
Abstract
Melanoma is an aggressive, rapidly developing form of skin cancer that affects about 22 per 100,000 individuals. Treatment options for melanoma patients are limited and typically involve surgical excision of moles and chemotherapy. Survival has been improved in recent years through targeted small molecule inhibitors and antibody-based immunotherapies. However, the long-term side effects that arise from taking chemotherapies can negatively impact the lives of patients because they lack specificity and impact healthy cells along with the cancer cells. Antibody–drug conjugates are a promising new class of drugs for the treatment of melanoma. This review focuses on the development of antibody–drug conjugates for melanoma and discusses the existing clinical trials of antibody–drug conjugates and their use as a melanoma treatment. So far, the antibody–drug conjugates have struggled from efficacy problems, with modest effects at best, leading many to be discontinued for melanoma. At the same time, conjugates such as AMT-253, targeting melanoma cell adhesion molecule, and mecbotamab vedotin targeting AXL receptor tyrosine kinase, are among the most exciting for melanoma treatment in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Antibody drug conjugates are a rapidly expanding type of cancer therapy. However, their development and application to advanced melanoma has lagged. Here, we summarize the clinical trials of antibody–drug conjugates in melanoma, finding that the existing clinical data are limited to approximately eight molecular targets, with melanoma cell adhesion molecule and AXL being the most promising. Generally, the antibody–drug conjugates are tolerable and safe, but currently progress suffers from lack of effective antibody–drug conjugate targets. |
1 Introduction: Overview of Melanoma
Melanoma is a type of skin cancer that occurs when melanocytes, or the cells that give skin its dark, brown color, exhibit unregulated proliferation. The first notable symptoms can be a new skin growth such as a mole or a change in an existing mole, such as it growing larger than 1 cm, taking an asymmetrical shape, or becoming discolored. When found early, melanoma is usually treatable, as the moles can be removed via surgery. However, when detected later in its stages, it is more likely to spread to other parts of the body, making it much more difficult to treat.
The risk of getting melanoma is 20 times higher in white individuals than in other ethnicities. The risk of melanoma also increases as individuals age, especially after the age of 65 years. Therefore, when it comes to development of drugs, one should consider both the efficacy as well as the possible side effects with these specific individuals. Many experts believe that melanoma occurs due to a combination of genetic and environmental factors, which include exposure to ultraviolet (UV) light, a history of sunburn, fair skin, and having more than 50 moles [1].
The Tumor, Nodes, and Metastasis (TNM) classification system of malignant tumors is used in melanoma to determine the stage of the cancer. Key factors include: (1) the tumor thickness and ulceration (T), (2) whether the cancer has spread to nearby lymph nodes (N), and (3) whether the cancer has metastasized to distant organs (M). Stages range from 0, or melanoma in situ, to stage IV, the most severe stage of melanoma. There are multiple types of melanomas that can manifest. The most common type is skin, or cutaneous, melanoma. This type of melanoma typically begins in the epidermis of the skin. Others, which are far less common, include ocular melanoma, mucosal melanoma, and pediatric melanoma. These are often seen in stage 0 because the cancer is confined to the epidermis. With each additional stage, the tumor size continues to grow, becomes ulcerated, and spreads to lymph nodes and other areas of the body.
Stage IV melanoma, known as advanced melanoma, indicates that the tumor has spread to nearby lymph nodes and organs such as the lungs, liver, brain, gastrointestinal tract, and bones. This is the most severe case of melanoma with a 5-year survival rate of only 22.5% [2]. Stage IV melanoma can also be characterized by the level of serum lactate dehydrogenase (LDH). It is an indicator of how aggressive the tumors are. The higher the amount of LDH, the more cancer there is in the body. The location at which the cancer has spread in the body can impact the symptoms an individual might face. Symptoms of metastatic melanoma include fatigue, swollen or painful lymph nodes, weight loss, loss of appetite, bone pain, headaches, seizures, and swelling of the liver. Stage 4 melanoma can be further staged as M1a, which is cancer that has metastasized to skin far from its original site, M1b, which is cancer that has spread to the lungs, M1c, which is cancer that has spread to parts of the body other than the central nervous system, and M1d, which is cancer that has spread to the central nervous system, including the brain and spinal cord [3]. It can be further staged as 0, where LDH levels are not elevated or 1, where LDH levels are elevated. With all the various therapies on the market, planned treatment should consider where the cancer has spread to fit the needs of the individual.
2 Search Methodology
Papers in this review were identified through searches of PubMed and clinicaltrials.gov. PubMed searches included the years 1998–2024, with Medical Subject Headings (MeSH) terms used including “melanoma,” “antibody–drug conjugates,” “pharmacokinetics,” and “clinical trial.” For articles that included information about clinical trials, more information was found detailing the trials through clinicaltrials.gov, including participant inclusion/exclusion criteria, study start/end date, and number of participants. Clinical trials that used antibody–drug conjugates for other indications with melanoma-relevant therapeutic targets were also searched for and included. This was done through PubMed, which was searched from the year 1998 to 2024 and using the terms “antibody–drug conjugates,” “clinical trial,” and “target.” For general information about antibody–drug conjugates, such as their design and pharmacokinetics, citation tracking was performed from the year 1980 to 2024, and background research was obtained from our prior review articles on antibody–drug conjugates [4, 5].
3 Current Treatments of Stage IV Melanoma
3.1 Overview
Current melanoma therapies can slow the progression of the disease. Prior to modern immune and targeted therapies in 2009, there was a 1-year survival rate for patients diagnosed with stage IV melanoma [6]. With advancements in therapies (Fig. 1), this survival rate has increased to 5 years. Due to the cancer being localized to multiple regions of the body, including the brain, lungs, and spine, it is one of the most difficult cancers to treat today. Immunotherapy-targeted drugs are often more effective than the traditional chemotherapy. Typically, checkpoint inhibitors are the first drugs tried. These include pembrolizumab (Keytruda), nivolumab (Opdivo), nivolumab combined with relatlimab (Opdualag), or pembrolizumab plus ipilimumab (Yervoy) [7], which are used in patients who do not possess the BRAF gene, which is a gene that encodes a protein that is involved in sending signals in cells and cell growth [8]. If the BRAF gene is found, patients are typically given a BRAF inhibitor and a mitogen-activated extracellular signal-regulated kinase (MEK) inhibitor. MEK is triggered by growth factors from BRAF or activating mutations of major oncogenic proteins such as Ras and Raf [9]. In any of these situations, patients must be watched for serious side effects due to off-target effects of the medication. Food and Drug Administration (FDA)-approved drugs for melanoma include aldesleukin, nivolumab, ipilimumab, pembrolizumab, and tebentafusp-tebn [8].

Timeline of FDA approval of current melanoma therapies. The mechanism of action for current melanoma therapies is highlighted [2]. The year of FDA approval for melanoma is listed when an agent was developed for melanoma. The overall approval years are given for Gleevec and temozolomide, which were developed for other indications. Gleevec is a BCR-Abl inhibitor that also inhibits c-Kit
3.2 Non-pharmacological Therapies
Non-pharmacological therapies are not typically used for treatment of stage IV melanoma, but rather for management of symptoms. Metastatic melanoma can be found using imaging tests such as computed tomography (CT) or magnetic resonance imaging (MRI) scans. With these tests, practitioners can see where the cancer has spread and determine their course of action. Surgery is not typically done in stage IV melanoma, but might be used to remove tumors to help relieve symptoms and to improve the quality of life. Similarly, radiotherapy can also be advised to patients to help alleviate symptoms. Commonly, radiotherapy is used with brain metastasis, as it is able to relieve symptoms such as headaches and prevent further tumor growth. It is also commonly used for patients whose melanoma has spread to the bones or parts of the body that are not accessible through surgery. Radiotherapy does damage both normal and cancer cells, which indicates the lack of specificity in this option [6].
