
Abstract
Objective
This study outlines the current regulatory review process and good review practices (GRevPs) at the Jordan Food and Drug Administration (JFDA) and compares them with those of regulatory agencies in Australia, Canada, Saudi Arabia and Singapore to gauge how well the JFDA is performing. We identify opportunities for further development of the JFDA as a key global reference agency.
Methods
Personnel within the JFDA completed a questionnaire comprising four sections: organisation, key milestones, review timelines, and GRevPs. The same questionnaire was used concurrently to gather information from Australia’s Therapeutic Goods Administration (TGA), Health Canada, the Saudi Food and Drug Authority (SFDA) and Singapore’s Health Sciences Authority (HSA).
Results
The JFDA conducts an abridged review for new active substances and requires a certificate of pharmaceutical product (CPP) at the time of submission and 6 months of pharmacovigilance data at the time of the final review as well as full pharmaceutical, chemistry, manufacturing and controls (CMC) and clinical data at the time of submission. A written summary and tabulated data are required for non-clinical data. The four comparator agencies conduct full assessments; the SFDA also requires a CPP, and the JFDA and SFDA both require pricing information at submission. All agencies have established target timelines, and the JFDA, SFDA, TGA and HSA currently exceed those targets. All agencies have also developed GRevPs as well as training and continuous-improvement processes.
Conclusions
The JFDA has achieved significant success in its role as a regulatory agency by setting and implementing clear regulations in line with international guidance. It is recognised as a training centre in the region, with considerable achievements in the development of its activities by simplifying and improving requirements, procedures and actions. It also publishes information regarding guidance, procedures, drug application submissions and registration dates for all new chemical entities on its website. The relationship between the JFDA and the pharmaceutical sector in Jordan has resulted in balanced, practical, internationally compatible regulations and demonstrates a viable model of collaboration. To assist the JFDA in its efforts to become a key global reference agency, it is suggested that the agency explore a risk-stratification approach to the regulatory review; accept CPPs after dossier submission or use alternatives to the CPP; conduct pricing evaluations in parallel with scientific assessments; establish defined target times for review milestones and improve internal tracking systems to monitor these milestones; and make certain information transparent to all stakeholders by publishing a summary basis of approval.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The Jordan Food and Drug Administration (JFDA) conducts an abridged review of new active substances and requires a certificate of pharmaceutical product (CPP) and 6 months of pharmacovigilance data at the time of final review. The Saudi Food and Drug Authority (SFDA), Australia’s Therapeutic Goods Administration (TGA), Health Canada and Singapore’s Health Sciences Authority (HSA) conduct full assessments; the SFDA also requires a CPP, and the JFDA and the SFDA both require pricing information at submission. |
The JFDA, SFDA, TGA, Health Canada and the HSA have all established target timing for reviews and have instituted good review continuous improvement and training practices. |
The JFDA could consider implementing a structured benefit–risk assessment framework, a risk-stratification-based regulatory review; accept CPPs after dossier submission and at the time of final review; conduct pricing evaluations in parallel with scientific assessments; and make certain information transparent to all stakeholders by publishing the summary basis of approval. |
1 Introduction
As the guidelines and requirements for the registration of medicines become regionally harmonised throughout the world, jurisdictions with emerging pharmaceutical markets are also looking to effectively and efficiently use resources, reduce duplication, facilitate review processes and expedite the availability of medicines through the development of programmes of cooperation and sharing [1]. However, these programmes require the identification, acknowledgement and application of mutual good review practices (GRevPs) that will result in timely, predictable, transparent and high-quality regulatory review [2]. In a recent review, Hashan et al. [3] stated that regulatory performance should be periodically assessed against established international standards, enabling agencies to appraise their performance, establish realistic strategies for improvement and assess the influence of those changes.
