
Abstract
Background
Against the background of increasing cost pressure in the German Health Care system German health policy introduced several law changes to increase competition within German health care system for both, payers and health care providers. At the same time law changes included centralization of decisions—counteracting a real competition. Both approaches are part of an austerity plan. The latest example for this approach is the new drug law (AMNOG) in 2011 with the core element of centralized early benefit assessment (§ 35a SGB V) for new drugs and therapies and price negotiations between federal association of health insurances and pharmaceutical company.
In this review we examine the implementation of the new drug law with respect to the achievement of political objectives: Ensure sustainable financing of innovations in the German health care system, provide innovations early to the patient, decrease overregulation and establish a transparent environment in which efforts of the pharmaceutical industry are being honored by fair prices.
Methods
We reviewed the new AMNOG process since its implementation on 1st January 2011 and first 64 values dossiers from pharmaceutical companies that have been evaluated by G-BA (Federal Joint Committee) between January 2011 and December 2013 with respect to the above mentioned political objectives. Parameters such as added value, determination of an adequate competitor, patient relevant endpoint surrogate parameter and subgroup analysis are being discussed.
Results
AMNOG process has been implemented as a learning system and indeed several issues have already been addressed, such as the determination of the adequate comparator by G-BA as well as the treatment of orphan drugs in this process. Basically implementation of AMNOG and early benefit assessment is a necessary step on the way to transparent priorisation of health care benefits. But the AMNOG process is one step further towards centralization of the German health care system and therefore contradicts a healthy and fair competition within the system. As a consequence the development of high quality solutions for patients might be hampered.
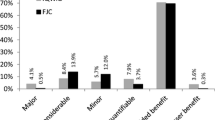
The analysis of the first 64 value dossiers shows that less of half of the affected patient populations (40 %) have access to new therapies being reimbursed by health insurances with a premium price. There is a major inbalance in assessment of drugs in different therapeutic areas. In combination with increased uncertainty for the pharmaceutical companies the AMNOG process on the middle and long-term might jeopardize the commitment of pharmaceutical industry in the German market. This in turn endagers the political objective to ensure patients early access to innovative therapies.
Besides this, centralization of the subjective parameter “added value” seems to be problematic, since value decisions should be taken by democratic processes.
We therefore suggest a model in which only objectively measurable value decisions are being taken centrally and subjective value decisions are as much as possible decentralized. This results in both a stronger competition of qualitatively best solutions for patients and in a higher fault tolerance. Instruments such as health care research and conditional reimbursement can help to enhance a fair competition for more quality in regionally organized health care and more economical allocation of short resources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Hintergrund und Fragestellung
Die kurzfristige Erholung der Budgets der gesetzlichen Krankenkassen (GKV) kann nicht darüber hinweg täuschen, dass die Ausgaben der GKV, die in den letzten Jahren nur marginal gestiegen sind, in Zukunft wieder steigen werden und sich damit die Budgetsituation verschärfen wird.
Allein auf Grund der demografischen Entwicklung ist abzusehen, dass der Kostendruck auf das deutsche Gesundheitssystem weiter zunehmen wird.
Vor diesem Hintergrund begrenzter Ressourcen und gleichzeitig steigender Ausgaben, ist die Einführung des Arzneimittel-Neuordnungsgesetztes (AMNOG) im Jahr 2011 mit der obligaten frühen Nutzenbewertung für neue Medikamente ein Schritt in die richtige Richtung. Sie liefert die Grundlage für eine transparente Priorisierung von Gesundheitsleistungen, um langfristig eine qualitativ hochwertige Patientenversorgung sicher zu stellen.
Der Gesetzgeber hatte hier in den letzten zwei Jahrzehnten eine Vielzahl an Gesetzen eingeführt, die sich im Spannungsfeld zwischen einem Mehr an Wettbewerb um mehr Qualität und Effizienz auf der einen Seite und einer stärkeren Zentralisierung bewegen. Letzteres führte u.a. zu einer kontinuierlichen Machtausweitung des Gemeinsamen Bundesausschusses (G-BA), dessen Einflussbereich durch die Politik fortlaufend erweitert wurde. Mit dem AMNOG wurde 2011 ein weiteres Kostendämpfungsgesetz in das deutsche Gesundheitssystem eingeführt, was zu einer verstärkten Zentralisierung beiträgt. Kernstück dieses Gesetzes ist die „Frühe Nutzenbewertung nach § 35a SGB V“, wobei zentral über den subjektiven Parameter „Nutzen/Zusatznutzen“ für rund 80 Millionen Menschen (gesetzliche und private Krankenversicherung) in Deutschland entschieden wird. Ziel der Politik sei es, „die rasant steigenden Arzneimittelausgaben der gesetzlichen Krankenkassen einzudämmen. Das AMNOG schaffe „die schwierige Balance zwischen Innovation und Bezahlbarkeit bei Arzneimitteln zu halten“ [1], womit schließlich die Beitragsstabilität gewährleistet werden soll. Das Einsparpotential soll 1,8 Milliarden Euro pro Jahr erreichen.
Die Entscheidungsprozesse sollen transparent und nachvollziehbar gestaltet werden und in faire Preisverhandlungen zwischen dem Spitzenverband Bund der gesetzlichen Krankenversicherungen (GKV-SpiBu) und der Pharmazeutischen Industrie münden, die die Innovationen entsprechend würdigen. Die Sonderstellung Deutschlands innerhalb Europas, Innovationen für den Patienten direkt nach Markteinführung verfügbar zu halten, soll beibehalten werden.
Dass nicht alle Anforderungen von Anfang an erfüllt werden können, war allen Beteiligten klar und ist an der Tatsache erkennbar, dass die Änderungen als „lernenden Systems“ implementiert wurden. Tatsächlich wurden bereits mit der Änderung der AMG Novelle erste Anpassungen vorgenommen [2].
Mit dem Koalitionsvertrag zwischen Union (CDU/CSU) und SPD wurde die Bewertung des Bestandsmarktes aufgegeben bevor die ersten Entscheidungen zum Bestandsmarkt (Gliptine) ihre Wirkung zeigen konnten [3].
1.1 Das AMNOG im Kontext gesetzlicher Kostendämpfungsmaßnahmen
Der Gedanke der zentralen Nutzenbewertung wurde bereits 2004 mit dem GMG vom Gesetzgeber verankert und das im Jahr 2004 hierfür gegründete Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) hatte den Auftrag, Nutzenbewertungen für neue Arzneimittel zu erstellen. Hier gab es bereits die Möglichkeit, Festbeträge für patentgeschützte Arzneimittel mit geringem therapeutischen Nutzen einzuführen. Diese Aufgaben wurden mit dem GKV-Wettbewerbsstärkungsgesetz, das zum 01.04.2007 in Kraft trat, um eine Kosten-Nutzen-Abwägung erweitert. In Konsequenz konnte dann erstmalig auf Basis dieser Bewertung von dem GKV Spitzenverband ein Höchstpreis für die Erstattung von patentgeschützten Arzneimitteln festgelegt werden. Insbesondere für solche Arzneimittel, die nicht bereits einem Festbetrag zugeordnet werden konnten. Diese Möglichkeiten bestanden zusätzlich auf Innovationen festgelegten Zwangsrabatten (6 % im Beitragssatzsicherungsgesetzt (2003), 16 % im GKV-Modernisierungsgesetz (2007)). Clopidogrel (Plavix® Iscover®, Sanofi, BMS) zum Beispiel wurde in 2008 durch eine Nutzen-Bewertung vom IQWiG in der Indikation Schlaganfallprophylaxe – trotz vorliegender Zulassung und überlegener Wirksamkeit gegenüber dem bisherigen Therapiestandard Azetylsalizylsäure – über einen Therapiehinweis von der Erstattung durch die GKV ausgeschlossen [4]. Paliperidon (Invega®, Janssen) wurde 2009 vom G-BA auf Festbetragsniveau eingestuft. Janssen entschied, das Medikament auf dem deutschen Markt zu lassen, sodass die Patienten die Differenz zwischen Erstattungspreis und Listenpreis selbst tragen mussten, aber das Arzneimittel weiter verfügbar war [5].