3.3 Immunotherapies
Immune checkpoint inhibitors are a preferred option for advanced melanoma. Immune checkpoint blockade allows immune cells to target the tumor and initiate an immune response. There are multiple types of immune checkpoint inhibitors, which target PD-1, PD-L1, CTLA-4, and LAG-3. Often these are antibodies that block extracellular interactions of these proteins with their ligands. Common immune-targeted inhibitors used for melanoma today can be seen in Table 1. Common side effects that can arise from taking these drugs include fatigue, cough, nausea, skin, rash, poor appetite, constipation, joint pain, and diarrhea [7]. Combination of checkpoint inhibitors are considered to be more effective, but they do lead to more side effects. Common combinations include using both Opdivo (nivolumab) with Yervoy (ipilimumab), or more recently, a single drug that combines nivolumab and relatlimab-rmbw, known as Opdualag.
3.4 Targeted Therapies
Targeted therapies, which are used as a second-line option, are helpful in treating melanomas that have certain genetic changes [7]. Specifically, melanoma can have a mutation in the BRAF gene where the altered BRAF protein is oncogenic. Presence of the BRAF mutation is determined by biopsy. Common BRAF inhibitors include vemurafenib (Zelboraf), dabrafenib (Tafinlar), and encorafenib (Braftovi). These are typically taken as pills once or twice per day. Side effects that usually occur include rash, itching, sensitivity to the sun, headache, fever, joint pain, hearing loss, and nausea. Serious side effects such as heart rhythm problems, liver problems, kidney failure, and severe allergic reactions can occur. MEK inhibitors are used alongside BRAF inhibitors because MEK and BRAF work in the same signaling pathway. Commonly used drugs include trametinib (Mekinist), cobimetinib (Cotellic), and binimetinib (Mektovi). These are pills that are taken once or twice per day and cause similar adverse effects as BRAF inhibitors. Using both inhibitors together might potentiate the adverse effects to a more severe degree. There are also drugs that target cells with c-Kit mutations. These are seen more commonly in melanomas that start in certain parts of the body, such as the hands, mucosal areas, or areas that have chronic sun exposure. Common drugs used include imatinib (Gleevec) and nilotinib (Tasigna). These are used less compared with the checkpoint inhibitors and BRAF/MEK inhibitors, but they are a viable option for patients who have failed certain drug therapies.
3.5 Chemotherapy
Chemotherapy is reserved for advanced melanoma after the previously mentioned treatments have been tried [7]. Chemotherapy is typically not as helpful for melanoma as compared with other forms of cancers, but it aids in shrinking tumors. Chemotherapy is chosen when a patient is unable to have a targeted drug or immunotherapy [11]. These are drugs that kill cancer cells by either being injected via the bloodstream or taken via a pill. Commonly used chemotherapy drugs include dacarbazine (DTIC), temozolomide, and nab-paclitaxel [6]. There are multiple side effects that can arise while taking chemotherapy drugs. These include hair loss, mouth sores, loss of appetite, and increased risk of infection. In severe cases, neuropathy can arise due to damage to the nerves. Typically, chemotherapy is taken along with trametinib. Due to the increase in side effects, providers may prefer to try other options first.
3.6 Clinical Trials
Clinical trials to identify new melanoma therapies, such as cell-based therapies and antibody drug conjugate-based therapies, are ongoing. For example, a recent clinical study performed in September 2022 using tumor-infiltrating lymphocytes showed promising results where progression-free survival was more than twice as long in the TIL group as in the ipilimumab group, and the hazard of disease progression or death was 50% lower [12]. These findings showed that this therapy can be effective as first-line treatment. At the same time, a number of antibody–drug conjugates are undergoing development, as discussed in the following sections.
4 Development of Antibody–Drug Conjugates for Melanoma
There are multiple options for available approaches to melanoma therapy, each with their own risks and benefits. Determination of the therapies mentioned previously that would be more effective for the patient is based on provider insight and patient input. It is worth discussing the advantages and disadvantages of each therapy with the patient and advocate for patients to enlist in a clinical trial when appropriate. There are multiple clinical trials being conducted today on drugs that are constantly being newly developed for melanoma, including trials for antibody–drug conjugates [13, 14]. This section reviews some of the general features of antibody–drug conjugate design.
4.1 What is an Antibody–Drug Conjugate?
Antibody–drug conjugates consist of an antibody framework, cytotoxic payload, and linker (Fig. 2). The antibody should be optimized to bind a good target antigen at the intended action site. The antibody should be able to perfuse through the tumor lesion and bind cancer antigens with a high affinity. The linker joins the cytotoxic drug with the antibody covalently, which allows for selective release of the drug. Common cytotoxic payloads include pyrrolobenzodiazepine (PBD) and monomethyl auristatin E (MMAE). The payloads allow for killing of tumor cells at intracellular concentrations that are achieved following antibody–drug conjugate internalization. Each of these factors contributes to the specificity of antibody–drug conjugates that allows for their promising effects on tumor eradication [14].

Generalized antibody–drug conjugate structure. The antibody–drug conjugate structure contains an antibody used for tumor cell targeting, a cytotoxic payload for tumor cell killing, and a linker that controls the payload release. Fab antigen binding fragment, Fc constant fragment, PBD pyrrolobenzodiazepine, PPD peptidyl-prolyl isomerase domain, MMAE monomethyl auristatin E, MMAF monomethyl auristatin F
4.2 Antibody–Drug Conjugate Design: Target Antigen
The target antigen is a key aspect of antibody–drug conjugate design, as it is crucial in the specificity of where the antibody–drug conjugate distributes. The ideal target antigen would be specifically expressed by tumor cells. If a target antigen is expressed on healthy cells, then there is a concern of off-target toxicity of the antibody–drug conjugate, leading to adverse effects. The target antigen must have a sufficient surface-retained profile to facilitate an adequate antibody–drug conjugate concentration within the tumor and be able to exhibit suitable target internalization following binding [15]. Target receptor density and internalization rates determine the target capacity of the receptor. It is desirable to have a high-target capacity, as it will allow sufficient concentration of payload to inhibit tumor progression.
4.3 Antibody–Drug Conjugate Design: Antibody
The antibody is the main component in an antibody–drug conjugate that attaches to the linker and the payload. The antibody provides a layer of specificity to cancer cells, perfusing the tumor lesion and binding cancer target antigens with a desirable affinity. The antibody usually consists of human or humanized immunoglobulin G (IgG) to avoid adverse immunogenic reactions in patients. In addition, IgG has a relatively long half-life [16]. The antibody should be able to be internalized via receptor-mediated endocytosis, as internalizing efficiency of an antibody–drug conjugate influences cellular payload levels. Once the cytotoxic payload has been delivered, the antibody can be recycled to the plasma membrane or degraded via lysosomal enzymes [17]. Specific antibodies with relevance to antibody–drug conjugates for melanoma will be discussed in the following sections.
