
Abstract
Pulmonary arterial hypertension is characterized by elevated blood pressure and pathological changes in the pulmonary arterioles, leading to the development of right-heart failure and potentially fatal outcomes if left untreated. This review aims to provide an overview of novel drugs or formulations and new drug indications for pulmonary arterial hypertension that are currently in phases II–III of randomized controlled trials, and describe the rationale for the use of these targeted therapies, as well as their efficacy, safety profile, and impact on quality of life and survival. The literature research was conducted using data from ClinicalTrials.gov for the period between 1 January 2016 up to 31 December 2022. The population of interest includes individuals aged ≥ 18 years who have been diagnosed with pulmonary arterial hypertension. The review selection criteria included trials with recruiting, enrolling by invitation, active, terminated or completed status in 2022 and 2023. A total of 24 studies were selected for evaluation based on the inclusion and exclusion criteria. This review summarizes the updated information from randomized clinical trials involving novel therapies for pulmonary arterial hypertension. However, larger clinical trials are required to validate their clinical safety and effects. In the future, clinicians should choose therapies based on the patient's individual situation and requirements when developing treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pulmonary artery hypertension is a chronic disease associated with poor prognosis and a 5-year survival rate of about 50–60%. |
Current treatments improve the prognosis, but they do not arrest the progression of disease and they are associated with significant adverse effects. |
There is an immediate need for new clinical trials involving new drugs with enhanced efficacy and milder adverse effects. |
1 Introduction
Pulmonary arterial hypertension (PAH) is a severe disease characterized by elevated blood pressure and remodeling of the pulmonary arterioles, which can rapidly progress to right ventricular failure and mortality if not treated [1, 2]. PAH is classified as a rare disease (ORPHA number 182090) with an estimated prevalence of approximately 5 cases per 1,000,000 individuals [3]. The 2022 guidelines established by the European Society of Cardiology and the European Respiratory Society define PAH by a resting mean pulmonary arterial pressure of 20 mmHg, a pulmonary artery wedge pressure of 15 mmHg or lower, and a pulmonary vascular resistance (PVR) of 3 Wood units or higher.
PAH is a heterogeneous condition, making it challenging to treat effectively. The World Health Organization (WHO) classifies PAH into five subtypes based on distinct pathophysiology, etiology and response to treatment. In WHO Group 1 PAH, there is a constriction or stiffening of the pulmonary arterioles, placing increased strain on the right side of the heart. It includes idiopathic PAH (IPAH), heritable PAH (HPAH), and PAH associated with conditions like congenital heart disease, liver disease, HIV, or connective tissue disorders. While treatments exist, there is no cure [4].
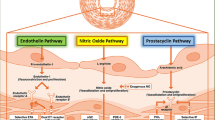
Prognosis varies among different subtypes of PAH but is generally unfavorable, and the estimated 5-year survival rate typically ranges from approximately 50–60%. However, early therapeutic intervention may improve survival rates [5]. Currently approved medications target well-known pathways: the endothelin pathway, the nitric oxide/cyclic guanosine monophosphate (cGMP) pathway, and the prostacyclin pathway [6]. Although these drugs improve the prognosis, they are vasodilators and are primarily focused on symptom management and slowing disease progression rather than providing curative solutions [7]. Many of the existing PAH drugs are associated with significant adverse effects, with an increased risk of side-effect amplification and drug–drug interactions when used in combination. Moreover, due to its rarity, conducting clinical trials for PAH proves challenging due to the limited number of eligible patients. This, consequently, leads to a lack of awareness among healthcare professionals and the general population, potentially resulting in delayed diagnoses and suboptimal treatments.
Based on recent advances in understanding the molecular pathophysiology of this complex and multifactorial disease, several new therapeutic targets are under development and undergoing testing, with the aim of modifying the disease process and ultimately enhancing long-term survival.
There is an immediate need for new clinical trials involving new drugs with enhanced efficacy and milder side effects. This entails targeting the underlying mechanisms of PAH more precisely and exploring innovative formulations, such as sustained-release or inhalable medications. Investigating optimal combinations of existing drugs or novel compounds can further enhance PAH management, potentially resulting in reduced adverse effects compared with high-dose monotherapies. This approach holds the potential to yield more effective and better tolerated treatments.
