hagemann-Madsen R, Byskov K, Orntoft TF, Andersen CL. MiRNA-362-3p induces cell cycle arrest through targeting of E2F1, USF2 and PTPN1 and is associated with recurrence of colorectal cancer. Int J Cancer. 2012. doi:10.1002/ijc.28010.
Decock A, Ongenaert M, Hoebeeck J, De Preter K, Van Peer G, Van Criekinge W, Ladenstein R, Schulte JH, Noguera R, Stallings RL, Van Damme A, Laureys G, Vermeulen J, Van Maerken T, Speleman F, Vandesompele J. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 2012;13(10):R95. doi:10.1186/gb-2012-13-10-r95.
Xu J, Shetty PB, Feng W, Chenault C, Bast RC Jr, Issa JP, Hilsenbeck SG, Yu Y. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer. 2012;12:243. doi:10.1186/1471-2407-12-243.
Vasiljevic N, Ahmad AS, Beesley C, Thorat MA, Fisher G, Berney DM, Moller H, Yu Y, Lu YJ, Cuzick J, Foster CS, Lorincz AT. Association between DNA methylation of HSPB1 and death in low Gleason score prostate cancer. Prostate Cancer Prostatic Dis. 2012. doi:10.1038/pcan.2012.47.
Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, Roberts WL, Fang JC, Samowitz WS, Heichman KA. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011;9:133. doi:10.1186/1741-7015-9-133.
Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Res. 2008;10(Suppl 1):S2. doi:10.1186/bcr1988.
Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA. 2000;97(26):14608–13. doi:10.1073/pnas.97.26.14608.
Magnowski P, Bochynski H, Nowak-Markwitz E, Zabel M, Spaczynski M. Circulating tumor cells (CTCs)—clinical significance in patients with ovarian cancer. Ginekol Pol. 2012;83(4):291–4.
Muller V, Riethdorf S, Rack B, Janni W, Fasching PA, Solomayer E, Aktas B, Kasimir-Bauer S, Pantel K, Fehm T. Prognostic impact of circulating tumor cells assessed with the Cell Search System and AdnaTest Breast in metastatic breast cancer patients: the DETECT study. Breast Cancer Res. 2012;14(4):R118. doi:10.1186/bcr3243.
Lucci A, Hall CS, Lodhi AK, Bhattacharyya A, Anderson AE, Xiao L, Bedrosian I, Kuerer HM, Krishnamurthy S. Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol. 2012;13(7):688–95. doi:10.1016/S1470-2045(12)70209-7.
Kołacińska APM, Morawiec Z. Novel diagnostic tests for breast cancer. Oncol Clin Pract. 2007;3(2):56–8.
Wang FB, Yang XQ, Yang S, Wang BC, Feng MH, Tu JC. A higher number of circulating tumor cells (CTC) in peripheral blood indicates poor prognosis in prostate cancer patients—a meta-analysis. Asian Pac J Cancer Prev. 2011;12(10):2629–35.
Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9(7):489–99. doi:10.1038/nrc2645.
Voon PJ, Kong HL. Tumour genetics and genomics to personalise cancer treatment. Ann Acad Med Singap. 2011;40(8):362–8.
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–32. doi:10.1056/NEJM198809013190901.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101(4):715–21. doi:10.1038/sj.bjc.6605177.
Lewandowska MA, Jóźwicki W, Starzynski J, Kowalewski J. Analysis of EGFR mutation frequency and coexistence of KRAS and EGFR mutations using RT-PCR in lung adenocarcinoma: may a clinical and pathological model of a patient’s qualification for targeted therapy have an impact on time to obtain genetic results? Pol J Cardio Thorac Surg. 2012;9(4):443–51.
Lewandowska MA, Żurawski B, Jozwicki W. KRAS and BRAF mutation analysis in colorectal adenocarcinoma specimens with a low percentage of tumor cells. Mol Diagn Ther. 2013;17(3):193–203. doi:10.1007/s40291-013-0025-8.
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–7. doi:10.1158/0008-5472.CAN-08-2466.
Jiang WQ, Fu FF, Li YX, Wang WB, Wang HH, Jiang HP, Teng LS. Molecular biomarkers of colorectal cancer: prognostic and predictive tools for clinical practice. J Zhejiang Univ Sci B. 2012;13(9):663–75. doi:10.1631/jzus.B1100340.
Gross E, Busse B, Riemenschneider M, Neubauer S, Seck K, Klein HG, Kiechle M, Lordick F, Meindl A. Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PLoS ONE. 2008;3(12):e4003. doi:10.1371/journal.pone.0004003.
Wei X, McLeod HL, McMurrough J, Gonzalez FJ, Fernandez-Salguero P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J Clin Invest. 1996;98(3):610–5. doi:10.1172/JCI118830.
Raida M, Schwabe W, Hausler P, Van Kuilenburg AB, Van Gennip AH, Behnke D, Hoffken K. Prevalence of a common point mutation in the dihydropyrimidine dehydrogenase (DPD) gene within the 5’-splice donor site of intron 14 in patients with severe 5-fluorouracil (5-FU)-related toxicity compared with controls. Clin Cancer Res. 2001;7(9):2832–9.