1.1 Jordan: A Thriving Healthcare Economy
In 2013, Jordan spent 7.2% of its gross domestic product on healthcare, equal to $Int761 for each of its 7,000,000 inhabitants; public spending accounted for 66% of this amount and private spending for the remaining 34% [4]. In 2012, $US627 million was spent on pharmaceuticals, which amounted to 27% of the expenditure for health, and Jordan’s High Health Council has instituted a plan to reduce drug spending as well as strategies to combat important health issues, including smoking and metabolic-related disorders [5]. In 2014, approximately 87% of Jordanians were covered by either public, private or military health insurance, and the Jordanian government plans to develop a comprehensive health insurance system in the next few years [5]. Jordan exported $Int722 million in pharmaceuticals in 2013, more than any other jurisdiction in the Arab World, and the country has also become a regional focus for pharmaceutical research [6].
The Jordan Food and Drug Administration (JFDA) is an independent national competent authority tasked with ensuring the safety and quality of food and the efficacy, quality and safety of medicines and related materials. The creation of the JFDA has been essential in the country’s efforts toward more efficient and transparent medicines regulation and supply. Its responsibilities also include product registration; pricing; licensing of pharmaceutical manufacturers, importers, wholesalers and establishments; promoting rational drug use (RDU); and post-marketing surveillance, including quality control and inspections [7]. Jordan is also participating in the Medicines Transparency Alliance (MeTA), which was formed to expedite access to needed medicines through the improvement of transparency and accountability in pharmaceutical development, manufacturing regulation, and access. The first phase of MeTA was launched in Jordan in 2009 and, after a successful pilot, the second phase of MeTA was hosted by the JFDA and guided by the World Health Organization (WHO) and Health Action International [8].
In recent years, Jordan’s leaders have achieved improvements in programmes for education, health, social welfare protection, infrastructure development and tax administration and created a climate for growth of public–private partnership. Despite unfavourable regional political conditions, Jordan’s economy is expanding and is expected to grow a further 3.6% in 2016 [9].
1.2 Study Rationale
As Jordan is an important economic force in the Middle East and an emerging centre for pharmaceutical research, it is appropriate that the JFDA achieve its long-term goal of becoming an established reference agency, facilitating regulatory review in emerging agencies in the region, thus expediting the availability of needed, innovative medicines to local populations. This study aimed to outline current JFDA review timelines, processes and procedures and to compare them with those of four international mid-sized regulatory agencies to identify areas of strength and areas requiring improvement to facilitate JFDA progress toward this goal.
2 Objectives
Specifically, the objectives of this study were as follows:
-
Characterise the current regulatory review processes used at the JFDA, identifying agency review models as well as key milestones and timelines.
-
Ascertain GRevP requirements and their implementation and measurement by the JFDA, including those to ensure consistency, transparency, timeliness, and predictability of the review process.
-
Compare JFDA review processes and practices with those of the regulatory agencies in Australia, Canada, Saudi Arabia and Singapore to identify areas in which the JFDA excels and to suggest opportunities for improvement to ultimately enhance patients’ access to new medicines.
3 Methods
The Centre for Innovation in Regulatory Science (CIRS) developed a three-part standardised questionnaire that was completed by JFDA personnel to determine agency regulatory review processes and practices. This instrument was originally developed to gather information about regulatory systems in jurisdictions with emerging pharmaceutical markets [10] and subsequently for a study of GRevPs in the Asia–Pacific Economic Cooperation (APEC) region [11] and in Saudi Arabia [3, 12].
The first part of the questionnaire established the JFDA’s structure, resources and review models. The second part of the questionnaire used regulatory process maps developed by CIRS through its work with regulatory agencies in mature and developing markets to identify JFDA review milestones and timelines. The third part examined JFDA activities that contribute to the quality of the decision-making process and GRevPs that have been adopted to improve consistency, transparency, timeliness and predictability in the regulatory review process. The results of the questionnaire were evaluated by JFDA study participants for potential additions, amendments or comments.
The same questionnaire was also completed concurrently and validated by Australia’s Therapeutic Goods Administration (TGA), Health Canada, Singapore’s Health Science Authority (HSA) and the Saudi Arabia Food and Drug Authority (SFDA). Using the same questionnaire for all agencies enabled the compilation of important information about the structure, processes and practices of, and accurate comparisons between, the international regulatory agencies. CIRS obtained pharmaceutical company data about agency approval times directly from pharmaceutical companies as part of its ongoing efforts to benchmark the approval times of global regulatory agencies.