The primary objective of this review was to conduct a comprehensive assessment of novel therapeutics, formulations, and emerging pharmacological indications for WHO Group 1 PAH. These interventions are currently undergoing phase II–III randomized controlled trials (RCTs), highlighting the need for timely insights.
Our research aims to offer a comprehensive overview of potential future therapies for PAH, providing detailed information on molecules, pathways, and new formulations currently under investigation for the treatment of this condition.
2 Drug Targets and Currently Approved Treatment for Pulmonary Arterial Hypertension (PAH)
PAH has a multifactorial pathology characterized by an increase in pressure in the pulmonary arteries and by a vascular remodeling. The aberrant growth of endothelial cells (ECs) and smooth muscle cells (SMCs) in normally non-muscular compartments or segments of vessel leads to vascular obstruction, causing an increase in right ventricular pressure and pulmonary artery pressure.
Vasoconstriction is an early event in pulmonary hypertension and it is related to an overexpression/function of the potassium channel and to endothelial dysfunction. This involves reduced production of vasodilators (nitric oxide and prostacyclin) and an overexpression of vasoconstrictors (endothelin 1) which also promote vascular remodeling [8].
These molecules and their pathways represent the targets to treat PAH. Prostacyclin, whose expression and synthesis are reduced in PAH, induces pulmonary vasodilatation and inhibits cell proliferation through the cyclic adenosine monophosphate (cAMP). Prostacyclin analogs (epoprostenol, iloprost, and treprostinil) and prostacyclin receptor agonist (selexipag) stimulate this pathway [9, 10].
Nitric oxide causes an increase of cGMP, through soluble guanylate cyclase (sGC), and, by linking with cAMP, promotes vasodilation and antiproliferative vascular effects. Also, the natriuretic peptide increases cGMP. In PAH, altered expression of cGMP is caused by reduced expression of nitric oxide synthase and is related to impaired vascular vasodilation. There are two classes of drugs that act on these pathways: phosphodiesterase type-5 inhibitors (PDE5i) and sGC activators. The former (such as sildenafil and tadalafil) inhibit the enzyme PDE5, preventing the breakdown and conversion of cGMP into GMP; the latter (such as riociguat) directly increase the activity of sGC [11].
Endotelin-1 is upregulated in vascular ECs of patients with PAH and plays an important role in vascular remodeling: binding to endothelin receptor A and B stimulates cellular proliferation, fibrosis, and vasoconstriction. Endothelin receptor antagonists (ERAs) (ambrisentan, bosentan, and macitentan) targeting this pathway cause vasodilation [12].
In some cases, a combination of different classes of drugs may be prescribed to manage PAH effectively. However, this approach increases the risk of experiencing adverse effects associated with each individual drug. For example, prostacyclin analogs often lead to common adverse effects like flushing, diarrhea, jaw pain, headache, and flu-like symptoms. ERAs can result in adverse effects such as liver function abnormalities, peripheral edema, nasal congestion, and headache. PDE-5i commonly induce adverse effects such as headache, flushing, nasal congestion, and digestive issues. Meanwhile, sGC stimulators may provoke adverse effects such as headache, diarrhea, and nausea [13].
Table 1 shows currently approved Food and Drug Administration medications for PAH along with their associated adverse reactions.
3 Search Methodology
A comprehensive research was conducted to identify novel treatment options for WHO Group 1 PAH currently in the pipeline using data available on ClinicalTrials.gov. The research strategy involved the use of the words “pulmonary arterial hypertension” in the field condition or disease. Through different searches, the following sentence was used in the field other terms: “treatment” or “novel therapy” or “drug, treatment, therapy” or “drug, new treatment, novel therapy”. The retrieved records were collected into an Excel reference list, and a screening process was performed to assess their relevance. Two independent researchers reviewed the relevant articles, resolving any discrepancies through discussion. The inclusion criteria were as follows:
-
English-language interventional studies from January 1, 2016, until December 31, 2022;
-
population of any ethnic group aged ≥18 years old;
-
phase II and III RCTs;
-
studies with a recruitment status of recruiting (currently recruiting participants), enrolling by invitation (the study is selecting its participants from a population or group of people decided on by the researchers in advance), active (the study is ongoing, but potential participants are not currently being recruited or enrolled), terminated (terminated early without subsequent restart), or completed (completed with no additional examinations or treatments on the enrolled patients).