Die forschende Pharmazeutische Industrie befindet sich im Hinblick auf die Entwicklung und Produktion innovativer Arzneimittel – ihrem bisherigen Kerngeschäft – in einer schwierigen Situation: Es wird von ihr zu Recht erwartet, „innovative Produkte“ für die Patienten auf den Markt zu bringen. Hierdurch trägt sie aber zu einer steigenden Nachfrage und damit zu höheren Ausgaben bei. Zudem stehen die monetären Zielrichtungen der Industrie dem ethischen Ansatz der zum Großteil solidarisch geförderten Gesundheitsversorgung vermeintlich entgegen, wodurch die Imageprobleme der Pharmazeutischen Industrie maßgeblich bestimmt werden. Vergessen wird in diesem Zusammenhang oft, dass eine wirtschaftlich starke Pharmazeutische Industrie nicht nur direkt über die Entwicklung und Produktion neuer Medikamente zur Gesundheit der Bevölkerung beiträgt, sondern auch indirekt durch sichere Arbeitsplätze und soziale Netzwerke, da sie allein in Deutschland mehr als 100.000 Mitarbeiter beschäftigt [6].
1.2 Die Regularien und beteiligte Institutionen
Seit dem 01.01.2011 müssen alle pharmazeutischen Hersteller für erstmals in den Verkehr gebrachte Wirkstoffe und Wirkstoffkombinationen ein Nutzen-Dossier für die frühe Nutzenbewertung nach § 35a SGB V einreichen, sofern sie für das neue Arzneimittel einen Zusatznutzen und damit einen aus ihrer Sicht akzeptablen Preis beanspruchen [7]. In Abb. 1 sind die Gesetzestexte, Verordnungen und Regularien aufgeführt, die bisher im Kontext der frühen Nutzenbewertung aufgestellt worden sind.

Regulationsdichte beim Verfahren der frühen Nutzenbewertung nach § 35a SGB V
Im SGB V sind die gesetzlichen Rahmenbedingungen verankert, die in der Arzneimittel-Nutzenverordnung des BMG konkretisiert werden [7]. Der G-BA hat in seiner Verfahrensordnung zur Nutzenbewertung die Struktur und die formalen inhaltlichen Anforderungen festgeschrieben, u.a. auch die Anforderungen an das Dossier eines pharmazeutischen Unternehmens (PU) [8].
Diese sind im fünften Kapitel der G-BA-Verfahrensordnung in vier Abschnitten festgelegt. Dabei geht es insbesondere auch um die Anforderungen an den Nachweis des so genannten „Zusatznutzens“ und die Bestimmung einer zweckmäßigen Vergleichstherapie.
Das IQWIG hat in seinen Allgemeinen Methoden auf 178 Seiten die wissenschaftlichen Kriterien für die Nutzenbewertung dargelegt [9]. Es fehlt nach wie vor ein wissenschaftlich anerkanntes Verfahren, wie ein Zusatznutzen quantifiziert werden soll. Das IQWiG hat im Anhang der Nutzenbewertung von Ticagrelor® ein Verfahren vorgestellt, das allerdings nicht international anerkannt ist und vom G-BA in seinen Entscheidungen nicht angewandt wird [10]. Es ergibt sich ein dringender Handlungsbedarf, dieses Defizit unter Beteiligung der medizinischen Fachgesellschaften (z. B. AWMF), Patientenvertretern, Krankenkassen und Pharmazeutischer Industrie zu beheben. Im Anschluss an die Nutzenbewertung durch den G-BA schließt sich die Verhandlung zwischen pharmazeutischem Hersteller und dem GKV-Spitzenverband über den Erstattungsbetrag für das neue Arzneimittel an.
In der Rahmenvereinbarung zu den Preisverhandlungen werden die Grundsätze der Preisverhandlungen zwischen pharmazeutischem Unternehmen und Spitzenverband Bund festgelegt und schließlich gibt es die Schiedsstellenordnung, die das Schiedsstellenverfahren zwischen dem Spitzenverband Bund (SpiBu) und dem pharmazeutischen Unternehmen regelt und zu einer zeitnahen Preisfestlegung führt.
Allein die Regulationsdichte zeigt, dass es sich um einen stark formalisierten Prozess handelt, wodurch die Verantwortlichen den Eindruck erwecken möchten, es handele sich um einen objektiv nachvollziehbaren Prozess.
Das Ziel der Deregulierung wurde, zumindest für alle, die sich mit der frühen Nutzenbewertung beschäftigen, nicht erreicht. Zusätzliche Administration für ein im Schnitt mehrere hundert Seiten langes Dossier ist nicht nur mit Bindung wichtiger zeitlicher Kapazitäten verbunden, sondern auch mit zusätzlichen Kosten. Laut ursprünglichen Schätzungen der Politik sollte ein Dossier ca. € 1.250 kosten.
Der VFA schätzt die tatsächlichen Kosten für ein Dossier dagegen auf ca. € 400.000–600.000. Die Kurzatmigkeit der Politik zeigt sich in dem aktuellen Koalitionsvertrag, indem die Bewertung des Bestandsmarktes komplett gekippt wurde, bevor die erste Bestandsmarktgruppe (Gliptine) in den laufenden Preisverhandlungen final entschieden worden ist [3]. Ob zukünftig Wettbewerbsverzerrungen aufgrund der Stichtagsregelung des AMNOGs vermieden werden können, wird sich im anstehenden Gesetzgebungsverfahren zur Änderung der frühen Nutzenbewertung herausstellen.
1.3 Timelines des Verfahrens der frühen Nutzenbewertung
Die Dossiereinreichung erfolgt spätestens mit der Meldung in der Lauertaxe zur Markteinführung durch das pharmazeutische Unternehmen, wobei der zeitliche Vorlauf von rund neun Monaten für das pharmazeutische Unternehmen beträgt. Das Dossier wird innerhalb von 3 Monaten durch IQWiG, G-BA oder Dritte bewertet und publiziert. Es folgt eine schriftliche und mündliche Anhörung innerhalb von drei Wochen und anschließend eine Beschlussfassung durch den G-BA.
1.4 Preisfestlegung
Wurde ein Zusatznutzen vom G-BA anerkannt, erfolgt in den nächsten sechs Monaten eine Preisverhandlung zwischen pharmazeutischem Hersteller und Spitzenverband Bund über einen Erstattungsbetrag für alle Krankenkassen als Rabatt auf den ursprünglichen, durch das pharmazeutische Unternehmen selbst festgelegten Abgabepreis (siehe SGB V §130b). Kommt es in der Verhandlung zu keiner Einigung, dann setzt eine „Schiedsstelle“ den Erstattungsbetrag fest. Ein Parameter soll dabei das „europäische Preisniveau“ sein.