We initially completed the standardised questionnaire for each of the agencies using public domain data. This was then reviewed by one senior representative of the regulatory authority in collaboration with their reviewing staff to confirm the data and to provide missing information that was not publicly available, such as GRevPs and timelines. This was then put into a standardised report format and returned to the agency for confirmation that the results had been correctly interpreted and appropriately documented before being incorporated into this review.
McAuslane et al. [10] identified three models for the scientific regulatory review of a product.
-
Type 1, the verification assessment model, requires that a product receives prior authorisation by two or more reference or competent regulatory agencies and employs a verification process to validate the product and ensure it conforms to the previously authorised product.
-
Type 2, the abridged assessment model, requires an abridged evaluation of a medicine relative to the local environment as well as pre-registration with a minimum of one reference or competent regulatory agency.
-
Types 3A and 3B, the full assessment models, require that the authority carries out a complete scientific review of a dossier’s scientific data for quality, safety and efficacy. For type 3A, pre-registration in another jurisdiction is required; for type 3B, registration by other regulatory agencies may be considered, but is not a prerequisite for authorisation.
4 Results
4.1 Processes
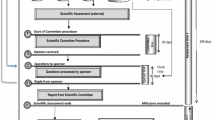
Figure 1 summarises the key features of the JFDA review process compared with those of the TGA, Health Canada, the HSA and the SFDA.

Summary comparison of key features of the pharmaceutical regulatory systems of Jordan, Saudi Arabia, Australia, Canada and Singapore. NAS new active substance
4.1.1 Review Model
The JFDA conducts an abridged (type 2) review of dossiers for new active substances and major line extensions. For these reviews, the reference country must be the country of origin, and a certificate of pharmaceutical product (CPP) is required with the application for final marketing authorisation. For some types of dossiers for which the JFDA conduct type 3A reviews, such as biologicals and biosimilars, 6 months of pharmacovigilance data from the CPP-issuing country are also required.
In comparison, the four other agencies utilise a full assessment model (type 3). The SFDA requires a CPP at the time of submission for final marketing authorisation (type 3A), whereas the TGA, Health Canada and the HSA do not (type 3B). However, to optimise resource use, the TGA can conduct an abridged review if requested by the sponsor and if the product has been approved by two or more reference agencies. Similarly, the HSA conducts an abridged review if the product has been approved by one or more reference agencies or a type 1 review (verification model) if the product has been approved by two or more reference agencies.
4.1.2 Data Requirements
The JFDA currently requires full pharmaceutical, chemistry, manufacturing and controls (CMC) and clinical data with submissions. The written summary and tabulated data are required for non-clinical data. The agency conducts a detailed assessment of the pharmaceutical CMC data but only reviews the non-clinical data if a query is raised. The clinical data are reviewed on the basis of the medicine’s benefit and risk factors, local medical practice, influence of national disease patterns and ethnic factors.
All the comparative authorities in this report require full datasets for the pharmaceutical CMC, non-clinical and clinical sections, and conduct a detailed assessment of the pharmaceutical CMC data. The HSA conducts an assessment based on the type of review it conducts, allowing resources to be conserved for the review of medicines associated with a high risk for their population. Whilst both the JFDA and the SFDA require information relating to pricing as part of the review process, this is carried out by a separate committee. A firewall is in place between the two committees, but disagreement on price may result in a negative decision. Pricing evaluations are not part of the technical review at the other comparative agencies.
Reviews of quality, safety and efficacy data are conducted in parallel at all five agencies in this study. At the JFDA, the primary scientific assessment is carried out by the agency’s technical committee, which includes both internal and external experts. The external experts are mainly responsible for providing a detailed assessment report, recommendations and a clinical opinion on the product. External experts are used ad hoc by the TGA, HSA and SFDA. Health Canada does not use external experts for dossier review.
4.1.3 Target and Approval Timing
The JFDA review process consists of the following common steps: validation of the submitted dossier, scientific assessment, company response and final authorisation. The JFDA have stipulated a target time of 30 calendar days (21 working days) for pricing and 180 calendar days (122 working days) for review, an overall target time of 210 calendar days (143 working days). The JFDA does not specify separate target times for the validation and scientific assessment processes.