The exclusion criteria were as follows:
-
studies focused on a different disease;
-
phase I studies;
-
pediatric RCTs;
-
completed or terminated before 2022;
-
results already published;
-
drugs and formulations already on the market for this indication.
4 Results
Initially, a total of 400 records were identified applying the aforementioned research query and inclusion criteria. Furthermore, after applying the exclusion criteria and eliminating duplicate trials, 143 studies were included. A full evaluation of the protocols of the RCTs was then conducted, and a total of 24 studies were finally analyzed. Of these, there were 1 active, 3 completed, 1 enrolling by invitation, 4 terminated, and 19 recruiting RCTs. The main characteristics of the selected studies are summarized in Table 2.
5 Discussion
This review describes new therapeutic approaches for the treatment of PAH currently undergoing phase II–III RCTs. In the 24 studies analyzed, new medications being investigated include bone morphogenetic protein receptor type 2 (BMPR2) signaling and activating receptor type IIA/B (Actr IIA/B) analogs, prostacyclin receptor agonists, tryptophan hydroxylase 1 (TPH1) inhibitors, vasoactive intestinal peptide (VIP) inhibitors, and IL-6 receptor modulators. The review also includes RCTs proposing innovative formulations for MK-5475, imatinib, L606, CS1, vardenafil, and a Smurf-1 inhibitor. PDE5i, kinase inhibitors, and estrogen receptor modulators are included.
Since this is a rapidly evolving field, a weakness of this review lies in the inclusion of RCTs whose results are not yet published. Consequently, efficacy and safety data for these novel therapies could not be obtained or reported. However, some of the included drugs work through innovative pathways or formulations that enhance their effectiveness, suggesting that the synergy of these medications could play a significant role in the treatment of PAH. The list of drugs included in our research is provided below.
5.1 BMPR2 Signaling and Actr IIA/B
5.1.1 Sotatercept
Sotatercept, acting by suppressing TGF-β signaling and rebalancing deficient BMPR2 signaling, has shown promising results in reversing pulmonary arterial wall and right ventricular remodeling in preclinical models of PAH. The transforming growth factor-beta (TGF-β) superfamily includes mediators that are implicated in cell proliferation, differentiation, and apoptosis. In particular, the TGF-β family includes the TGF-β group (TGF-β isoforms, activins, nodals, and some growth and differentiation factors [GDFs]) and the bone morphogenetic protein (BMP) group (BMPs and most GDFs). The mechanism of signaling requires that TGF-β family members bind to transmembrane receptors. The activated receptors transduce the signal to the nucleus by phosphorylating Smad (canonical signaling) and binding DNA proteins mediate the transcription of target genes. The TGF-β group acts through SMAD 2 and 3, and the BMP group acts thought SMAD 1, 5, and 8 [60, 61,62,].
In PAH, an imbalance between anti-proliferative signaling and pro-proliferative signaling induces uncontrolled proliferation. The ActrIIs are upregulated in pulmonary arteries instead of BMPR2, the expression of which is reduced. The dysfunction of these pathways is associated with hyperproliferation and apoptosis resistance of pulmonary arterial ECs in PAH [61,62,64,63,].
In order, in pulmonary arteries the mutant form of BMPR2 stimulates the TGF-β into the medium, thereby accelerating SMC growth and hypertrophy. These mutations are associated with an increased risk of death, need for transplantation, and overall mortality in individuals with PAH [65, 66].
The published results of PULSAR (a phase II trial) and STELLAR (a phase III trial) have shown improvement in exercise capacity compared with placebo and the longer-term safety and durability of clinical benefit of sotatercept for PAH.
In the PULSAR trial, the safety and efficacy of sotatercept were studied using the primary outcome of change from baseline to week 24 in PVR. Sotatercept or placebo was administered to participants with PAH of WHO Group 1, functional class II–III plus background therapy. Participants received placebo, sotatercept 0.3 mg/kg, or sotatercept 0.7 mg/kg by subcutaneous injection every 21 days for a period of 24 weeks. For both doses it was noted that there was a reduction in PVR greater than placebo. The most common adverse event was thrombocytopenia without bleeding [67].