4.4 Antibody–Drug Conjugate Design: Linker
The linker is a key component of the antibody–drug conjugate that is designed around the payload and drug. It accounts for stability in circulation, maintaining positive pharmacokinetic properties, and release of the payload at the desired tumor site. Early linkers are derived from acid-labile hydrazones, which are designed to be cleaved in cancer cells. They are noneffective if they cleave at non-target sites. An antibody–drug conjugate payload may be internalized by an adjacent cell that does not express the antigen, which leads to cytotoxicity of these cells. This is known as the bystander effect, and in antibody–drug conjugate design, it is important to limit this effect to enhance therapeutic efficacy. Enzyme-labile linkers were also created, which link a drug to an antibody via a peptide linkage. The free drug can be cleaved from the antibody–drug conjugate through lysosomal proteases, which are present in tumors. Linkers must be stable in circulation until the conjugate reaches the desired target cell to reduce the number of side effects a patient can experience from premature cleavage of the payload.
The most stable of the linkers in use today are the non-cleavable linkers. With this type of linker, the drug is released through internalization of the antibody–drug conjugate, which is quickly followed by degradation of the antibody component in the lysosome. This causes the release of the drug with the linker attached. Similar to the enzyme-labile linkers, the specificity of the antibody–drug conjugate has led to a decrease in the bystander effect. When the released drug is charged, it is less able to diffuse into adjacent antigen-negative cells. Unlike cleavable linkers, which may be released despite being poorly internalized, a non-cleavable linker only becomes active when it is internalized and degraded within the cell. With the variations of linkers available, one can select the best linker option after considering the response needed to create a specific delivery system to target the tumor cells [18].
4.5 Antibody–Drug Conjugate Design: Payload
When deciding the appropriate payload for antibody–drug conjugates, potency, solubility, conjugation, and stability are principal factors to consider. Payloads are often highly potent molecules that lack sufficient therapeutic indices to be used in non-conjugated forms. Lipophilic drugs are able to pass through cell membranes. While this can be seen as a benefit to cell entry, it does have a greater potential to be released from the target cell and cause bystander effects. The payload must also be soluble enough to allow for conjugation to the antibody in different conditions that would prevent antibody denaturation. If a payload has low solubility, then a hydrophilic linker can be used. This can allow a higher drug–antibody ratio and can lead to more clearance out of the body. Furthermore, payloads must be stable enough to reach the target. If a drug is acid-sensitive, such as disulfide, alkene, or epoxide drugs, they may degrade in lysosomes and must be protected [19].
One of the cytotoxic payloads used in an antibody–drug conjugates is pyrrolobenzodiazepine (PBD). When the antibody–drug conjugate is internalized by an antigen expressing cell, PBD is released and binds to the minor groove of DNA. The imine can alkylate or crosslink the two DNA strands and block DNA replication, which ultimately would reduce growth of the tumor [20]. Similar to PBD payloads, a DNA minor groove-binding agent that belongs to the pyrridinobenzodiazepine class (PDD) also has been reported. Unlike PBD dimer payloads, which crosslink DNA, PDD are mono-alkylating agents that have similar efficacy and enhanced tolerability compared with PBD [21]. Another common payload seen in about one-third of antibody conjugates is monomethyl auristatin E (MMAE), which works by inhibiting the polymerization of tubulin [22]. With the variety in payloads available with antibody conjugates, further research must be done to select the best payload for a given application. A recent preclinical study looked into the use of PBD versus MMAE payloads in overall tumor control. It discovered that, while both payloads inhibited growth of CD276 (an antigen expressed in tumor cells and tumor endothelial cells) expressing xenograft models, the PBD conjugate was effective in vitro against tumor endothelial cells and expressed better overall in vivo tumor control than the MMAE conjugate [23]. While PBD did show better efficacy in killing tumor cells, there was no further research on the difference in payloads in melanoma cells. There are more drugs against melanoma tumors that use MMAE compared with PBD (or PBD derivatives), but often no specific reason is given as to why these payloads were chosen.
There have been thoughts of introducing multiple payloads onto one antibody–drug conjugate. In an in vitro study, DNA crosslinking agent PNU-159682 and tubulin polymerization inhibitor MMAF were conjugated to a HER2-targeting antibody. The dual conjugate showed selective and potent cytotoxicity against HER2 cell lines with both mechanisms. Specifically, PNU-159682 caused S-phase cell cycle and MMAF caused G2/M-phase cell cycle arrest. These two payloads did not work synergistically with each other [24]. While this idea does seem good in theory, there is not enough information to know whether this truly would be effective in practice. It is unclear how dual payloads would maximize the synergism ratio [4, 5]. Furthermore, it is hard to determine the desired concentration of payloads needed for this to be effective.
Determination of the concentration of a payload needed to obtain the desired effect varies between studies in both in vitro and in vivo studies. The amount of payload must be considered along with the linker being used, as studies have shown that the linker and dose together impact payload delivery to tumors. The level of payload in the tumor is also determined by the amount of conjugate entering the tissue, local antibody–drug conjugate catabolism rate, and the payload tissue-retention properties. There are “plateau” effects with payload efficacy, which is the maximum concentration of payload to tumors that does not enhance efficacy [25]. After the plateau has been determined, it should not be exceeded, as doing so might increase adverse effects.
5 Pharmacokinetics of Antibody–Drug Conjugates
After designing an antibody–drug conjugate, it is important to understand the various impacts each component has pharmacokinetically in the body (Fig. 3).

Desired and undesired kinetics of antibody–drug conjugates. The composition of the antibody–drug conjugate influences its desired uptake by the tumor cells, non-desired uptake by clearance organs such as the liver, and premature payload release in the blood, leading to systemic toxicity
5.1 Absorption
Absorption properties of antibody–drug conjugates are similar to unconjugated antibodies. Antibody–drug conjugates are administered primarily via IV injection with 100% bioavailability. As for subcutaneous administration, the bioavailability ranges from ~ 50 to 80%. Subcutaneous administration is typically not seen due to the potential reactions to cytotoxic payloads and off-target toxicity by immune cells in the skin, leading to deposits of cytotoxic material [26].
5.2 Distribution
Antibody–drug conjugates traffic throughout the vascular and lymphatic system at a slow rate. They tend to have a lower volume of distribution compared with small molecules. When a small molecule is conjugated to the antibody, it lowers the volume of distribution of the small molecule. This allows for the small molecule to reach cells and tissues that are not accessible by free diffusion due to the specificity of antibody to antigen. It is equally important to consider the small molecule distribution after degradation of the antibody–drug conjugate in tissues. Small molecule drugs are often widely distributed when they are unconjugated. While high amounts of antibodies can be seen in the liver, lungs, kidneys, and skin, the small molecule can be seen in multiple other tissues, such as thyroid small intestine, thymus, spleen, lung, liver, and pancreas. Specifically, the small molecule MMAE has higher concentrations than the antibody in the liver. This difference in distribution can be due to the clearance function of the liver [27].
Antibody–drug conjugates are used owing to their specificity to target cells from the antibody binding. The internalization rate is usually determined by the properties of the antigen. Antibody–drug conjugate antibodies have two recognition sites that bind to the target epitope. The antibody Fab segment binds to the antigen epitope of the tumor cell, while the Fc segment may bind to the FcR on the surface of NK cells, provided the payload conjugate does not inhibit interactions with the FcR. This can lead to a direct killing effect in some antibody–drug conjugates, provided an additional mechanism of action. Binding to the antigen also allows for the antibody–drug conjugate to come into the cell via an endosome where the payload is released from linkers and inhibits growth of the tumor cell. This is the desired distribution of classic antibody–drug conjugates. With biepitope antibodies, there are two antigen recognition sites that can bind two different epitopes on the same antigen. Binding to different epitopes can change the internalization rate and promote internalization of antibody–drug conjugate into tumor cells. In both situations, the importance of the antigen allows for specificity of the target cancer cells to be attacked and destroyed.