Als einziges Druckmittel um Einfluss auf einen aus Sicht des pharmazeutischen Unternehmens fairen Preis zu nehmen, bleibt dem PU die Option „Out“, d.h. die Rücknahme des Arzneimittels aus dem deutschen Markt. Bisher wurden 6 Arzneimittel vom Markt genommen bzw. in Deutschland nicht eingeführt. Es ist sicherlich noch zu früh, um abschließend zu bewerten, ob dadurch die Therapiemöglichkeiten in Deutschland signifikant beeinträchtigt werden.
Diese Entwicklung zeigt, dass eine der Zielsetzungen der Politik, weiterhin in Europa Innovationen für Patienten schnell zugänglich zu halten, zumindest einmal in Frage gestellt ist. Bei nicht anerkanntem Zusatznutzen erfolgt eine Eingruppierung in eine Festbetragsgruppe, sofern möglich. Ist dies nicht möglich, dürfen die Jahrestherapiekosten nicht höher sein als die der zweckmäßigen Vergleichstherapie (ZVT) (s. Abb. 2).

Timelines der frühen Nutzenbewertung nach § 35 a SGB V
Spätestens nach 12 Monaten nach Einreichen eines Nutzendossiers soll ein Erstattungsbetrag festgelegt sein, der rückwirkend wirksam wird (s. Abb. 2). Im Anschluss kann das Arzneimittel in den vom G-BA anerkannten Zusatznutzen-Indikationen verordnet werden, ohne dass, im Falle einer Richtgrößenprüfung, die Verordnungen zu einer Belastung des individuellen Arzneimittelbudgets führen. Diese Verordnungen können als Praxisbesonderheit anerkannt werden, wie im Falle von Ticagrelor® [11] oder nicht, wie bei Incivo® [12].
Ein großer Diskussionspunkt war und ist hier die Offenlegung des verhandelten Rabatts. Diese ist politisch gewollt und wurde bereits für das erste abgeschlossene Nutzenbewertungsverfahren von Ticagrelor® praktiziert.
Auch die 16. AMG-Novelle brachte hier keine Änderung [13]. Für die Pharmazeutische Industrie wird durch eine Offenlegung eine negative Preisspirale innerhalb Europas initiiert, da Deutschland für einen Großteil der europäischen Länder als Preisreferenzland dient. Dieser Umstand trägt, ebenso wie die fehlende Transparenz in den Entscheidungsprozessen, dazu bei, dass sich die pharmazeutischen Unternehmen zukünftig genau überlegen werden, ob sie ein neues Medikament im deutschen Markt einführen.
Nach der Festlegung des Erstattungsbetrages können dezentrale Verträge zwischen pharmazeutischen Hersteller und Krankenkassen geschlossen werden. Der hier vereinbarte Preis sollte jedoch den zentral festgelegten Preis nicht überschreiten. Dies dürfte die Motivation der Pharmazeutischen Industrie, ihr Know-How und ihre Kapazitäten für eine bessere Versorgung der Patienten einzubringen schmälern, wodurch der ursprünglich politisch gewollte Wettbewerb um mehr Qualität in der Versorgung kontakariert wird.
Ebenso kann von beiden Verhandlungsseiten eine Kosten-Nutzen-Bewertung beantragt werden, wobei die Frage, wie man einem Nutzen eine quantifizierbare Kostendimension zuordnen kann, derzeit Gegenstand einer Methodenerarbeitung durch das IQWiG ist. In Erarbeitung befinden sich derzeit die Methodik der Conjoint Analysis und der Analytic Hierachic Analysis [14]. Diese Methoden bieten gegenüber den bisherigen Methoden den Vorteil, dass erstmalig Patientenpräferenzen bezüglich der Endpunkte berücksichtigt werden und hiermit der Patientennutzen tatsächlich in den Mittelpunkt rückt.
1.5 Bedeutung von G-BA und IGWiG im Rahmen der frühen Nutzenbewertung
Der Gemeinsame Bundesausschuss (G-BA), das oberste Beschlussgremium der gemeinsamen Selbstverwaltung der Ärzte, Zahnärzte, Psychotherapeuten, Krankenhäuser und Krankenkassen in Deutschland, wurde am 1. Januar 2004 durch das GKV-Modernisierungsgesetz (GMG) gegründet. Ihm obliegt die Ausgestaltung der gesundheitlichen Versorgung der Bevölkerung. Die vom G-BA beschlossenen Richtlinien haben den Charakter so genannter „untergesetzlicher Normen“, d. h. sie sind für die gesetzlichen Krankenkassen und die behandelnden Ärzte sowie andere Leistungserbringer verbindlich.
Diese Machtfülle wurde mit der Arzneimittel-Nutzenverordnung vom 28.12.2010 deutlich erweitert, in der der Gesetzgeber den G-BA als durchführendes Organ der frühen Nutzenbewertung nach § 35a SGB V benannt hat. Hiernach entscheidet der G-BA zentral, wie hoch die subjektive Größe „Nutzen“ eines Arzneimittels für Patienten in Deutschland ist. Hinter dem Begriff „Nutzen“ stehen gesellschaftliche Werteentscheidungen (bekennende Werteentscheidung), die hiermit zentralisiert werden.
Bereits im Jahr 2004 hatte der Gesetzgeber den G-BA beauftragt, ein unabhängiges, wissenschaftliches Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) zu errichten, das den medizinischen Nutzen, die Qualität und die Wirtschaftlichkeit von Leistungen, diagnostischer und therapeutischer Verfahren bei ausgewählten Krankheiten sowie den Nutzen von Arzneimitteln in der gesetzlichen Krankenversicherung bewerten soll.
Der G-BA erteilt dem IQWiG zwar Aufträge, dieses arbeitet aber eigenständig anhand des aktuellen medizinischen Wissenstandes (evidenzbasierte Medizin). So sind die Nutzenbewertungen des IQWiG für den G-BA eine wichtige Entscheidungshilfe und müssen nach dem Willen des Gesetzgebers in die Richtlinien mit einfließen. Die abschließende Entscheidung des G-BA wird aber nicht vorweg genommen. Die Trägerorganisationen des G-BA, Krankenkassen, Krankenhäuser und Vertragsärzte, sind hierfür nicht demokratisch legitimiert. Werteentscheidungen erfordern ein breiteres Mandat. Daher sollten Vertreter der medizinischen Wissenschaft und der Selbsthilfe wirksam mit eingebunden werden. Der BPI forderte daher schon früh eine Reform des G-BA und schlug u.a. einen wissenschaftlichen Beirat und eine wissenschaftliche Schiedskommission vor. Außerdem sollten die Vertreter der jeweiligen Selbsthilfeorganisation Stimmrecht erhalten vor [15].
2 Aktueller Status nach drei Jahren
Bis zum 31.12.2013 wurden vom G-BA für 77 Arzneimittel mit neuen Wirkstoffen ein Nutzenbewertungsverfahren eröffnet, von denen 64 bereits abgeschlossen waren.