For the period 1 January 2014 through 5 March 2015, the actual median approval time in Jordan for 76 products was 417 days (283 working days). Although it would be beneficial to have company-reported timelines for comparison, as these usually represent only a proportion of the total number of products reviewed, these data can be misleading, whereas the dataset from the JFDA is comprehensive and complete. In comparison, the overall target approval time for the SFDA, which conducts a type 3A review, is 420 calendar days (290 working days). The overall target approval times for the TGA, Health Canada and the HSA, which all conduct type 3B reviews, are 305 calendar days (209 working days); 355 calendar days (243 working days); and 395 calendar days [270 working days (mainly for abridged reviews)], respectively. The review times for the SFDA were lower than the target time, whereas the review times from 2011 to 2013 for the TGA exceeded the specified target. Approval times for Health Canada were approximately on target during the same time period (Fig. 2).

Regulatory approval time from date of submission to date of approval for new active substances approved 1 January 2011–31 December 2013 in Saudi Arabia, Australia, Canada and Singapore and 1 January 2014–3 September 2015 in Jordan. Figures in parentheses are the number of drug applications and number of companies providing data. Boxes represent the 25th and 75th percentiles and whiskers represent the 5th and 95th percentiles. Diamonds indicate company-provided data, triangles indicate agency target time and circles indicate agency-provided data (from agency and public domain). NA not available. Singapore company data shows approval time for all review types, but the majority of the applications were an abridged review. Asterisk new active substances approved in Jordan from 1 January 2013–3 September 2015, double asterisk new active substance approval time in Saudi Arabia for local and international companies, triple asterisk Singapore target time shown for type 3B review (full review)
4.2 Good Review Practice
We assessed a variety of metrics to determine how the five agencies compared with regard to practices in place to ensure quality, transparency and predictability of the process. Health Canada has a programme for formal use in place, whereas the other agencies reported having GRevPs in place but no formal implementation.
4.2.1 Quality Measures
The quality measures evaluated in this comparative study included the availability and use of internal quality policies, GRevPs, standard operating procedures (SOPs) for assessors, assessment templates, the existence of a quality assurance department, the use of scientific committees and the use of shared and joint reviews with other agencies. Six of these seven measures are in place within the JFDA: an internal quality policy, GRevP system, SOPs for assessors, assessment templates, a scientific committee, and a quality assurance department to monitor the agency’s processes is under development. In comparison, six of the seven measures are also in place in the SFDA and TGA, whereas Health Canada and HSA employed five each (Fig. 3).

Regulatory quality measures at regulatory agencies in Jordan, Saudi Arabia, Australia, Canada and Singapore. SOP standard operating procedure, asterisk as part of the Gulf Cooperation Council process; double asterisk agency has internal audit quality programme
4.2.2 Transparency and Communication
Information communicated by regulators to stakeholders could include feedback on submitted dossiers, technical staff contact information, pre-submission scientific advice, official guidelines, ability to track the progress of applications, summary of the grounds of approval, approval times, advisory committee meeting dates and the approval of products. The JFDA have eight of these nine parameters in place, the SFDA five, the HSA six, Health Canada eight and the TGA all nine. The JFDA, SFDA and HSA do not publish a summary basis of approval; the SFDA, Health Canada, and the HSA do not supply advisory meeting dates or give feedback to the industry on the submitted dossier. The SFDA is the only agency that does not share information on how to contact their technical staff during the review (Fig. 4).

Transparency and communication measures at regulatory agencies in Jordan, Saudi Arabia, Australia, Canada and Singapore. Asterisk all approved drugs are published on the agency website in addition to a separate list for data exclusivity; approval of products is only published if there is data exclusivity
4.2.3 Continuous Improvement Initiatives
The continuous improvement initiatives assessed in this study included external and internal quality audits, tracking systems and review of assessor and stakeholder feedback. Four of these continuous improvement processes are in place in the JFDA, TGA and HSA, and Health Canada has three. Only the SFDA engages in all five continuous improvement processes.