The hydrophobicity of small molecules is another factor to take into consideration. If the small molecule is highly hydrophilic, then it is less likely to access tissues compared with a highly lipophilic small molecule drug. Lipophilic small molecules such as MMAE allow cytotoxicity toward the tumor and can diffuse into neighboring cells that do not express the target antigen due to heterogeneity of tumors [28].
5.3 Metabolism
The metabolism of antibody–drug conjugates depends on linker stability, site of conjugation, and total drug load. Antibody–drug conjugates with disulfide and hydrazone bonds are more susceptible to enzymatic or chemical cleavage. Deconjugation is seen with drugs AXL-107-MMAE and DEDN6526A, as they both contain protease viable linkers. The antibody backbone is preserved. Payloads that are released by deconjugation also impact metabolism. Once released from the antibody–drug conjugate, the small molecule payloads can be metabolized by cytochrome P450 enzymes and have drug–drug interactions [29].
Catabolism can also occur simultaneously with deconjugation. This is driven by receptor-mediated endocytosis or fluid-phase pinocytosis with trafficking to the lysosome, which is soon followed by enzymatic degradation [30]. Cytotoxic drug-containing catabolites are produced by cleavage of the linker or catabolism of antibodies with further intracellular processing. Some catabolic products are able to diffuse from the antigen-expressing cell to neighboring cells, causing their death, which is advantageous in heterogeneous tumors. However, catabolites are able to diffuse into plasma and lead to systemic effects that include toxicity and drug interactions [27].
With metabolism of antibody–drug conjugates, there is always a balance between desired and undesired metabolism. In many pre-clinical studies of antibody–drug conjugates, experiments were done on mice instead of humans. This is important because these species can metabolize drugs in diverse ways. For example, in mice, dipeptide linkers are unstable in plasma due to the CES1C hydrolase enzyme, which is an enzyme humans do not have [31]. This type of undesired metabolism would not be seen in humans. Furthermore, it is vital to consider the characteristics of the linker, as this can have an impact on plasma stability. A linker must be stable in blood and only cleave when it is at the targeted tumor cell. Improving the hydrophilicity of a linker can improve plasma stability. For example, one experiment tested a variation of a more hydrophilic linker known as the 2-(maleimidomethyl)-1,3-dioxane linker compared with the commonly used linker N-succinimidyl-4-(maleimidomethyl) cyclohexanecarboxylate. They found that the 2-(maleimidomethyl)-1,3-dioxane was more stable in human plasma with only a degradation of 3% in 120 h than the N-succinimidyl-4-(maleimidomethyl) cyclohexanecarboxylate with degradation of 38% in 120 h [32]. Deconjugation of the payload off the antibody via the linker is the desired metabolism to see at the target site.
5.4 Elimination
Elimination also differs between antibody–drug conjugates. The elimination of each component (antibody, linker, and payload) will be discussed.
5.4.1 Antibody Elimination
The elimination of the antibody from an antibody–drug conjugate usually occurs through target-mediated clearance, which is when the antibody binds to the target cell and is internalized and degraded. This pathway can lead to nonlinear pharmacokinetics. The antibodies also bind to the Fc-γ receptors, which can prevent elimination. The liver, spleen, and lungs can also participate in non-desirable internalization and catabolism of the antibody [33].
5.4.2 Linker Elimination
Hydrophobicity of the linker influences its elimination. Low hydrophilicity of the linker can lead to undesirable pharmacokinetics via rapid elimination from the plasma [34]. Linkers that include maleimide have an increased chance of retro-Michael elimination, which is irreversible [35]. Those that contain thiol-maleimide are more susceptible to retro-Michael reactions, which can lead to instability of antibody–drug conjugates in the circulatory system and a low therapeutic index [36]. N-Phenyl maleimide-Val-Cit linkers are favorable, as they are more stable and have more antitumor activity than antibody–drug conjugates containing N-alkyl maleimide [34, 37]. Therefore, they are less likely to be eliminated as quickly as N-alkyl maleimide linkers.
5.4.3 Payload Elimination
Conjugation has been shown to increase in the clearance of the antibody, specifically conjugation with a higher drug–antibody ratio than with a lower drug–antibody ratio. Antigens can be shed by the tumor and end up in the systemic circulation. This can cause antibody–drug conjugates (specifically those with cleavable linkers) to attach to the circulating antigen, affecting its distribution to the targeted tumor. This binding can result in the clearance by the liver, leading to liver toxicity due to the high amount of cytotoxic drug at the liver. As such, liver toxicity is a common side effect of antibody–drug conjugates [27]. For example, MMAE is eliminated predominately through the CYP3A4/5-mediated metabolic pathway and biliary/fecal excretion and is eliminated through hepatic clearance from the liver interstitial space [38]. As mentioned earlier, at times more MMAE than antibody can be seen in the liver. This can lead to liver toxicity and more unfavorable side effects.
6 Trials of Antibody–Drug Conjugates in Melanoma
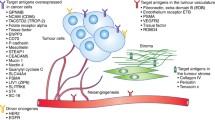
There are multiple antigens that are unique to melanocytes and/or melanoma cells and therefore are potential targets for antibody–drug conjugates (Fig. 4; Tables 2, 3, 4). While there are currently no FDA-approved antibody–drug conjugates in the treatment of melanoma, there are at least 14 FDA-approved antibody–drug conjugates for other various cancers. This indicates the emergence of antibody–drug conjugates in the future of oncology. The next section will discuss various antibody–drug conjugates that have been investigated in preclinical and clinical studies for the treatment of stage IV melanoma starting from 1998.

Melanoma targets of antibody–drug conjugates and selected developmental candidates. Desirable targets in melanoma include the ganglioside GD3; melanocyte specific transmembrane proteins such as GPNMB, PMEL, and MELTF; adhesion molecules expressed in melanoma, such as MUC18; receptor tyrosine kinases upregulated in tumors such as AXL, HER2, and c-Kit; and the GPCR that is increased in malignant melanocytes, EDNRB. GD3 disialoganglioside, GPNMB glycoprotein NMB, PMEL premelanosome protein, MUC18 melanoma cell adhesion molecule, MELTF melanotransferrin, EDNRB endothelin receptor type B, HER2 erb-b2 receptor tyrosine kinase 2
6.1 Membrane Target: GPNMB
Glycoprotein NMB (GPNMB) is a transmembrane protein expressed by metastatic melanoma [40]. Glembatumumab vedotin (CR011-vcMMAE), which utilizes this target, reached a Phase II clinical trial. In mice, the drug inhibited the growth of GPNMB-positive melanoma cells at doses as low as 1.25 mg/kg and caused complete regressions in tumor growth [41]. In the clinical trials, Phase I/II studies were associated with an encouraging overall response rate of 25–33%, though three treatment-related deaths occurred at concentrations above the MTD [42]. In a Phase II study, this drug was given to 62 patients who were refractory to checkpoint inhibitors and MEK/BRAF inhibition. Adverse effects that arose were alopecia, neuropathy, rash, fatigue, and neutropenia. Despite this, there was a modest improvement in progression-free survival, which was elevated in patients who had a rash during the first cycle [43]. This drug also was beneficial to uveal melanoma patients who were heavily pretreated with a median progression-free survival of 3.1 months and median overall survival of 11.9 months [44]. Although the objective response rate was low, the disease control rate was high and sustained. However, the skin toxicities observed suggested that the antibody was also binding to the target in normal epithelial tissues. The development was discontinued in 2018 for failure to meet the primary endpoint according to a company press release.