Konfliktfrei verliefen die bisherigen Verfahren naturgemäß nicht. Umstritten war und ist bei den Bewertungen insbesondere die Frage nach der Operationalisierung des Zusatznutzens und eng damit verbunden die Frage nach der geeigneten zweckmäßigen Vergleichstherapie. Besonders problematisch war dies in denjenigen Fällen, in denen die Anforderungen des G-BA nicht in Einklang standen mit den Anforderungen der Zulassungsbehörden für die Durchführung von pivotalen Studien. Ende März 2012 trafen sich unter anderem Vertreter der Hersteller, der Krankenkassen und des G-BA im Bundesgesundheitsministerium (BMG), um eine für alle Seiten akzeptable Lösung zu finden. Erstes Ergebnis des Treffens: Der G–BA soll künftig ausführlich begründen, warum er eine bestimmte Vergleichstherapie gewählt hat [16]. Dies wurde in den nachfolgenden Verfahren vom G-BA transparent umgesetzt.
2.1 Erstattungsbetrag für innovative Arzneimittel
Nach den bereits drei Hürden, die ein pharmazeutischer Unternehmer für die Zulassung eines Arzneimittels in Deutschland nehmen muss (therapeutische Wirksamkeit, toxikologische Unbedenklichkeits- und pharmazeutische Qualitätsprüfung), muss er nun verpflichtend zur Markteinführung den Zusatznutzen gegenüber einer zweckmäßigen Vergleichstherapie belegen, sofern er einen höheren Preis erstattet haben möchte [17]. Wie bereits dargestellt, ist der Zusatznutzen als Bewertungskriterium für ein Arzneimittel mit der Möglichkeit, den Erstattungspreis zu regulieren, nicht die erste Möglichkeit um den Preis für innovative Arzneimittel zu begrenzen.
Neu ist aber die verbindliche Terminierung, sowie dass ein genau vorgegebenes Dossier mit der Markteinführung dem G-BA vorzulegen ist, als auch, dass nach der Entscheidung über einen Zusatznutzen das pharmazeutische Unternehmen zentrale Preisverhandlungen mit dem GKV Spitzenverband beginnt, die in der Festlegung eines Erstattungsbetrages münden. In den bisher 40 abgeschlossenen Verfahren wurden bei 32 Arzneimitteln eine direkte Einigung erzielt, in 5 Fällen wurde die Marktrücknahme (Opt-out) bzw. keine Markteinführung vorgenommen und in 3 Fällen wurde das Ergebnisse der Schiedsstelle angenommen [Stand: 01.01.2014]. Ob und inwieweit dies in Zukunft zu fairen Arzneimittelpreisen von innovativen Arzneimitteln führen wird, bleibt abzuwarten. Die ersten Verfahren sind bereits durch die Schiedsstelle entschieden worden. Die freie Preisbildung für ein innovatives Arzneimittel in Deutschland, die bisher den deutschen Markt so attraktiv für Arzneimittelhersteller gestaltet hat, ist endgültig Geschichte. Erschwerend für die Pharmazeutische Industrie kommt hinzu, dass der Ausgang des Bewertungsverfahrens ungewiss ist und somit die Planungsunsicherheit für ein pharmazeutisches Unternehmen weiter ansteigt.
2.2 Ausmaß und Operationalisierung des Zusatznutzens
Der Begriff des Nutzens und Zusatznutzens ist von dem der Wirksamkeit, die im Zulassungsverfahren belegt worden ist, abzugrenzen und wird in der Verfahrensordnung des G-BA in § 3 wie folgt definiert: „Der Nutzen eines Arzneimittels ist der patientenrelevante therapeutische Effekt insbesondere hinsichtlich der Verbesserung des Gesundheitszustandes, der Verkürzung der Krankheitsdauer, der Verlängerung des Überlebens, der Verringerung von Nebenwirkungen oder einer Verbesserung der Lebensqualität“ (…) „Der Zusatznutzen eines Arzneimittels ist ein Nutzen, der qualitativ oder quantitativ höher ist als der Nutzen, den die ZVT aufweist.“ [8] Anlass zu den aktuellen Diskussionen liefert insbesondere die Quantifizierung des Zusatznutzens im Verhältnis zu einer ZVT. Ausschlaggebend ist hier, dass es sich bei dem „Nutzen“ um eine subjektive Größe handelt, die nicht objektiv messbar ist und daher nicht zentral festgelegt werden sollte.
Aber auch, wenn man es akzeptieren würde, dass der Nutzen zentral festgelegt würde, bleibt eine Vielzahl an Unklarheiten.
Trotz aller Regulierungen ist das Ausmäß eines Zusatznutzens in den bisherigen Methodenpapieren des IQWiG noch nicht definiert worden, sondern ergibt sich lediglich aus dem Anhang der ersten Bewertung des Arzneimittels Ticagrelor (s. Tab. 1). In den entsprechenden Tabellen werden die patientenrelevanten Endpunkte: Mortalität, Morbidität, Nebenwirkungen und Lebensqualität qualitativ und quantitativ beschrieben (siehe dort). Die quantitative Beschreibung bezieht die Festlegung willkürlicher Obergrenzen für Konfidenzintervalle ein. Darüber hinaus sind die 95 % Konfidenzintervallobergrenzen sowie die relativen Risikoreduktionen extrem ambitioniert, so dass – insbesondere wenn schon wirksame Behandlungsregime zur Verfügung stehen – diese Grenzen in der Praxis kaum unterschritten werden können.
Diskussionswürdig scheint auch die Nicht-Berücksichtigung absoluter Risiken, da dies zu einer Verzerrung bei der Bewertung des Ausmaßes des Zusatznutzens, der für den Patienten relevant ist, führt. Eine Risikoreduktion der Mortalität von 80 % auf 40 % ist für den Patienten relevanter als von 0,8 % auf 0,4 %, auch wenn in beiden Fällen eine Risikoreduktion von 50 % zu verzeichnen ist.
Darüber hinaus scheint es fraglich das Ausmaß des Zusatznutzens vom Streuungsmaß des Effektschätzers abzuleiten, da das Streuungsmaß maßgeblich von der Fallzahl der eingeschlossenen Studienpopulation abhängt und nicht von der Qualität und Stärke des beobachteten Effekts. Es ist medizinisch nicht sinnvoll einem Arzneimittel einen höheren Zusatznutzen beizumessen nur weil eine Studie mit höherer Fallzahl verfügbar ist und deshalb das Streuungsmaß geringer ist. Dies benachteiligt Indikationsgebiete, bei denen nur eine begrenzte Anzahl an Patienten zur Verfügung steht. Hauptkritikpunkt an diesem Vorgehen ist – abgesehen von der fehlenden Rationale für die Grenzsetzungen – die Tatsache, dass diese Operationalisierung zahlreiche implizite Werteentscheidungen enthält, die zentral getroffen werden und über die objektiv messbaren Werteentscheidungen hinaus gehen.
Es ist fraglich, ob sich Werteentscheidungen mit Hilfe von mathematischen Modellen quantifizieren lassen, oder ob sie nicht auf Einzelfallentscheidungen in den jeweiligen Indikationsgebieten beruhen sollten. Hier ist eine wissenschaftliche Diskussion um Werteentscheidungen notwendig, bei der die am Bewertungsprozess betroffenen Parteien beteiligt werden sollten (Tab. 1).