4.2.4 Training and Education
The types of training and continuing education that can enhance the review process include international workshops, external and in-house courses, on-the-job training, lectures by external speakers, induction training, sponsorship of postgraduate degrees and placements and secondments. The JFDA have five of the eight training measures in place but lack induction training, sponsorship of postgraduate degrees or placements and secondments, whereas the TGA, Health Canada and HSA employ all eight and the SFDA seven.
4.2.5 Contributors and Barriers to Good-Quality Decision Making
The responses from the JFDA indicated that the major contributors to an effective and efficient regulatory authority are guidelines, high-calibre employees and good scientific committee experts. The other agencies indicated a diverse set of enablers, but agreement was universal among all five agencies that good-quality procedures and processes and qualified staff are necessary.
The JFDA listed limited resources, incomplete dossier submissions, review procedures and readiness for electronic submission of the common technical document (e-CTD) as barriers to quality review. Similarly, the comparative agencies reported that incomplete submissions and lack of experienced staff were barriers to effective quality decision making.
5 Discussion
The JFDA currently employs a type 2, abridged review, method to assess new chemical entities, and this method of assessment is appropriate given their resources, requirements and staff competency levels. However, to evolve its competencies to be able to assess new molecules or biosimilars that are not approved by a reference agency (referring to the country of origin), the JFDA would need to more efficiently utilise and conserve constrained resources. A risk-stratification approach to reviews is one way to conserve regulatory resources [12]. With this approach, agencies such as the HSA and the TGA perform verification reviews of products that have been approved by two or more reference agencies, only using resources to conduct full reviews of products that have not been previously reviewed. Additionally, the JFDA currently use the same review process for major line extensions and for new chemical entities. Different processes for these two types of applications, as well as delineation between major and minor product variations, would further enable prioritisation of resources.
One of the main requirements for a type 2 or abridged review is the provision of a CPP. Whereas the JFDA requires a legalised CPP for regulatory submissions, the WHO guidelines indicate this is no longer mandatory. The SFDA also requires a legalised CPP at the time of application; however, if the product has not been approved elsewhere and is a priority product, the CPP can be submitted at the time of authorisation. The TGA, Health Canada and the HSA do not require a CPP. For jurisdictions that require proof of prior marketing authorisation before regulatory review, strategies that could be considered to expedite the availability of medicines include the acceptance of a CPP after dossier submission, but before marketing authorisation, such as is practiced in Mexico, Chinese Taipei and Turkey [2]. Other agencies use alternate evidence of market authorisation such as information from the websites of other agencies [10].
The JFDA requirement for 6 months of pharmacovigilance data from the country issuing a CPP at submission could delay the availability of medicines for local patients. One long-term solution to this would be via strengthening of the JFDA pharmacovigilance system by implementing the Guideline on Good Pharmacovigilance Practices for Arab Countries [13] to eliminate reliance on data from other agencies. Like the SFDA, the JFDA require pricing information as part of the review process, including the reference agencies’ price lists for the drug product. In comparison, price evaluation is not part of the TGA, Health Canada or the HSA review process. The JFDA currently conduct the pricing assessment at the end of the scientific review; we suggest that performing these two functions in parallel would accelerate marketing authorisation.
The JFDA approval times of 417 days (210 working days) between January 2014 and March 2015 exceeded the agency’s overall target time of 210 calendar days (143 working days), suggesting room for improved timeliness, consistency and process predictability. Some of the delays could be attributed to the time taken by the sponsoring company to respond to agency questions. Continuous dialogue and transparency between the JFDA and industry will improve the quality of dossier submissions and reduce the number of agency questions. Approval times at the SFDA, TGA and HSA from 2011 to 2013 also exceeded agency target times. Health Canada approval times were approximately the same as target times during this time period, most likely because the agency has made concerted efforts to abide by target times to avoid penalties of up to 50% of user fees as mandated by the User Fees Act [14].
The JFDA has established target times for the authorisation procedure and overall approval timing, whereas the SFDA, the TGA, Health Canada and the HSA set separate target times for validation, scientific assessment, authorisation and overall approval times. Defining target timing for individual milestones within the review facilitates planning for both agencies and sponsoring companies and permits the identification of the most appropriate areas for improvement. The JFDA have a manual system but are converting to an electronic internal tracking system to monitor the various milestone timelines and make them available to stakeholders. This will also enable the JFDA to observe the lag time between first-in-world approval and approval in Jordan to monitor treatment gaps and access to medicines for Jordanian patients.