6.2 Membrane Target: Melanotransferrin
Seagen was planning a clinical trial with L49-vcMMAF. L49 is used to target melanotransferrin (MELTF, p97), a glycosylphosphatidylinositol-anchored glycoprotein as a cell-surface marker in melanomas. L49 is conjugated through a valine-citrulline (vc) linker to the antimitotic drug MMAF. In animals, this antibody–drug conjugate was effective at killing cancer cells that expressed elevated levels of MELTF while sparing normal cells that had low levels [45]. Further publications on why this drug never continued to progress in clinical trials did not arise. However, a recent study found that SGN-CD288A, an antibody–drug conjugate targeting melanotransferrin with a novel glucuronide linker, exhibited better anti-melanoma activity relative to the vc linker control in animals. This antibody was proceeding to a Phase I clinical trial [46], but subsequently it was cancelled in 2023 due to portfolio prioritization.
6.3 Membrane Target: MUC18
Using a proteomics approach to discover tractable cell surface targets, a recent study identified MUC18 [melanoma cell adhesion molecule (MCAM)] as a potential antibody–drug conjugate target for melanoma therapy [47]. The same study described the development of AMT-253, which contains an antibody targeting MUC18 conjugated to exatecan, a topoisomerase inhibitor, via a self-immolative T-moiety linker. AMT-253 was effective in multiple xenograft models of melanoma. Interestingly, the group also prepared a control antibody using the common vcMMAE as the linker-payload combination. AMT-253 was more effective than the control antibody–drug conjugate. The group plans to advance AMT-253 to clinical trials, and two trials are recruiting as of 2024.
6.4 Membrane Target: PMEL
An earlier study described an antibody–drug conjugate known as 17A9-vcMMAE [48]. The target of this antibody–drug conjugate is PMEL, which is a pigment-forming gene that encodes a melanocyte type I transmembrane glycoprotein [49]. At 6 mg/kg, this antibody–drug conjugate showed excellent inhibition of tumor growth in a xenograft model [48]. It is not clear whether this antibody–drug conjugate progressed clinically, though these in vivo results suggest that PMEL may be a promising therapeutic target.
6.5 G Protein-Coupled Receptor Target: Endothelin B Receptor
The endothelin B receptor (EDNRB) is essential for development of melanocytes via G-coupled signaling. In melanoma, EDNRB is upregulated, which causes an alteration of tumor–host interactions leading to the progression of melanoma. Investigational drugs that target this receptor inhibit melanoma cell growth and increase the apoptosis of melanoma cells, leading to some stabilization of melanoma patients [50, 51]. Two antibody–drug conjugates have shown beneficial effects when targeting EDNRB.
One preclinical study targeting EDNRB focused on the 5E9 monoclonal antibody conjugated with MMAE. This antibody–drug conjugate has a high rate of cellular internalization, which with the MMAE payload, will lead to destruction of tumor cells and stabilization in cancer growth. Specifically, a melanoma tumor xenograft was slowed by a dose of 6 mg/kg [52]. However, the study mentions that the density of EDNRB influences the efficacy of this antibody–drug conjugate, indicating that this conjugate has variable efficacy. They also found that EDNRB was not downregulated with prolonged antibody–drug conjugate exposure. This indicates that this antibody–drug conjugate does not inhibit the receptor for a long duration. Therefore, while this preclinical study has shown a positive effect in inhibiting tumor growth, it is not any more effective than other antibody–drug conjugates in long-term tumor growth.
Another antibody–drug conjugate that also has this target, DEDN6526A, advanced to clinical trials. Similar to 5E9-vcMMAE, DEDN6526A is also conjugated to MMAE. This drug was given every 3 weeks with varying doses of 0.3–2.8 mg/kg during Phase I. Following Phase II, the dosage recommended was 2.4 mg/kg every 3 weeks. The drug-related adverse effects that occurred included fatigue, neuropathy, nausea, diarrhea, alopecia, and chills, with severe cases leading to drug-induced liver injury. Of the 53 patients who received this antibody–drug conjugate, 32% had a stable disease for more than 6 months, with 11% only having a partial response [53]. However, this outcome was not different than what would be expected with a standard chemotherapy drug. Therefore, continuation of this clinical trial came to a halt in June 2015 [54]. From these two studies, it can be concluded that EDNRB is a target for treating melanoma, but efficacy in the clinic is lacking. Further research must be done to see the impact that this target could have in reducing tumor growth.
6.6 Receptor Tyrosine Kinase Targets: AXL
Heterogeneity of tumors is a leading cause of failure of therapeutics, and it allows cancers to continue to grow. In melanoma, AXL levels are high, which causes these cells to be resistant to MAPK pathway inhibitors, but other cells have low levels of AXL, which leads to MAPK inhibitor sensitivity. Clinical trials have evaluated the use of AXL antibody–drug conjugates as a sole therapeutic or in addition to other treatments such as MAPK pathway inhibitors.
One of the developmental candidates targeting ACL is AXL-107-MMAE (enapotamab vedotin). Its payload consists of MMAE with a protease-cleavable linker. In animals, the effective dose is 0.3–1 mg/kg both used as a single agent or combined with an MAPK pathway inhibitor. In humans, there were two different dosing schedules: 1 dose given every 3 weeks and 3 doses given every 4 weeks. The study showed that more benefit was seen in cooperatively using AXL-107-MMAE and MAPK pathway inhibitors, as the MAPK inhibitors potentiated the efficacy of AXL-107-MMAE. The specific mechanism of using both these treatments simultaneously allowed for targeted therapy in both high and low levels of AXL expressed tumor cells. AXL-107-MMAE specifically eradicated AXL-high cells, while MAPK inhibitors selectively reduced the AXL-low population [55]. This candidate was reported to advance to Phase II clinical trials [56], but was discontinued in 2020 according to a company press release.
BioAtla is developing CAB-AXL-ADC (BA3011, mecbotamab vedotin), and it has reached a Phase II trial. This clinical trial consists of 120 participants who have advanced solid tumors. In Phase I, the safety, tolerability, pharmacokinetics, and antitumor activity were evaluated, whereas the Phase II trial that is currently underway will assess whether BA3011 performs better alone or with a PD-1 inhibitor. What makes this antibody–drug conjugate different from AXL-107-MMAE is that anti-AXL CAB (conditionally active biologic) antibodies reversibly bind to AXL and EXL expressing cells that are in the tumor microenvironment only, while no binding occurs in normal tissues [57]. This suggests it has good specificity for tumor cells versus normal cells. Further information will be provided as more data arise from the clinical trial, as BioAtla, Inc. is currently in their recruiting stage for Phase II of clinical trials.
Tyrosine kinase AXL is a receptor target that should be further researched in antibody–drug conjugate development, as promising results have been observed in trials that are currently underway. In both of the studies mentioned, combination therapy via an antibody–drug conjugate and a current therapy of melanoma treatment, whether that is a MAPK or PD-1 inhibitor, have shown positive effects.