2.3 Zusatznutzen für Orphan Drugs
Bei einem Arzneimittel gegen eine seltene Erkrankung, einem so genannten „Orphan Drug“, gilt nach der gesetzlichen Regelung der Zusatznutzen durch die Zulassung als belegt [18]. So reichen für die frühe Nutzenbewertung die Zulassungsstudien der Hersteller aus sowie ein sogenanntes „schlankes Nutzendossier“. Die im Rahmen der Zulassung bei der Europäischen Arzneimittelagentur (EMEA) gewählten Vergleichstherapien werden vom G-BA auch für die frühe Nutzenbewertung akzeptiert. Deshalb ist eine Einschaltung des IQWiG zur Erstellung einer Nutzenbewertung anhand einer anderen Vergleichstherapie nicht erforderlich [19]. Dennoch wird auch für Orphan Drugs das „Ausmaß des Zusatznutzens“ durch den G-BA quantifiziert. So wurde z. B. für Pirfenidon der Zusatznutzen als „nicht quantifizierbar“ eingestuft und für Tafamidis als „gering“.
Nicht transparent ist hier die Festlegung, dass für Orphans Drugs ab einem Umsatzvolumen von 50 Mio € pro Jahr doch ein vollständiges Nutzendossier eingereicht werden muss.
2.4 Heilung als erheblicher Zusatznutzen
Nach §5 Zusatznutzen der AM-NutzenV Absatz 7 Satz 1 liegt eine erheblicher Zusatznutzen immer dann vor, wenn insbesondere eine Heilung der Erkrankung gegenüber der ZVT erreicht wird.
Das IQWiG lässt dies willkürlich mit der Begründung „Heilung könne per se mit Mortalität abgeglichen werden“ in seiner Nutzenkonkretisierung weg.
Aus der Sicht der Autoren ist dies nicht korrekt. Eine Heilung liegt immer dann als gesicherte Erkenntnis auf Basis der EBM vor, wenn in RCT eine signifikant höhere Heilungsrate erreicht wird, unabhängig von arbiträren Hazard Ratio und oberen 95 % Konfidenzintervallen.
Für die Nutzenbewertung der neuen HCV-Therapien Telaprevir (Janssen) und Boceprevir (MSD) spielt dies eine wesentliche Rolle, da mit der Viruslast ein patientenrelevanter Endpunkt zur Verfügung steht, der nach nationalen und internationalen Leitlinien eine „Heilung“ der infizierten Patienten ermöglicht [19], der aber zunächst vom IQWiG nicht anerkannt wurde und im weiteren Verlauf vom G-BA als „nicht quantifizierbar“ eingestuft wurde [20]. Die Rationale für diese Entscheidungen bleibt unklar, da die genauen Ergebnisse zur Verbesserung der Heilungsraten in den Dossiers nachgewiesen worden sind. Hier wäre nach Arzneimittel-Nutzenverordnung die Kategorie erheblich zutreffender, die bisher in keinem der 64 abgeschlossenen Verfahren vergeben worden ist.
2.5 Wahrscheinlichkeit des Zusatznutzens
Das IQWiG hat folgende Anforderungen an die Aussagen zur Beleglage festgelegt und eine Klassifizierung in Beleg, Hinweis und Anhaltspunkt vorgenommen. Die Kriterien sind in Tab. 2 aufgeführt [21].
Dies bedeutet, dass, wenn z. B. auf Grund niedriger Patientenzahlen nur eine Studie möglich ist, der Hersteller maximal einen „Hinweis“ auf einen Zusatznutzen erzielen kann.
2.6 Zweckmäßige Vergleichstherapie
Die Festlegung einer zweckmäßigen Vergleichstherapie (ZVT) durch den G-BA hat sich als besonders problematisch in den ersten anderthalb Jahren des AMNOG erwiesen, da sie nicht mit den Vergleichstherapien aus den Zulassungsstudien abgestimmt werden musste. Somit konnte zum Zeitpunkt der Markteinführung eines neuen Arzneimittels das u.U. umfangreiche Studienprogramm im Vergleich zu der ZVT rein zeitlich nicht vorliegen. Dieser Kritikpunkt wurde im Sinne eines lernenden Systems bereits mit der aktuellen AMG Novelle aufgegriffen [22].
Für die frühe Nutzenbewertung ist die Festlegung der zweckmäßigen Vergleichstherapie und deren Anforderung von entscheidender Bedeutung. Die zweckmäßige Vergleichstherapie ist Dreh- und Angelpunkt der frühen Nutzenbewertung. Die Auswahl durch den G-BA bestimmt maßgeblich die Möglichkeit, einen Zusatznutzen nachzuweisen und auch die Höhe des zu verhandelnden Erstattungspreises. Dabei sind folgende Anforderungen des G-BA zu berücksichtigen [23]:
Die zweckmäßige Vergleichstherapie muss im Anwendungsgebiet grundsätzlich eine Zulassung haben. Sie muss nach den Maßstäben der internationalen Standards der evidenzbasierten Medizin ermittelt werden. Bei mehreren Alternativen ist die wirtschaftlichere Therapie zu wählen, vorzugsweise mit Festbetrag, und es sollten Therapien bevorzugt werden, deren patientenrelevanter Vorteil durch den G-BA bereits festgestellt worden ist.
Der pharmazeutische Unternehmer hat von Anfang an die Option von der zweckmäßigen Vergleichstherapie, die vom G-BA vorgeschlagen wurde, abzuweichen, muss dies aber begründen. In den bisherigen Verfahren hatte das IQWiG bzw. G-BA jedoch in ihren Entscheidungen (z. B. bei Linagliptin, Retigabin und Bromfenac) keine Abweichungen von den Vorgaben akzeptiert und das Dossier als ungültig eingestuft.
2.7 Patientenrelevante Endpunkte
Studienendpunkte werden im Rahmen der Nutzenbewertung nur dann berücksichtigt, wenn sie gemäß dem Methodenpapier des IQWiG patientenrelevant sind [21]:
-
Mortalität
-
Morbidität (Beschwerden und Komplikationen)
-
Gesundheitsbezogene Lebensqualität
-
Heilung wurde wie oben diskutiert weggelassen
Auch die Entscheidung über patientenrelevante Endpunkte (patient-related outcome PRO) beinhaltet erneut subjektive Werteentscheidungen. Dies erscheint problematisch, da bisher in keiner Weise die Patientenreferenzen zu Endpunkten ermittelt wurden. In der Nutzenbewertung zu Abirateronacetat (Onkologie) wurde z. B. trotz Vorliegen signifikant verbesserter Werte des chronischen Müdigkeitssyndroms Fatique unter der neuen Therapie dieser Aspekt nicht als PRO anerkannt. Da Fatique die Lebensfreude- und -qualität deutlich einschränkt, ist diese Entscheidung nicht unumstritten.
Zu diskutieren ist in diesem Kontext auch, dass etliche patientenrelevante Aspekte einer Therapie in RCT nicht messbar sind, wie z. B. Art der Applikation, Häufigkeit der Einnahme, Dauer der Therapie. Alle diese Faktoren haben einen signifikanten Einfluss auf die Therapiecompliance eines Patienten und damit auf die Versorgungsqualität aber auch auf die Kosten einer Therapie. Auch die Erfassung sehr seltener Nebenwirkungen z. B. Heparin-induzierte Thrombozytopenie, die bei synthetischen Wirkstoffen aufgrund der Molekülstruktur nicht zu erwarten sind, können in RCT grundsätzlich nicht geprüft werden und beinhalten dennoch einen relevanten Vorteil für die Patienten.