This study is supported by data from the primary phase of the PULSAR study (Phase II, ClinicalTrials.gov identifier: NCT03496207), in which participants taking any approved single or combination therapy for PAH were randomized to receive additional sotatercept or placebo for 24 weeks. The PULSAR study demonstrated a statistically significant improvement in its primary endpoint, PVR. The study duration for a given participant will be approximately 4 years with a screening period, treatment period and a follow-up period.
In the STELLAR trial, the safety and efficacy of sotatercept was studied using the primary outcome of 6-minute walk distance (6MWD) at 24 weeks. Randomly, 324 adults with PAH (WHO functional class II or III), who were already being treated with stable background therapy, received subcutaneous sotatercept at a starting dose of 0.3 mg/kg, 0.7 mg/kg, or placebo every 3 weeks. The median change from baseline in the 6MWD was 34.4 m (95% confidence interval [CI] 33.0–35.5) in patients treated with sotatercept and 1.0 m (95% CI −0.3 to 3.5) in those treated with placebo. The most common adverse events were epistaxis, dizziness, telangiectasia, increased hemoglobin levels, thrombocytopenia, and increased blood pressure which appeared more frequently in the sotatercept group [68].
Currently, there are three other ongoing trials: HYPERION, SOTERIA, and ZENITH. Phase III trial HYPERION (NCT04811092) will compare sotatercept with placebo (in addition to background PAH therapy) in newly diagnosed PAH patients at intermediate or high risk of disease progression, with the time to clinical worsening as the primary outcome. A total of 662 participants will be divided into two groups; sotatercept and placebo will be administered at a starting dose of 0.3 mg/kg to a final dose of 0.7 mg/kg every 21 days [69]. The SOTERIA trial (NCT04796337) will analyze the long-term safety, tolerability, and efficacy of sotatercept, with primary outcomes including the incidence of adverse events (AEs) with detectable anti-drug antibodies, abnormal laboratory test results, and assessments of blood pressure and electrocardiogram at 200 weeks. Seven hundred patients that had completed prior sotatercept studies will be treated with a target dose of 0.7 mg/kg plus their background therapy [70]. In the ZENITH trial (NCT04896008), the primary outcome is the time to first confirmed morbidity or mortality event (at 46 months), including all-cause death, lung transplantation, or PAH worsening-related hospitalization of at least 24 hours. Two hundred patients at high risk of mortality will be treated with sotatercept (0.7 mg/kg every 21 days) when added to maximum tolerated background PAH therapy [71].
5.2 Prostacyclin Receptor Agonists
5.2.1 Ralinepag
Ralinepag is a new oral agonist of non-prostanoid prostacyclin (IP) receptor that has been shown to reduce PVR in patients with PAH who are already receiving single or dual combination therapy. Its mechanism of action involves acting as a prostacyclin receptor agonist, mimicking the effects of endogenous prostacyclin. This leads to vasodilation of pulmonary and systemic arterial vessels, inhibition of SMCs proliferation, prevention of platelet aggregation, and reduction of inflammation. These pharmacological effects collectively contribute to the improvement of hemodynamic parameters in PAH patients [72]. The results already published of the trial NCT02279160 showed that ralinepag reduced PVR compared with placebo in PAH patients with background therapy. In particular, 61 PAH patients received ralinepag at a starting dose of 20 μg to a maximum total dose daily of 600 μg for 22 weeks. Ralinepag decreased PVR by 163.9 dyn·s·cm−5 (compared with an increase of 0.7 dyn·s·cm−5 with placebo) and increased 6MWD from baseline by 36.2 m (compared with 29.4 m with placebo) [73]. There are three trials ongoing: ADVANCE OUTCOMES (ROR-PH-301), AVDANCE CAPACITY3 (ROR-PH-302), and ADVANCE EXTENSION (ROR-PH-303). The ADVANCE OUTCOMES trial (NCT03626688), a phase III trial, will study the efficacy of ralinepag via the primary endpoint of time-to-clinical events, defined as mortality, unplanned PAH-related hospitalization, disease progression, unsatisfactory treatment response, and need for intravenous prostacyclin therapy. In this trial, 1000 participants will receive 50, 250, and 400 μg of the extended-release tablets once daily titrated to the highest tolerated dose [74]. The ADVANCE CAPACITY 3 (NCT04084678) [75] trial aims to assess the impact of ralinepag therapy on exercise capacity by measuring the change in VO2 consumption obtained from cardiopulmonary exercise testing. The primary endpoint for this trial is the change in baseline VO2 at week 28. Ralinepag will be administered once a day at 50, 250, and 400 μg doses up to the individual maximum tolerated dose (maximum dose of 1400 μg). Participants who successfully complete this 28-week study will have the opportunity to receive ralinepag in the ADVANCE EXTENSION (NCT03683186) open-label extension study [76]. In this extension trial, the long-term safety and tolerability of ralinepag will be evaluated by monitoring the number of subjects experiencing treatment-related AEs per expected 1000 participants over a period of up to 6 years.