6.7 Receptor Tyrosine Kinase Targets: c-Kit
The c-Kit protein is another receptor that is overexpressed in melanomas. Previous treatment with small molecule inhibitors such as imatinib resulted in resistance or limited activity due to c-Kit mutations. The antibody–drug conjugate LOP628 was evaluated against mutant c-Kit cells. This drug worked by being rapidly internalized and processed to release active catabolite to create a cytotoxic response in tumor cells. In animal models, its dosing varied by model, with some models showing tumor regression at 2.5 mg/kg and others at 10 mg/kg [58]. While this study did show slowed tumor growth in animals, in the clinic acute hypersensitivity reactions due to mast cell degranulation were observed. This caused cessation of the Phase I clinical trial. The cause of this hypersensitivity was due to the co-engagement of the FcγR and c-Kit. The study found that interference of the c-Kit antibody to engage Fc receptors by pre-incubation with IgG or engineered Fc silencing mutations reduced degranulation. Therefore, more research should be done in identifying the class of Fc γ receptor that is causing the mast cell degranulation, as it would provide more information on developing an antibody–drug conjugate that would prevent further hypersensitivity reactions.
6.8 Membrane Target: HER2
The tyrosine kinase receptor HER2 (erb-b2 receptor tyrosine kinase 2) is a well-known therapeutic target in breast cancer. A recent study evaluated the therapeutic potential of a HER2 targeted antibody–drug conjugate for use in cutaneous melanoma [59]. The group evaluated RC48 (disitamab vedotin), a vcMMAE containing antibody–drug conjugate, which showed excellent growth inhibition in a melanoma xenograft model. The growth inhibition was enhanced by the addition of BRAF inhibitors in a BRAF mutant model. These data suggest that HER2-targeted antibody–drug conjugates may also find use in melanoma therapy. A number of clinical trials are ongoing with disitamab vedotin, though we are not aware of any specifically targeting melanoma.
6.9 Melanoma Lipid Markers: GD3
A study drug known as PF-06688992 was being tested on patients with unresectable stage III or stage IV malignant melanoma. This antibody–drug conjugate linked a chemotherapy drug PF-06464368 to an antibody that binds GD3, or disialoganglioside. GD3 is a marker that is found on most melanomas but less so on normal cells in the body, which adds to the antibody–drug conjugate specificity. The therapeutic agent kills the GD3-positive cells after internalization. While the study was completed on 10 January 2020, no further data were released from the clinical trial [60]. It is possible the trial was halted due to the mode of action, as the payload may not have been as effective compared with the MMAE or PBD payloads.
6.10 Immunotherapy Target: CD25
ADCT-301 (camidanlumab tesirine) has been tested in a clinical trial in patients with selected advanced solid tumors. This drug, also known as camidanlumab tesirine, is given through intravenous infusion to those that have CD25+ regulatory T cell (Treg) content in tumors. After positive results from a trial in lymphoma patients, it was further evaluated in melanoma patients. This trial tested the use as both a monotherapy and a combination therapy to find the efficacious dose, adverse side effects, effects on cardiac function via ECG monitoring, progression-free survival, and overall survival. Unlike other PBD dimer-containing antibody–drug conjugates that target specific antigens expressed in tumors, this trial is targeting tumor cells that contain immune cells, specifically CD25+ Treg cells, via antitumor immunity. Tregs suppress immune responses, which can be a major obstacle for tumor eradication via immunotherapies [61]. Being able to target tumors with high Tregs would allow for slower tumor growth as killing Tregs would allow for other immune cells to attack the tumor. However, recruitment was terminated for melanoma as of December 2022, as the signals of immunomodulatory activity shown were insufficiently compelling at the tested dose to justify continuation of the study [62], though trials continue for other indications.
7 Discussion
Antibody–drug conjugates are an exciting new drug therapy for a variety of cancers, including melanoma. While many of the antibody–drug conjugates have shown varying results on the efficacy, safety, and tolerability of that specific antibody–drug conjugate, they are an alternative therapy for poorly responding patients on immune checkpoint inhibitors, BRAF/MAPK inhibitors, chemotherapy, and c-Kit inhibitors.
At this time, it appears that AMT-253 is the most exciting antibody–drug conjugate in development for melanoma. It is one of only four melanoma-relevant antibody–drug conjugates we identified with ongoing clinical trials (Table 2), and the only one specifically being developed for melanoma. Additional clinical trials are proceeding on the other three conjugates, but do not appear to be focused on melanoma despite their efficacy in preclinical models and melanoma-relevant targets.
In each of the antibody–drug conjugates discussed, there have been both benefits and drawbacks. This should not discourage researchers from developing novel new therapies for treatments of melanoma. Rather, it is critical to consider the specific challenges of antibody–drug conjugates when it comes to resistance and toxicities. For example, melanoma heterogeneity makes it hard to find a common tumor antigen. Immunomodulation can occur, which limits finding the optimal antigen target. Target downregulation can lead to resistance, which is associated with decreased cytotoxic payload internalization, ineffective payload delivery, and poorer clinical response. P-glycoprotein can lead to efflux of antibody–drug conjugate payloads such as MMAE. There are multiple factors to consider in antibody–drug conjugate design when it comes to reaching its optimal efficacy.
Antibody–drug conjugates are susceptible to multiple mechanisms of resistance. These include decreasing antigen levels on tumor cells or overexpression of the P-glycoprotein pump or MDR1 drug transporter. Since lysosomes are vital in the function of antibody–drug conjugates, having a defect in the lysosomal function can also lead to resistance. This is due to the decrease in proteolytic activity in the lysosome to cleave the acid sensitive linkers. Further resistance can occur in the signaling pathways. This leads to changes in signaling pathways, which can activate the anti-apoptotic factors, such as the PI3K/AKT pathways and JAK/STAT pathways [63].
With these resistance mechanisms available, it is vital to determine what can be done to antibody–drug conjugates to prevent resistance. This can be done by looking at each functional component of antibody–drug conjugates, such as the linker. The linker is a key factor in antibody–drug conjugates that can impact payload distribution. However, this is difficult, as there is already a small therapeutic window, and there have not been enough studies to see the impact of various linker types conjugated with the same payload on resistance [64]. One group patented a targeting moiety that would result in early dissociation of the antibody–drug conjugate from its target, which can be recycled to the cell surface and internalize more antibody–drug conjugate molecules. Using this would help to diminish cytosolic escape of the payload, as it enhances more of a target-mediated payload and decreases bystander effect [65]. This can potentially lower resistance of antibody–drug conjugates.
Antibody–drug conjugates have been shown to be safe in terms of their adverse effects. However, there are concerns of toxicity with antibody–drug conjugates, which is typically due to the payload. In this review on melanoma, the most common payload discussed was MMAE. A general review of payload toxicities found that grade 3 and 4 anemia, neutropenia, and peripheral neuropathy are typically seen with MMAE payloads. Thrombocytopenia resulting from MMAF payloads was more likely to occur compared with other payloads such as DM1 and MMAE [66]. This toxicity occurs due to off-target toxicity, which is when the payload is released prematurely and causes unwarranted toxicity. While many can conclude that toxicity is caused by the payload, it is important to consider the linker to prevent premature release of the payload. As mentioned previously, the linker should be designed to be specific in its release at the target cell to prevent the payload from killing normal cells over the cancer cells. Therefore, further studies must be done for careful consideration of linker design when it comes to not only resistance of antibody–drug conjugates but also limiting toxicities.