2.8 Surrogatparameter
Bisher hat das IQWiG in fast allen Fällen die Anerkennung von Surrogatparametern als patientenrelevante Endpunkte abgelehnt. Surrogatparameter sollen nur dann berücksichtigt werden, wenn nach Überprüfung der Operationalisierung, Patientenrelevanz und Validität auf Endpunktniveau nachgewiesen ist.
Insgesamt ist die Diskussion um die Validierung von Surrogatparametern schwer nachvollziehbar vor dem Hintergrund, dass selbst das IQWiG schreibt, dass es „für die Validierung eines Surrogatendpunktes weder ein Standardverfahren noch eine allgemein beste Schätzmethode noch ein allgemein akzeptiertes Kriterium, dessen Erfüllung den Nachweis der Validität bedeuten würde, gibt“ [24]. Auch in der praktischen Durchführung bleibt das Vorgehen des IQWiG im Hinblick auf die Anerkennung von Surrogatparametern intransparent und inkonsequent. Teilweise werden als patientenrelevant deklarierte Endpunkte als Surrogatparameter abgelehnt, wenn sie nicht ausreichend validiert worden sind, wie z. B. schwere, asymptomatische Beinvenenthrombosen. Zum Teil validiert das IQWiG aber Surrogatparameter z. B. SVR (Sustained Viral Response) bei HBV-Infektion und lässt sie zu, obwohl die Kriterien der Validierung nicht hinreichend erfüllt sind. Der G-BA hat den Endpunkt SVR gemäß den Vorgaben der nationalen und internationalen Leitlinien als patientenrelevant eingestuft und damit im Rahmen der gesetzlichen Vorgaben den internationalen Standard der EBM zu berücksichtigen. Der Entscheidungsprozess des G-BA basiert auf Einzelfallentscheidung und folgt nicht einem bestimmten Algorithmus.
Wichtig in der Diskussion um die Anerkennung von Surrogatendpunkten ist auch der Umstand, dass hier wiederum bekennende, d.h. subjektive Werteentscheidung getroffen werden.
2.9 Subgruppenanalysen
Subgruppenbildungen in klinischen Studien können nur deskriptiver Natur sein, da die Studien in der Regel auf die Gesamtpopulation bezogen sind. Wenn der G-BA mit seiner Forderung nach Subgruppen zu Alter, Geschlecht und Krankheitsstadium einen engeren Anwendungsbereich des Arzneimittels erreichen möchte, dann wäre dies besser mit einem Therapiehinweis möglich. Wenn Subgruppen dennoch a posteriori durchgeführt werden, sollten sie nur bei positivem Interaktionstest gegenüber der Gesamtpopulation berücksichtigt werden und hier auch nur explorativ und nicht konfirmatorisch interpretiert werden.
Die Subgruppen haben aufgrund der eingeschränkten Fallzahl eine geringere Genauigkeit des Ergebnisses und damit größere Konfidenzintervalle. Die Anwendung der oberen Konfidenzintervallgrenze auf Subgruppen führt zu einer systematischen Herabstufung des Zusatznutzens, da die Subgruppen definitionsgemäß immer kleinere Fallzahlen aufweisen als die der ursprünglichen Studienpopulation, auf welche die Studie gepowert wurde.
2.10 Anteile der gewährten Nutzenkategorien
Fasst man die bis Ende Dezember 2013 durchgeführten 64 abgeschlossenen Nutzenbewertungen zusammen und errechnet die Patientenzahlen, die bisher von einer positiven Nutzenbewertung durch den G-BA profitieren dürfen, so sieht man, dass nur für ca. 40 % der vom G-BA festgelegten Subpopulationen ein Zusatznutzen nachgewiesen werden konnte. (s. Tab. 3).
Die Kategorie „erheblicher Zusatznutzen“ erreichte keines der bisherigen Arzneimittel.
Dies liegt insbesondere darin begründet, dass der G-BA die vom IQWIG neu eingeführte Methodik zur Quantifizierung des Zusatznutzens konsequent nicht anwendet, als auch darin, dass der G-BA der starken Zersplitterung des zugelassenen Anwendungsgebietes durch das IQWiG in den ersten Verfahren widerstanden hat und so auf das klinisch relevante Maß zurückgeführt hat. Dadurch hat sich die Diskussion um Subpopulationsbildung im lernenden System weitgehend erledigt (Tab. 4).
Es zeigt sich, dass Orphan Drugs, Onkologika und Antiinfektiva mit jeweils geringen Patientenpopulationen eher gute Bewertungen erhalten, während Indikationsgebiete mit großen Patientenpopulationen (z. B. kardiovaskuläre Indikationen wie Herzinfarkt- oder Schlaganfallprophylaxe, Diabetes mellitus oder Arzneimittel im ZNS-Bereich bzw. sonstige Indikationen).
3 Fazit und Ausblick
Vor dem Hintergrund knapper werdender Ressourcen im Gesundheitswesen ist es nicht nur ökonomisch sinnvoll, sondern geradezu unerlässlich, sich über die Allokation der knappen Ressourcen Gedanken zu machen und eine öffentliche Diskussion um die Priorisierung von Leistungen anzustoßen, auf deren Basis eine transparente Rationierung von Gesundheitsleistungen stattfinden könnte.
Mit Hilfe des AMNOG möchte der Gesetzgeber die Basis schaffen, eine Priorisierung von Leistungen vorzunehmen, was grundsätzlich zu begrüßen ist. Ebenso zu begrüßen ist die Tatsache, dass der G-BA den AMNOG-Prozess der frühen Nutzenbewertung als lernendes System implementiert hat.
Die kritischen Details in der Umsetzung wurden diskutiert und einige dieser Probleme konnten durch die letzte AMG-Novelle teilweise behoben werden [25]. Dazu gehören die Bewertung von Orphan Drugs, die Begründung der zweckmäßigen Vergleichstherapie, die obligatorische Beteiligung der Zulassungsbehörden an der Frühberatung (vor Phase-III-Studien) und die sofortige erneute Einreichungsmöglichkeit bei Ablehnungen des Zusatznutzens aus formalen Gründen.
Folgende Punkte bleiben aber kritisch:
-
(1)
Zentralisierung bekennender (subjektiver) Werteentscheidungen Da der Nutzen keine objektiv messbare Größe darstellt, sondern von den Bedürfnissen und Präferenzen der Betroffenen abhängt und damit eine Werteentscheidung der Betroffenen darstellt, sollte eine Quantifizierung des Nutzens oder des Zusatznutzens nicht zentral „von oben“ für alle durchgeführt werden. Zentral sollten nur erkennende Werteentscheidungen, d.h. objektiv messbare Ergebnisse, wie z. B. die Wirksamkeit aufgearbeitet werden.
Das IQWiG sollte sich daher auf die transparente Darstellung der wissenschaftlichen Datenlage beschränken und keine Werteentscheidungen treffen, da es dafür nicht legitimiert ist.
-
(2)
Implizite Werteentscheidungen durch Operationalisierung des Zusatznutzens Die Operationalisierung des Zusatznutzens beinhaltet immer implizite Werteentscheidungen, die derzeit zentral und intransparent vom G-BA getroffen werden. Eine Methodik für eine Kosten-Nutzen-Bewertung der Leistungen, auf Grundlage derer eine Diskussion auf breiterer Basis um eine Priorisierung starten könnte, wurde bisher nicht etabliert.