5.2.2 Treprostinil
Treprostinil is a synthetic analog of prostacyclin that causes direct vasodilation in the pulmonary and systemic arterial circulation, as well as inhibiting platelet aggregation. Currently, treprostinil is authorized for the treatment of PAH in many countries in subcutaneous/continuous formulations and in the US it is available in an inhalation solution and extended-release oral tablet. Despite the effectiveness of the drug, frequent administrations or continuous infusion are necessary due to its rapid elimination [77].
Formulations capable of guaranteeing a slower release of the drug and a longer duration of action are being studied.
Treprostinil palmitil (TP) is hydrolyzed into treprostinil by endogenous esterases and it was study designed to provide slow release of treprostinil in the lung [78]. In a phase I study, treprostinil palmitil inhalation powder (TPIP) demonstrated a safety profile and good tolerability. Also, pharmacokinetic studies indicated the potential for once-daily administration [79].
The phase IIb clinical trial NCT04791514 [80], which terminated in August 2022 because of low enrollment, aimed to evaluate the efficacy, safety, and pharmacokinetics of a single dose (112.5 μg) of TPIP for the treatment of PAH. The primary objective of the study was to assess the incidence of AEs from baseline to day 30 of treatment.
Another trial, NCT05147805 [81], is also evaluating the effectiveness, safety, and pharmacokinetics of TPIP in 99 participants with PAH. Participants will receive the powder once per day, starting at 80 μg and up-titrated to a maximum dose of 640 μg based on individual tolerance during the first 3 weeks of treatment. The primary outcome measure of the study is the change in PVR from baseline to week 16 of treatment.
A phase III trial (NCT04691154) will be conducted to demonstrate the short-term safety and tolerability of L606 in patients who are switching to the treprostinil inhalation solution dose [82]. This study will monitor the incidence of AEs for up to 12 months, as well as evaluate the steady-state pharmacokinetics, effects on 6MWD, quality of life (QoL), and treatment satisfaction.
5.3 Serotonin (5-HT) Signaling Pathway
5.3.1 Rodatristat Ethyl
Rodatristat ethyl acts as a potent peripheral TPH1inhibitor , an enzyme involved in the biosynthesis of serotonin. In PAH, TPH1 is overexpressed in the arterial ECs. The excessive serotonin production in PAH contributes to the uncontrolled growth of SMCs, leading to pulmonary vascular remodeling and a reduction in arterial lumen diameter. By inhibiting TPH1 and subsequently lowering serotonin levels, rodatristat ethyl aims to halt or reverse this pathological process. Importantly, it has a low potential for drug–drug interactions with other approved PAH drugs, making it suitable for evaluation in combination with existing therapies [83].
Its clinical efficacy, safety, pharmacokinetics, and pharmacodynamics will be studied in the ELEVATE 2 trial (NCT04712669) [84]. It is a phase IIb trial in which 90 patients will be randomized to placebo or oral rodatristat ethyl 300 mg or 600 mg twice daily. All subjects will be receiving background standard-of-care treatment. The primary outcome is percentage change from baseline of PVR from initiation of treatment to week 24.