While conducting this research, we found all data on antibody–drug conjugate clinical trials to be more recent than 1990. Articles that were used as resources for the general information of antibody–drug conjugates, such as their design and pharmacokinetics, were articles that were created earlier than the 1990s. While antibody–drug conjugates at that time were a new phenomenon, they provide foundational knowledge to elucidate clinical progress. Furthermore, we included clinical trials that had an indication for melanoma, but experimented on other indications such as lymphoma. This is notable, as while some of the clinical trials mentioned in the review article were discontinued owing to lack of efficacy or safety, this might be disproven if the antibody–drug conjugates in the study were to be studied more in melanoma patients.
A common theme seen throughout the research on clinical trials was that most of the antibody–drug conjugates presented with a payload of MMAE, while one used the PDD payload. The PDD payload was unfortunately discontinued, whereas there was an inconsistency when it came to which therapies were discontinued with the MMAE payload. With this observation, it can be concluded that PDD payloads have been shown to be ineffective in antibody–drug conjugate efficacy in melanoma so far. However, further research on PDD payloads should be done to better understand this observation. In contrast, MMAE has been most effective as a payload. There was only one example, AXL-107-MMAE, that presented with a possible MMAE toxicity, leading to neuropathy, neutropenia, and anemia. However, the other discontinued antibody–drug conjugates with this specific payload were not discontinued due to MMAE intolerability, but rather due to the ineffectiveness of the antibody–drug conjugate as a whole.
We did not find many studies that evaluated antibody–drug conjugates in combination with other therapies, such as an immune checkpoint inhibitor. All but two of the studies discuss antibody–drug conjugates used as a monotherapy after first-line treatments have been used. One of the antibody–drug conjugates discussed that is currently in progress as a combination therapy is CAB-AXL-ADC (BA3011). While there is no data on this trial as of now, it is important to keep an eye on this to determine its impact, as a previous antibody–drug conjugate trial, in camidanlumab tesirine (ADCT-30), failed when experimenting on use of dual-therapy. However, the trial was discontinued due to the signals of immunomodulatory activity present. It is not known whether that was due to the antibody–drug conjugate itself or in combination with the anti-PD-1 antibody. The trial does show promising news, as synergistic activity can be seen with combination of antibody–drug conjugates with immune checkpoint inhibitors.
Some of the examples mentioned in this paper were not only studied with melanoma but also showed effects with other cancers. These include ADCT-301 (camidanlumab tesirin), LOP628, and CAB-AXL-ADC (BA3011). Having this information will allow researchers to look into what worked from each clinical trial and how this can be implemented in melanoma therapy. “Melanoma” in this review indicates any stage of melanoma unless specified. There was little to no research done on antibody–drug conjugates specific to stage IV melanoma. Many of the clinical trials that were conducted for melanoma studied patients who had stage 1–3, and fewer focused on stage IV. There is an urgent need for further studies to be done on antibody–drug conjugates for use in patients with stage IV melanoma, as the number of patients who have been diagnosed with this cancer continues to grow. It is vital that other therapies are available for patients who progress to this stage to improve their quality of life.
For the majority of our studies, there were resources readily available on the background information of antibody–drug conjugates. However, some of the clinical trials mentioned were able to be traced on clinicaltrials.gov but had no further articles describing the research. When a clinical trial mentioned that it had been discontinued, there was little to no information on why that trial was discontinued. This element should be considered when looking into experimental therapeutics so we can understand why the drug was discontinued and what can be done to address the reason it was discontinued, and eventually re-try the improved version. Of the adverse effects that were reported, most of them were mild, such as fatigue, nausea, diarrhea, alopecia, and chills. However, reactions such as the hypersensitivity observed with LOP628 had a major adverse effect that led to the discontinuation of the clinical trial. If more researchers were able to have that information, the drug components could be modified to become safer, more tolerable, and more effective.
8 Conclusions
Today, further research should be done to continue to increase our knowledge of the drug conjugates and their implementation in the treatment of melanoma. Antibody–drug conjugates have more specificity to target tumors than normal cells, which can reduce the toxicity of the drug candidate. Antibody–drug conjugates have been shown to be promising candidates in the treatment of cancers by allowing patients to have another option for therapy after previous measures have failed. With more antibody–drug conjugates making their way onto the market, favorable results regarding tumor eradication for melanoma patients will lead to better health outcomes and longer life expectancy.
References
Moffitt Cancer Center. Melanoma Cancer Treatment Information. 2023. https://www.moffitt.org/cancers/melanoma/. Accessed 27 Sep 2023.
Melanoma Research Alliance. About Melanoma. n.d. https://www.curemelanoma.org/about-melanoma. Accessed 14 Apr 2023.
Cancer Treatment Centers of America. Metastatic melanoma. 2022. https://www.cancercenter.com/cancer-types/melanoma/types/metastatic-melanoma#what-is-metastatic-melanoma. Accessed 14 Apr 2023.
Jin Y, Edalatian Zakeri S, Bahal R, Wiemer AJ. New technologies bloom together for bettering cancer drug conjugates. Pharmacol Rev. 2022;74:680–711.
Jin Y, Schladetsch MA, Huang X, Balunas MJ, Wiemer AJ. Stepping forward in antibody-drug conjugate development. Pharmacol Ther. 2022;229: 107917.
AIM at Melanoma Foundation. FDA approved drugs. 2022. https://www.aimatmelanoma.org/how-melanoma-is-treated/fda-approved-drugs/#Chemotherapy. Accessed 14 Apr 2023.
American Cancer Society. Chemotherapy for Melanoma Skin Cancer. 2019. https://www.cancer.org/cancer/melanoma-skin-cancer/treating/chemotherapy.html. Accessed 14 Apr 2023.
National Cancer Institute. Treatment for cancer. n.d. https://www.cancer.gov/about-cancer/treatment. Accessed 14 Apr 2023.
Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141:160–71.
Understanding cancer immunotherapy research. Immunotherapy Drugs. 2020. https://www.ucir.org/immunotherapy-drugs. Accessed 5 Dec 2022.
Cancer Research UK. Melanoma skin cancer. 2020. https://www.cancerresearchuk.org/about-cancer/melanoma. Accessed 14 Apr 2023.
Rohaan MW, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. N Engl J Med. 2022;387:2113–25.
Goodman R, Johnson DB. Antibody-drug conjugates for melanoma and other skin malignancies. Curr Treat Options Oncol. 2022;23:1428–42.
Anderson TS, Wooster AL, La-Beck NM, Saha D, Lowe DB. Antibody-drug conjugates: an evolving approach for melanoma treatment. Melanoma Res. 2021;31:1–17.
Tabrizi M, Funelas C, Suria H. Application of quantitative pharmacology in development of therapeutic monoclonal antibodies. AAPS J. 2010;12:592–601.
Hock MB, Thudium KE, Carrasco-Triguero M, Schwabe NF. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015;17:35–43.
Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs. 2013;5:13–21.
Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5–13.
McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J. 2015;17:339–51.
Tiberghien AC, et al. Design and synthesis of tesirine, a clinical antibody-drug conjugate pyrrolobenzodiazepine dimer payload. ACS Med Chem Lett. 2016;7:983–7.
Hoffmann RM, et al. A novel antibody-drug conjugate (ADC) delivering a dna mono-alkylating payload to chondroitin sulfate proteoglycan (CSPG4)-expressing melanoma. Cancers (Basel). 2020;12:1029.
Wang Y, et al. Antibody-drug conjugate using ionized Cys-linker-MMAE as the potent payload shows optimal therapeutic safety. Cancers (Basel). 2020;12:1.