-
(3)
Fehlende Berücksichtigung von Patientenpräferenzen Für eine Nutzenbewertung gilt es insbesondere die Patientenpräferenzen zu berücksichtigen; solange dies nicht geschieht, kann nicht von einem belegten Zusatznutzen für Patienten gesprochen werden.
-
(4)
Fehlender Anreiz für dezentrale Versorgungsverträge Dezentrale Versorgungsverträge, die politisch gewollt waren und sind, um einen Wettbewerb um ein Mehr an Qualität in der Patientenversorgung zu initiieren, werden für die Pharmazeutische Industrie, die hier als Geld- aber auch Ideengeber fungieren könnte, nicht attraktiver, da der nach §130b verhandelte Erstattungspreis als Höchstpreis für nachfolgende Verhandlungen fest gelegt wird. Damit wird der Wunsch des Gesetzgebers, pharmazeutische Unternehmen als Vertragspartner für integrierte Versorgungsverträge nach § 140b SGB V stärker in den Wettbewerb um eine qualitativ hochwertige Versorgung zu integrieren, kontakariert.
-
(5)
Formalisierung und Administration Die extreme Formalisierung des Prozesses birgt die Gefahr der Verselbstständigung, d.h. die Nutzenbewertung wird zum Selbstzweck und unterstützt nicht mehr die ursprünglichen Zielsetzungen der Politik. Daher sollten sich die Verantwortlichen immer wieder hinterfragen, ob der Patient noch im Mittelpunkt des Interesses steht. Dies führt zu immer umfangreicheren Dossiers mit z.T. über 1.000 Subgruppenanalysen pro Indikationsgebiet, die oft ohne jede klinische Relevanz sind. Für die Dossiereinreichung von Bestandsmarktprodukten wurde keine geeignete Methodik entwickelt, so dass die Dossiers kaum vollständig erstellbar sind. Sowohl die Politik wie auch der G-BA haben die Problematik erkannt und wollen den Bestandsmarktaufruf nicht weiter verfolgen.
-
(6)
Fehlende Integration der ambulanten und stationären Versorgung Die Bewertung der medizinischen Innovationen erfolgt heute durch den AMNOG Prozess nur für den ambulanten Bereich und trägt damit nicht dem Erfordernis Rechnung, die Ressourcen sektorübergreifend ökonomisch sinnvoll einzusetzen.
-
(7)
Wettbewerbsverzerrung durch Stichtagregelung des AMNOG Durch die Stichtagregelung des AMNOG zum 01.01.2011 kann es zu massiven Wettbewerbsverzerrungen kommen, z. B. wenn Wirkstoffe wie z. B. Prasugrel einer Festbetragsgruppe zugeordnet werden, die Generika wie z. B. Clopidogrel enthalten, während andere Wirkstoffe wie z. B. Ticagrelor der gleichen ATC-Klasse sich durch nachgewiesenen Zusatznutzen der Festbetragsgruppenbildung entziehen können. Dies ist insbesondere dann wettbewerbsverzerrend, wenn die Wirkstoffe (z. B. Ticagrelor vs. Prasugrel) aufgrund der klinischen Studienergebnisse als gleichwertig zu beurteilen wären. Es sind hier kreative, aber rechtsverbindliche Lösungen gefragt, z. B. eine optionale Bestandsmarktbewertung für den pharmazeutischen Hersteller, um einen Zusatznutzen nachzuweisen.
-
(8)
Große indikationsspezifische Unterschiede in den G-BA Bewertungen Es muss genau analysiert werden, warum in den großen Indikationsgebieten (Herzinfarkt-, Schlaganfallprophylaxe, Diabetes mellitus) die Bewertungen deutlich schlechter ausfallen als in den onkologischen Indikationen oder bei den Antiinfektiva.
Im Folgenden soll ein Vorschlag dargestellt werde, wie die künftige Versorgung und Finanzierung von Innovationen gestaltet werden kann.
Die gedankliche Überschrift für ein solches Modell lautet, alle subjektiven Werteentscheidungen, wie z. B. über den Nutzen für Patienten, so weit wie möglich dezentral zu fällen und lediglich die objektiv messbaren Werteentscheidungen durch zentrale Institutionen aufzubereiten. Ebenfalls kann zentral eine Methodik für eine Kosten-Nutzen-Analyse als Diskussionsgrundlage erarbeitet werden. Dieser Ansatz würde nicht nur den politischen Wunsch nach mehr Wettbewerb im Gesundheitssystem unterstützen, sondern hätte auch den Vorteil einer deutlich höheren Fehlerfreundlichkeit.
In Anlehnung an den Vorschlag der Bioethik-Kommission der Bayrischen Staatsregierung im Positionspapier über die zukünftige Finanzierung des medizinischen Fortschritts im solidarisch finanzierten Gesundheitssystems [26], können folgende Schritte definiert werden:
-
(1)
Bewertung des Regelleistungskatalogs mit Hilfe einer Kosten-Nutzen-Analyse und anschließender Priorisierung. Die nicht vermeidbare Rationierung von Leistungen muss transparent erfolgen.
-
(2)
Innovationen: Ein wissenschaftliches Institut erarbeitet auf Basis objektiv messbarer Kriterien einen Vorschlag zu einer Kosten-Nutzen-Analyse. Diese dient als Diskussions- und Entscheidungsgrundlage für dezentrale Verhandlungen zwischen Herstellern, Leistungserbringern und Kostenträgern unter Beteiligung der Patientenvertretung.
-
(3)
Die Innovationen müssen unter Alltagsbedingungen evaluiert werden und können bei entsprechender Tauglichkeit als Satzungsleistung von den Krankenkassen übernommen werden. Hiermit würde die Versorgungsforschung in Deutschland ein solides Fundament erhalten.
-
(4)
Um die Add-on Problematik zu vermeiden, müssen regelmäßig weniger taugliche Methoden wieder aus den Satzungsleistungen ausgeschlossen werden.
Bis zu diesem Zeitpunkt, kann von dem „conditional reimbursement“ (vorläufiger Erstattung) verstärkt Gebrauch gemacht werden, um eine flächendeckende Versorgung mit dem neuen Arzneimittel sicherzustellen.
-
(5)
Der geplante Wegfall des Bestandsmarktaufrufs sollte nicht zu Wettbewerbsverzerrungen führen. Die pharmazeutischen Hersteller sollten die Möglichkeit einer optionalen Bestandsmarkteinreichung erhalten, um einen Zusatznutzen nachzuweisen, sofern ein neues Arzneimittel nach §35a SGB V bewertet worden ist.
Unter diesen Bedingungen wären dezentrale Verhandlungen zwischen den Beteiligten, wie sie ursprünglich von der Gesundheitspolitik erwünscht waren, wieder attraktiv und sinnvoll.
Die zahlreichen positiven Ansätze zwischen der Pharmazeutischen Industrie und den Krankenkassen, die Versorgung chronisch kranker Patienten zu verbessern, könnten hier aufgegriffen und weiter entwickelt werden.