5.4 VIP Pathway
5.4.1 Pemziviptadil
Pemziviptadil, also known as PB1046, is a prolonged-release analog of VIP that has been developed for the treatment of PAH. VIP is a neuroendocrine hormone involved in regulating water and electrolyte secretion in the intestine. In addition to its intestinal functions, VIP is also present in the lungs, where it induces vasodilation of the pulmonary arteries. In patients with IPAH, lower serum concentrations of VIP and an overexpression of its receptor have been observed. By binding to these receptors, pemziviptadil mimics the action of VIP and promotes vasodilation of the pulmonary arteries, improving cardiac function and hemodynamic parameters such as PVR [85]. Based on these promising results, pemziviptadil received FDA approval as an orphan drug for the treatment of WHO Group 1 PAH in 2015. Ongoing clinical trials, such as the phase II trial (NCT03556020), aim to further evaluate the efficacy and safety of pemziviptadil, with a focus on assessing its impact on PVR [86]. Patients have been randomized in a 2:1 ratio to one of two parallel dose groups, a high-dose group where the drug is up-titrated from a 0.2 mg/kg minimally effective starting dose to a target high dose level of at least 1.2 mg/kg or higher to a maximally tolerated dose. The period of treatment consisted of two phases, an initial 10-week dose titration phase to the target dose of at least 1.2 mg/kg and a maintenance phase following week 11 and continuing for 6 weeks. The study was terminated in January 2022 because of delayed drug resupply (as a result of the Covid-19 pandemic).
These trials also include follow-up studies, such as the extended phase II trial (NCT03795428), to monitor the long-term tolerability, incidence, and severity of AEs, as well as to assess patients' QoL [87]. This trial was also terminated for the same reason.
5.5 SGC Stimulators
5.5.1 MK-5475
MK-5475 is a novel, dry-powder formulation of a small-molecule stimulator of sGC that has been specifically designed for inhaled delivery. This formulation aims to enhance deep-lung deposition and enable direct delivery to the site of action. By delivering the medication directly to the lungs, the inhaled formulation offers several advantages over systemic therapy. These advantages include optimized drug delivery to the pulmonary arterial circulation at lower doses, reduced systemic vasodilation, and minimized AEs.
A phase I trial was conducted to estimate safety, tolerability, pharmacokinetics, and pharmacodynamics of a single dose of MK-5475 administered to patients with PAH. The treatment resulted in improved pulmonary circulation and the safety and tolerability of the drug didn’t show dose-limiting systemic toxicities, suggesting a selective action on the lungs. Despite several limitations of the study (small sample size, short duration of treatment and the potential reporting bias of AEs during the open-label portion of the study), this trial laid the foundation for the subsequent trials [88].
The INSIGNIA-PAH trial (NCT04732221) [89] is a two-part study consisting of a phase II and phase III evaluation. The trial aims to assess the efficacy and safety profile of inhaled MK-5475 compared with placebo. In the first part (phase II), the efficacy of three different inhaled doses of MK-5475 (32 μg, 100 μg, and 380 μg) will be evaluated in comparison with a placebo. The primary endpoint of this phase is the reduction in PVR at week 12 compared with baseline. Based on the safety profile observed, the dose with the most favorable safety profile will be selected for the second part (phase III) of the study. Phase III aims to confirm the efficacy (measured by the 6MWD), safety, and tolerability of MK-5475 versus placebo.
5.6 Kinase Inhibitors
5.6.1 Imatinib
Imatinib is a drug that exerts its therapeutic effects by targeting kinases involved in PAH and, in particular, it acts on PDGFR, discoidin domain receptor (DDR), KIT proto-oncogene receptor tyrosine kinase (KIT), colony stimulating factor 1 receptor (CSF1R), and Abelson murine leukemia viral oncogene homolog (ABL) kinases. In this way, imatinib blocks remodeling and fibrotic signaling, resistance to apoptosis of vascular SMCs, and immune dysregulation [90,91,92].
In 2010, a phase II clinical trial suggested the tolerability of imatinib in patients with PAH who have not achieved a sufficient response to treatment with prostacyclin analogs, ERAs, PDE5 inhibitors, or a combination of these. The objectives were to evaluate safety using the 6MWD test and to study the changes in hemodynamic parameters and functional classes. Although no improvements in 6MWD compared with placebo were observed, probably due the background therapy reducing this functional capacity, there was a decrease in PVR and an increase in cardiac output [93].
The published results of IMPRES, a terminated phase III clinical trial, show the ability of the drug to improve exercise capacity and hemodynamics in patients with advanced PAH already under treatment with a combination of two to three drugs. Due to serious adverse events, treatment had to be discontinued for several patients [94].