The WHO has stated that GRevPs facilitate timely and high-quality regulatory reviews and enable regulatory convergence and resource conservation [15]. The JFDA, like the comparator agencies in this study, employs many of the essential elements of GRevPs. However, it is recommended that the agency formalise its good review practice system and ensure its full implementation to maintain consistency and process predictability.
The JFDA should consider providing a summary of the grounds on which approval was granted, that is, a Summary Basis of Approval or similar documentation, as this would provide transparency to patients and healthcare providers regarding internal agency decision making. In providing a rationale for the publication of such summaries, the Health Canada website states that it “improves the transparency of the drug and medical device regulatory review processes. They also give Canadians improved access to information about decisions to authorize products for sale in Canada” [16].
Recognising that training is a key requirement for the establishment of regulatory quality [17], the JFDA have the majority of necessary training measures in place to develop the expertise of its staff. Induction courses for new assessors could be a valuable addition to agency training resources, and induction courses by industry in areas such as biosimilars and biotechnology products could supplement agency expertise where needed.
If secondments or outside placements are not feasible because of resource limitations, the development of a robust ‘on-the-job training’ programme would help ensure the development of competent reviewers. In addition, the JFDA could consider developing a relationship with reference agencies that might be willing to second one of their reviewers to the JFDA, thereby providing significant training and educational value for new assessors.
Study participants recognised the need to build a robust, flexible information technology infrastructure at the JFDA, facilitate the sharing of post-marketing surveillance information within the region or even from reference countries, which would set the stage for the agency’s leadership in the region.
Finally, as reported by Leong et al. [18], it has been established that a structured framework for the evaluation of benefits and risks is needed for the effective review of new medicines. The JFDA may consider using the Universal Methodology for Benefit-Risk Assessment (UMBRA) framework, which has been positively assessed by several mature agencies [18]. A summary template derived from the framework has also been favourably reviewed by regulators in jurisdictions with emerging pharmaceutical markets [19].
6 Recommendations
Our comparison of the JFDA processes and practices with those of similar medium-size regulatory agencies has enabled the development of several proposals to assist the agency in its efforts to become an internationally recognised reference agency, as follows:
-
1.
The JFDA could conserve resources by
-
employing a risk-stratification approach (verification review) to the regulatory review of products that have been approved by two or more reference agencies and conducting an abridged review for products that have been reviewed by only one reference agency,
-
developing separate review processes for new chemical entities and major line extensions.
-
-
2.
The JFDA could expedite local patient access to medicines by
-
accepting CPPs after submission and before final review or accepting other evidence of prior regulatory approval such as agency website information,
-
conducting price assessments in parallel with scientific reviews,
-
enhancing its internal pharmacovigilance system as an alternative to waiting for 6 months of pharmacovigilance data from other jurisdictions.
-
-
3.
The JFDA could enhance review quality, transparency and predictability by
-
establishing defined target times for review milestones in addition to the predefined overall authorisation procedure approval timing, improving internal tracking systems to monitor these milestones, and making this information available to all stakeholders;
-
providing publicly available summaries of the basis for approval;
-
developing a relationship with one or two reference agencies to encourage training through secondments and job shadowing.
-
References
Institute of Medicine Forum on Drug Discovery, Development, and Translation. Transforming Clinical Research in the United States: Challenges and Opportunities: Workshop Summary. Washington (DC): National Academies Press; 2010.
Patel P, McAuslane N, Liberti L, et al. Workshop report: focus on Latin America: building quality submission and review processes and practices—overcoming challenges and meeting expectations, Lima, Peru: 23–24 January 2014. http://cirsci.org/sites/default/files/CIRS_January_2014_Workshop_Report.pdf. Accessed 16 Mar 2016.
Hashan H, Aljuffali I, Patel P, et al. The Saudi Arabia Food and Drug Authority: an evaluation of the registration process and good review practices in Saudi Arabia in comparison with Australia, Canada and Singapore. Pharm Med. 2016;30:37–47.