Ein wesentlicher Faktor für die Finanzierbarkeit dieser wettbewerblichen Aktivitäten sind transparente und faire Preisverhandlungen, die einen adäquaten Preis für Innovationen ermöglichen. Damit bleibt nicht nur die Arzneimittelforschung auch in Zukunft attraktiv, sondern auch die finanziellen Spielräume der Pharmazeutischen Industrie bleiben erhalten. In diesem Zusammenhang wäre eine Geheimhaltung der verhandelten Preisrabatte hilfreich, um einer negativen Preisspirale innerhalb Europas für das Pharmazeutische Unternehmen vorzubeugen.
Literatur
Das Gesetz zur Neuordnung des Arzneimittelmarktes (AMNOG). http://www.bgbl.de/Xaver/start.xav?startbk=Bundesanzeiger_BGBl&bk=Bundesanzeiger_BGBl&start=//*[@attr_id=’bgbl110s2262.pdf’]#__Bundesanzeiger_BGBl__%2F%2F*
Bundestag hat AMG-Novelle beschlossen. Abgerufen am 07.01.2014
Koalitionsvertrag zwischen CDU, CSU und SPD, 17. Legislaturperiode – Deutschlands Zukunft gestalten. 2013. Abgerufen am 07.01.2014] www.spd.de/linkableblob/112790/data/20131127_koalitionsvertrag.pdf
Arzneimittelrichtlinie Clopidogrel. Erhältlich unter: http://www.g-ba.de/downloads/39-261-634/2008-02-21-AMR10-Clopidogrel-ASS_BAnz.pdf. Abgerufen am 22.12.2013
Aufzahlung für GKV Patienten? Ärztezeitung vom 03.09.2009 http://www.aerztezeitung.de/politik_gesellschaft/berufspolitik/article/563936/aufzahlung-gkv-patienten.html?sh=3&h=1888414273. Abgerufen am 22.12.2013
Efpia, Pressemitteilung Nr. 17/2012; vom 8.6.2012. Abgerufen am 22.12.2013
Arzneimittel-Nutzenbewertungsverordnung vom 28. Dezember 2010 (BGBl. I S. 2324). Abgerufen am 07.01.2014. http://www.gesetze-im-internet.de/bundesrecht/am-nutzenv/gesamt.pdf
Verfahrensordnung des gemeinsamen Bundesausschusses. Erhältlich unter: http://www.g-ba.de/downloads/62-492-634/VerfO_2012-06-21.pdf. Abgerufen am 27.12.2013
Allgemeine Methoden IQWIG (Version 4.0) https://www.iqwig.de/download/IQWiG_Methoden_Version_4_0.pdf. Abgerufen am 27.12.2013
Ticagrelor Nutzenbewertung. Erhältlich unter: http://www.g-ba.de/downloads/92-975-5/2011-01-01-D-001_Ticagrelor_IQWiG-Nutzenbewertung.pdf. Abgerufen am 27.12.2013
Ticagrelor Anzeige, Ärzte Zeitung, Ausgabe 145, 21.08.2012
Ein Preis, aber keine Praxisbesonderheit. Erhältlich unter: www.aerztezeitung.de/news/article/824636/telaprevir-preis-aber-keine-praxisbesonderheit.html. Abgerufen am 26.10.2012
Deutsches Ärzteblatt: Bundestag verabschiedet AMG Novelle. Erhältlich unter: https://www.aerzteblatt.de/nachrichten/50712. Abgerufen am 08.11.2012
Mühlbacher, A. Suche nach dem Maß des Gesamtnutzen. In Monitor Versorgungsforschung. 2012. Ausgabe 04/12, S. 12–16
Stellungnahme des BPI zum AMNOG. Erhältlich unter: http://www.bpi.de/fileadmin/media/bpi/Downloads/Internet/Geschaeftsfelder/Gesundheitspolitik/2010-09-22%20BPI-AMNOG%20Stellungnahme%20Anh%C3%B6rung%20AfG.pdf. Abgerufen am 22.12.2013
Protokoll Expertengespräch am 22. März Die frühe Nutzenbewertung von Arzneimitteln Umsetzungen Erfahrungen Folgerungen. Erhältlich unter: http://www.g-ba.de/downloads/17-98-3240/Expertengespr%C3%A4ch%20am%2022-03-2012_Protokoll%20G-BA.pdf. Abgerufen am 22.12.2013
Tunder R. Market Access im Spiegel der Zeit. In: Ecker T, PreußKJ, Tunder R, Hrsg. Handbuch Market Access. Fachverlag Handelsblatt, Düseldorf. 2011. S. 2.
vfa Pressemitteilung 002/2012. Erhältlich unter: http://www.vfa.de/de/presse/pressemitteilungen/pm-002-2012-seltene-erkrankungen-vertragen-keine-unverbindlichen-vorschlaege.html. Abgerufen am 25.10.2012
vfa Pressemitteiliung 007/2012. Erhältlich unter: http://www.vfa.de/de/presse/pressemitteilungen/pm-007-2012-pharmaforschung-bringt-ueber-20-neue-medikamente-gegen-seltene-erkrankungen-in-die-zulassung.html. Abgerufen am 25.10.2012
http://www.dgvs.de/media/Leitlinie_Hepatitis_C_2010_ZfG.pdf. Abgerufen am 8.11.2012
Allgemeine Methoden IQWIG. Version 4.1 vom 28.11.2013. Abgerufen 07.01.2014. https://www.iqwig.de/download/IQWiG_Methoden_Version_4-1.pdf
Pressemitteilung Bundesministerium für Gesundheit 2012/02. Erhältlich unter: http://www.bmg.bund.de/ministerium/presse/pressemitteilungen/2012-02/bundestag-hat-amg-novelle-beschlossen.html. Abgerufen am 9.11.2012
Verfahrensordnung des Gemeinsamen Bundesausschusses. Erhältlich unter: http://www.g-ba.de/downloads/62-492-548/VerfO_2011-01-20_2011-08-04.pdf?. Abgerufen am 22.12.2013
Bundesrat billigt Novelle des Arzneimittelgesetzes. Erhältlich in: http://www.aerzteblatt.de/nachrichten/51751. Abgerufen am 18.11.2012
Hankowitz, J, Pirk, O. AMNOG-Monitor – Überblick über die frühe Nutzenbewertung. (2014) http://www.diapharm.com/deutsch/newsletter/value-dossier-monitor.html. Abgerufen 07.01.2014
Positionspapier über die zukünftige Finanzierung des medizinischen Fortschritts im solidarisch finanzierten Gesundheitssystem. Abgerufen 07.01.2014. http://www.bioethik-kommission.bayern.de/Anlage10369034/anlage.pdf
Interessenkonflikte:
Dr. Johannes Hankowitz erhielt für Beratungen und Vorträge von folgenden pharmazeutischen Unternehmen Honorare: BMS, Daiichi, GSK, Gilead, Sanofi, Sanofi Pasteur, Winthrop, Zentiva
Open Access
Dieser Artikel unterliegt den Bedingungen der Creative Commons Attribution License. Dadurch sind die Nutzung, Verteilung und Reproduktion erlaubt, sofern der/die Originalautor/en und die Quelle angegeben sind.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Höhle-Pasques, S., Hankowitz, J. & Oberender, P. Drei Jahre frühe Nutzenbewertung nach §35a SGB V – kritische Würdigung und Lösungsvorschlag. PharmacoEcon. Ger. Res. Artic. 11, 13–24 (2013). https://doi.org/10.1007/s40275-014-0007-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40275-014-0007-4