Drake PM, Rabuka D. Recent developments in adc technology: preclinical studies signal future clinical trends. BioDrugs. 2017;31:521–31.
Nilchan N, Li X, Pedzisa L, Nanna AR, Roush WR, Rader C. Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antibody Ther. 2019;2:71–8.
Zhang D, et al. Exposure-efficacy analysis of antibody-drug conjugates delivering an excessive level of payload to tissues. Drug Metab Dispos. 2019;47:1146–55.
Lucas AT, Price LSL, Schorzman AN, Storrie M, Piscitelli JA, Razo J, Zamboni WC. Factors affecting the pharmacology of antibody-drug conjugates. Antibodies (Basel). 2018;7:1.
Lin K, Tibbitts J. Pharmacokinetic considerations for antibody drug conjugates. Pharm Res. 2012;29:2354–66.
Okeley NM, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–97.
Han TH, Zhao B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab Dispos. 2014;42:1914–20.
Erickson HK, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426–33.
Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs. 2016;8:659–71.
Dovgan I, Kolodych S, Koniev O, Wagner A. 2-(Maleimidomethyl)-1,3-dioxanes (MD): a serum-stable self-hydrolysable hydrophilic alternative to classical maleimide conjugation. Sci Rep. 2016;6:30835.
Sadekar S, Figueroa I, Tabrizi M. Antibody drug conjugates: application of quantitative pharmacology in modality design and target selection. Aaps J. 2015;17:828–36.
Su Z, et al. Antibody-drug conjugates: recent advances in linker chemistry. Acta Pharm Sin B. 2021;11:3889–907.
Szijj PA, Bahou C, Chudasama V. Minireview: addressing the retro-Michael instability of maleimide bioconjugates. Drug Discov Today Technol. 2018;30:27–34.
Hamblett KJ, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70.
Christie RJ, et al. Pyrrolobenzodiazepine antibody-drug conjugates designed for stable thiol conjugation. Antibodies (Basel). 2017;6:20.
Chang HP, Cheung YK, Shah DK. Whole-body pharmacokinetics and physiologically based pharmacokinetic model for monomethyl auristatin E (MMAE). J Clin Med. 2021;10:1332.
Lambert JM, Morris CQ. Antibody-Drug dConjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv Ther. 2017;34(5):1015–35. https://doi.org/10.1007/s12325-017-0519-6.
Qian X, Mills E, Torgov M, LaRochelle WJ, Jeffers M. Pharmacologically enhanced expression of GPNMB increases the sensitivity of melanoma cells to the CR011-vcMMAE antibody-drug conjugate. Mol Oncol. 2008;2:81–93.
Tse KF, et al. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin Cancer Res. 2006;12:1373–82.
Ott PA, et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J Clin Oncol. 2014;32:3659–66.
Ott PA, et al. A Phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer. 2019;125:1113–23.
Hasanov M, et al. A phase II study of glembatumumab vedotin for metastatic uveal melanoma. Cancers (Basel). 2020;12:1.
Smith LM, Nesterova A, Alley SC, Torgov MY, Carter PJ. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol Cancer Ther. 2006;5:1474–82.
Mazahreh R, et al. SGN-CD228A is an investigational CD228-directed antibody-drug conjugate with potent antitumor activity across a wide spectrum of preclinical solid tumor models. Mol Cancer Ther. 2023;22:421–34.
Shi J, et al. A cell surface-binding antibody atlas nominates a MUC18-directed antibody-drug conjugate for targeting melanoma. Cancer Res. 2023;83:3783–95.
Chen Y, et al. The melanosomal protein PMEL17 as a target for antibody drug conjugate therapy in melanoma. J Biol Chem. 2012;287:24082–91.
Esnault C, et al. Antibody-drug conjugates as an emerging therapy in oncodermatology. Cancers (Basel). 2022;14:778.
Saldana-Caboverde A, Kos L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:160–70.
Kefford R, et al. A Phase II study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage IV metastatic melanoma. Invest New Drugs. 2007;25:247–52.
Asundi J, et al. An antibody-drug conjugate targeting the endothelin B receptor for the treatment of melanoma. Clin Cancer Res. 2011;17:965–75.
Sandhu S, et al. Phase I study of the anti-endothelin B receptor antibody-drug conjugate DEDN6526A in patients with metastatic or unresectable cutaneous, mucosal, or uveal melanoma. Invest New Drugs. 2020;38:844–54.
Genentech. A study of DEDN6526A in patients with metastatic or unresectable melanoma. ClinicalTrials.gov identifier: NCT01522664. https://classic.clinicaltrials.gov/show/NCT01522664. Accessed 1 May 2024.
Boshuizen J, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24:203–12.
Genmab. Enapotamab vedotin (HuMax-AXL-ADC) safety study in patients with solid tumors. ClinicalTrials.gov identifier: NCT02988817. https://classic.clinicaltrials.gov/show/NCT02988817. Accessed 12 Nov 2023.
Sharp LL, et al. Abstract 827: anti-tumor efficacy of BA3011, a novel conditionally active biologic (CAB) anti-AXL-ADC. Cancer Res. 2018;78:827–827.
Abrams T, et al. Preclinical antitumor activity of a novel anti-c-KIT antibody-drug conjugate against mutant and wild-type c-KIT-positive solid tumors. Clin Cancer Res. 2018;24:4297–308.
Li W, et al. Combined therapy of dabrafenib and an anti-HER2 antibody-drug conjugate for advanced BRAF-mutant melanoma. Cell Mol Biol Lett. 2024;29:50.
Pfizer. Pfizer PF-06688992 in patients with stage III or stage IV melanoma. ClinicalTrials.gov identifier: NCT03159117. https://classic.clinicaltrials.gov/show/NCT03159117. Accessed 10 Jan 2024.
Zammarchi, F, Havenith, K, Bertelli, F, Vijayakrishnan, B, Chivers, S, van Berkel, PH. CD25-targeted antibody-drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J Immunother Cancer. 2020;8.
ADC Therapeutics. Study of ADCT-301 in patients with selected advanced solid tumors. ClinicalTrials.gov identifier: NCT03621982. https://classic.clinicaltrials.gov/show/NCT03621982. Accessed 8 Dec 2023.
McKertish CM, Kayser V. Advances and limitations of antibody drug conjugates for cancer. Biomedicines. 2021;9:872.
Collins DM, Bossenmaier B, Kollmorgen G, Niederfellner G. Acquired resistance to antibody-drug conjugates. Cancers (Basel). 2019;11:394.
Ward E.S., OR, Kang J., Sun W. (2018). Endolysosomal targeting conjugates for improved delivery of cargo molecules to the endolysosomal compartment of target cells WO2018136455A1.
Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018;36:121–35.
Acknowledgements
We appreciate critical proofreading and comments from Christine Hsiao. A.W. was supported by 1R21CA273042-01 from the National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
A.W. was supported by 1R21CA273042-01 from the National Cancer Institute.
Conflict of interest
The authors have no financial interest in the material discussed in this work.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data, material and/or code availability
Not applicable.
Author contributions
Conceptualization, literature search, writing, and image preparation: I.L. Conceptualization, reviewing, image preparation, and editing: A.W. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lami, I., Wiemer, A.J. Antibody–Drug Conjugates in the Pipeline for Treatment of Melanoma: Target and Pharmacokinetic Considerations. Drugs R D 24, 129–144 (2024). https://doi.org/10.1007/s40268-024-00473-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-024-00473-7