Van Kuilenburg AB, Meinsma R, Zoetekouw L, Van Gennip AH. Increased risk of grade IV neutropenia after administration of 5-fluorouracil due to a dihydropyrimidine dehydrogenase deficiency: high prevalence of the IVS14+1g>a mutation. Int J Cancer. 2002;101(3):253–8. doi:10.1002/ijc.10599.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–57. doi:10.1056/NEJMoa022289.
Bras-Goncalves RA, Rosty C, Laurent-Puig P, Soulie P, Dutrillaux B, Poupon MF. Sensitivity to CPT-11 of xenografted human colorectal cancers as a function of microsatellite instability and p53 status. Br J Cancer. 2000;82(4):913–23. doi:10.1054/bjoc.1999.1019.
Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(3 Pt 1):839–44. doi:10.1158/1078-0432.CCR-05-1846.
Krawczyk P, Ramlau R, Powrózek T, Wojas-Krawczyk K, Sura S, Jarosz B, Walczyna B, Pankowski J, Szumiło J, Dyszkiewicz W, Woźniak A, Milanowski J. Wykrywalność mutacji w genie EGFR u chorych na niedrobnokomórkowego raka płuca w wybranych ośrodkach w Polsce zaangażowanych w diagnostykę molekularną. Pol J Cardio Thorac Surg. 2012;9(4):431–8. doi:10.5114/kitp.2012.32680.
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi:10.1371/journal.pmed.0020073.
Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105(6):2070–5. doi:10.1073/pnas.0709662105.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104(52):20932–7. doi:10.1073/pnas.0710370104.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi:10.1038/nature05945.
Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115(8):1723–33. doi:10.1002/cncr.24181.
Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, Haruta H, Hamada T, Yamashita Y, Ishikawa Y, Sugiyama Y, Mano H. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA. 2008;105(50):19893–7. doi:10.1073/pnas.0805381105.
Soda M, Isobe K, Inoue A, Maemondo M, Oizumi S, Fujita Y, Gemma A, Yamashita Y, Ueno T, Takeuchi K, Choi YL, Miyazawa H, Tanaka T, Hagiwara K, Mano H. A prospective PCR-based screening for the EML4-ALK oncogene in non-small cell lung cancer. Clin Cancer Res. 2012;18(20):5682–9. doi:10.1158/1078-0432.CCR-11-2947.
Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi:10.1056/NEJMoa062867.
Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, Begg CB, Agarwala SS, Schuchter LM, Ernstoff MS, Houghton AN, Kirkwood JM. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745–51.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi:10.1038/nature00766.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi:10.1056/NEJMoa1103782.
Spittle C, Ward MR, Nathanson KL, Gimotty PA, Rappaport E, Brose MS, Medina A, Letrero R, Herlyn M, Edwards RH. Application of a BRAF pyrosequencing assay for mutation detection and copy number analysis in malignant melanoma. J Mol Diagn. 2007;9(4):464–71. doi:10.2353/jmoldx.2007.060191.
Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–61. doi:10.1158/0008-5472.CAN-07-6787.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, MacConaill LE, Zhang J, Gray NS, Sellers WR, Dummer R, Garraway LA. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106(48):20411–6. doi:10.1073/pnas.0905833106.
Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–6. doi:10.1200/JCO.2006.06.2984.
Lim JS, Chen XA, Singh O, Yap YS, Ng RC, Wong NS, Wong M, Lee EJ, Chowbay B. Impact of CYP2D6, CYP3A5, CYP2C9 and CYP2C19 polymorphisms on tamoxifen pharmacokinetics in Asian breast cancer patients. Br J Clin Pharmacol. 2011;71(5):737–50. doi:10.1111/j.1365-2125.2011.03905.x.
Lim HS, Ju Lee H, Seok Lee K, Sook Lee E, Jang IJ, Ro J. Clinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancer. J Clin Oncol. 2007;25(25):3837–45. doi:10.1200/JCO.2007.11.4850.
Xu Y, Sun Y, Yao L, Shi L, Wu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, He L, Li P, Xie Y. Association between CYP2D6 *10 genotype and survival of breast cancer patients receiving tamoxifen treatment. Ann Oncol. 2008;19(8):1423–9. doi:10.1093/annonc/mdn155.
Abraham JE, Maranian MJ, Driver KE, Platte R, Kalmyrzaev B, Baynes C, Luccarini C, Shah M, Ingle S, Greenberg D, Earl HM, Dunning AM, Pharoah PD, Caldas C. CYP2D6 gene variants: association with breast cancer specific survival in a cohort of breast cancer patients from the United Kingdom treated with adjuvant tamoxifen. Breast Cancer Res. 2010;12(4):R64. doi:10.1186/bcr2629.
Toyama T, Yamashita H, Sugiura H, Kondo N, Iwase H, Fujii Y. No association between CYP2D6*10 genotype and survival of node-negative Japanese breast cancer patients receiving adjuvant tamoxifen treatment. Jpn J Clin Oncol. 2009;39(10):651–6. doi:10.1093/jjco/hyp076.
Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–62. doi:10.1158/1535-7163.MCT-09-0295.
Bassullu N, Turkmen I, Dayangac M, Yagiz Korkmaz P, Yasar R, Akyildiz M, Yaprak O, Tokat Y, Yuzer Y, Bulbul Dogusoy G. The predictive and prognostic significance of c-erb-B2, EGFR, PTEN, mTOR, PI3K, p27, and ERCC1 expression in hepatocellular carcinoma. Hepat Mon. 2012;12(10 HCC):e7492. doi:10.5812/hepatmon.7492.
Cittelly DM, Das PM, Spoelstra NS, Edgerton SM, Richer JK, Thor AD, Jones FE. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol Cancer. 2010;9:317. doi:10.1186/1476-4598-9-317.
Nordentoft I, Birkenkamp-Demtroder K, Agerbaek M, Theodorescu D, Ostenfeld MS, Hartmann A, Borre M, Orntoft TF, Dyrskjot L. miRNAs associated with chemo-sensitivity in cell lines and in advanced bladder cancer. BMC Med Genomics. 2012;5:40. doi:10.1186/1755-8794-5-40.
Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012;108(1):11–27. doi:10.1007/s11060-011-0793-0.
van den Bent MJ, Hegi ME, Stupp R. Recent developments in the use of chemotherapy in brain tumours. Eur J Cancer. 2006;42(5):582–8. doi:10.1016/j.ejca.2005.06.031.
Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N, Stupp R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10(6):1871–4.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi:10.1056/NEJMoa043331.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4. doi:10.1056/NEJM200011093431901.
Zawlik I, Vaccarella S, Kita D, Mittelbronn M, Franceschi S, Ohgaki H. Promoter methylation and polymorphisms of the MGMT gene in glioblastomas: a population-based study. Neuroepidemiology. 2009;32(1):21–9. doi:10.1159/000170088.
Watts GS, Pieper RO, Costello JF, Peng YM, Dalton WS, Futscher BW. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol Cell Biol. 1997;17(9):5612–9.
Patel SA, Graunke DM, Pieper RO. Aberrant silencing of the CpG island-containing human O6-methylguanine DNA methyltransferase gene is associated with the loss of nucleosome-like positioning. Mol Cell Biol. 1997;17(10):5813–22.
Pieper RO, Patel S, Ting SA, Futscher BW, Costello JF. Methylation of CpG island transcription factor binding sites is unnecessary for aberrant silencing of the human MGMT gene. J Biol Chem. 1996;271(23):13916–24.
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59(4):793–7.
Nakada M, Furuta T, Hayashi Y, Minamoto T, Hamada J. The strategy for enhancing temozolomide against malignant glioma. Front Oncol. 2012;2:98. doi:10.3389/fonc.2012.00098.
Karakousis G, Yang R, Xu X. Circulating melanoma cells as a predictive biomarker. J Invest Dermatol. 2013;133(6):1460–2. doi:10.1038/jid.2013.34.
Magbanua MJ, Sosa EV, Scott JH, Simko J, Collins C, Pinkel D, Ryan CJ, Park JW. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer. 2012;12:78. doi:10.1186/1471-2407-12-78.
Reinholz MM, Kitzmann KA, Tenner K, Hillman D, Dueck AC, Hobday TJ, Northfelt DW, Moreno-Aspitia A, Roy V, LaPlant B, Allred JB, Stella PJ, Lingle WL, Perez EA. Cytokeratin-19 and mammaglobin gene expression in circulating tumor cells from metastatic breast cancer patients enrolled in North Central Cancer Treatment Group trials, N0234/336/436/437. Clin Cancer Res. 2011;17(22):7183–93. doi:10.1158/1078-0432.CCR-11-0981.
Lewandowski R, Roszkowski K, Lewandowska MA. Personalized medicine in oncology: vision or realistic concept? Contemp Oncol. 2011;15(1):1–6.
Poznanski M, Lewandowska MA. Application of high-throughput technologies in cancer molecular diagnostics. Oncol Clin Pract. 2013;9(2):70–7.
The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–68. doi:10.1038/nature07385.
Lewandowska MA, Furtak J, Szylberg T, Roszkowski K, Windorbska W, Rytlewska J, Jozwicki W. An analysis of the prognostic value of IDH1 (isocitrate dehydrogenase 1) mutation in polish glioma patients. Mol Diagn Ther. 2013 Aug 10. doi:10.1007/s40291-013-0050-7.
Acknowledgments
This work is supported by a Foundation for Polish Science, co-financed from the European Union Regional Development Fund (HOMING PLUS/2010-2/7). The authors have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Nalejska, E., Mączyńska, E. & Lewandowska, M.A. Prognostic and Predictive Biomarkers: Tools in Personalized Oncology. Mol Diagn Ther 18, 273–284 (2014). https://doi.org/10.1007/s40291-013-0077-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-013-0077-9