World Health Organization. Country profile: Jordan. http://www.who.int/countries/jor/en/. Accessed 16 Mar 2016.
Oxford Business Group. The Report: Jordan 2015. http://www.oxfordbusinessgroup.com/jordan-2015-0. Accessed 16 Mar 2016.
World Trade Organization. International trade statistics. https://www.wto.org/english/res_e/statis_e/its2013_e/its2013_e.pdf Accessed 16 Mar 2016.
Baghdadi-Sabeti G, Serhan F. WHO Good Governance for Medicines programme: An innovative approach to prevent corruption in the pharmaceutical sector. 2010. http://www.who.int/healthsystems/topics/financing/healthreport/25GGM.pdf. Accessed 16 Mar 2016.
Naffa S, Nuseirat A. Medicine transparency alliance Jordan Workplan. http://www.medicinestransparency.org/fileadmin/uploads/Renee/MeTA__Jordan_Workplan.pdf. Accessed 16 Mar 2016.
The World Bank. Jordan. http://data.worldbank.org/indicator/NY.GDP.PCAP.PP.CD. Accessed 16 Mar 2016.
McAuslane N, Cone M, Collins J, et al. Emerging markets and emerging agencies: a comparative study of how key regulatory agencies in Asia, Latin America, the Middle East and Africa are developing regulatory processes and review models for new medicinal products. Drug Info J. 2009;43:349–59.
Liu LL, McAuslane N, Tzou M-C, et al. Characterizing good review practices: A survey report among agencies of APEC member economies. Ther Innov Reg Sci. 2013;47:678–83.
Alsager SSN, Hashan H, Walker S. Evaluation of the regulatory review system in Saudi Arabia. Pharm Med. 2015;29:93–103.
The League of Arab States. The guideline on good pharmacovigilance practices (GVP) for Arab countries. http://www.who-umc.org/graphics/29613.pdf. Accessed 9 Nov 2016.
Government of Canada. User Fees Act S.C. 2004, c.6. http://laws-lois.justice.gc.ca/eng/acts/U-3.7/page-1.html#h-7. Accessed 9 Nov 2016.
World Health Organization. Good review practice guidelines for regulatory authorities. http://apps.who.int/medicinedocs/documents/s21799en/s21799en.pdf. Accessed 16 Mar 2016.
Health Canada. Summary basis of decision. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/index-eng.php. Accessed 9 Nov 016.
Al-Essa R, Al-Rubaie M, Walker S, et al. Pharmaceutical regulatory environment: Challenges and opportunities in the Gulf region. Cham: Springer International Publishing; 2015.
Leong J, Walker S, Salek S. A practical approach to communicating benefit-risk decisions of medicines to stakeholders. Front Pharmacol. 2015;6:99. doi:10.3389/fphar.2015.00099.
Walker S, McAuslane N, Liberti L, et al. Implementing an internationally acceptable framework for the benefit-risk assessment of medicines: how close are we to this objective? CIRS Workshop Report, June 2013. http://www.cirsci.org/sites/default/files/CIRS_June_2013_Workshop_low-res.pdf. Accessed 16 Mar 2016.
Acknowledgements
The authors gratefully acknowledge the input of Sami Alsager (SFDA), Jason Ferla (TGA), Barbara Sabourin and Jeanne Siegert (Health Canada) and Jalene Poh (HAS) to construct this comparative study and the writing and editorial assistance of Patricia Connelly (CIRS), in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This independent research study was conducted by CIRS as part of its ongoing initiatives to understand pharmaceutical development and regulatory activities in emerging markets. No funding was received for the development of this manuscript.
Conflict of interest
Wesal Salem Al Haqaish is the Head, Drug Registration, JFDA. Hayel Obeidat is the Director General, JFDA. Prisha Patel is employed by, and Stuart Walker is the founder of, CIRS, London, UK, which conducted the research described in this report.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Haqaish, W.S.A., Obeidat, H., Patel, P. et al. The Jordan Food and Drug Administration: Comparison of its Registration Process with Australia, Canada, Saudi Arabia and Singapore. Pharm Med 31, 21–30 (2017). https://doi.org/10.1007/s40290-016-0172-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-016-0172-4