Currently, several trials are underway. The NCT04416750 trial is a phase II study that aims to determine the highest tolerated dose of imatinib (ranging from 100 to 400 mg) using a Bayesian continual reassessment method. Over a period of 12 months, the trial will identify the maximum tolerated dose and assess the PVR after 24 months [95]. In the IMPAHCT trial (NCT05036135), which is a phase IIb/III trial, dry powder inhaled imatinib will be administered. The phase IIb portion of the trial will focus on determining the optimal dosage. Subsequently, in the phase III trial, the safety and efficacy of inhaled imatinib will be evaluated compared with placebo. The primary endpoints for the respective phases are the change from baseline in PVR (phase IIb) and the change from baseline in the 6MWD at 24 weeks (phase III) [96].
The IMPAHCT-FUL trial (NCT05557942) [97] is a follow-up study to the previous IMPAHCT trial, investigating the safety and efficacy of inhaled imatinib in treating PAH. The trial will follow 462 participants who completed the previous study for an additional 48 weeks. The primary outcome measure is the incidence of AEs.
5.6.2 Seralutinib
Similarly to imatinib, seralutinib (formerly known as GB002) is a kinase inhibitor that targets multiple receptors including PDGFR, colony stimulating factor 1 receptor (CSF1R), and c-KIT.
It is the first tyrosine kinase inhibitor specifically designed for inhalation therapy in the treatment of PAH. This approach allows for potential optimization of therapeutic efficacy while minimizing systemic exposure and associated adverse effects [98].
Two phase Ia randomized, double-blind, placebo-controlled studies involving healthy volunteers were conducted to assess the safety and tolerability of various doses of seralutinib [99]. These single and multiple-ascending dose studies demonstrated a dose-dependent increase in seralutinib exposure. Seralutinib exhibited rapid absorption and clearance from the systemic circulation. Doses of seralutinib, including the highest tested dose of 90 mg twice daily, were well tolerated.
A phase II trial (NCT04816604) will analyze change from baseline in the 6MWD and emergent AEs from first dose of study drug up to 148 weeks [100].
5.7 Estrogen Receptor Modulators
5.7.1 Tamoxifen
Tamoxifen, a selective estrogen receptor modulator (SERM), exerts its mechanism of action in PAH by binding to estrogen receptors and blocking their activation. By inhibiting estrogen signaling, tamoxifen helps counteract the enhanced estrogen signaling observed in PAH patients. Furthermore, it inhibits SMCs proliferation and migration and exerts anti-inflammatory effects, which can help mitigate inflammation and tissue damage in PAH [101]. The T3PAH study (NCT03528902) is a phase II clinical trial comparing the effects of tamoxifen 20 mg daily with placebo in patients with PAH. All subjects will also be treated with background standard-of-care therapy at the discretion of their PAH care physician. The study will assess changes in tricuspid annular plane systolic excursion and other parameters measured through transthoracic echocardiography. Additionally, the study will evaluate changes in the 6MWD test, assessments of QoL, and hormone levels [102].
5.8 PD5i
5.8.1 TPN171H (simmerafil)
TPN171H (simmerafil) is a novel and selective PDE5i designed for the treatment of PAH. PDE5, the primary isoform in the lung, is upregulated in conditions associated with PAH. Simmerafil exerts its therapeutic action by selectively inhibiting PDE5, which leads to an increase in intracellular levels of cGMP. Elevated cGMP levels promote vasodilation of the pulmonary arteries and relaxation of the PASMC within the vessel walls. By enhancing the NO/cGMP signaling pathway, simmerafil promotes pulmonary vasodilation and reduces PVR [103]. Preclinical studies have demonstrated that TPN171H exhibits good selectivity, satisfactory safety and pharmacokinetic profiles, and a longer-lasting effect compared with sildenafil in animal models [12].
In a phase IIa trial (NCT04483115) included in our research, the effect of single-dose TPN171H tablets on acute hemodynamic parameters will be studied. The trial is expected to include 60 participants, divided 1:1:1:1:1:1 into six groups: placebo; the test drugs 2.5 mg, 5 mg, and 10 mg; and tadalafil tablets 20 mg and 40 mg, with each group comprising 10 cases. The primary outcome of the trial is the percentage of the maximum change in PVR [104].
5.8.2 Vardenafil
Vardenafil is another selective PDE5i that has demonstrated efficacy in improving exercise capacity in patients with PAH when used as oral monotherapy for a duration of 12 weeks. RT234 (vardenafil inhalation powder) possesses a slow dissociation rate and rapid clearance, which make it well suited for as-needed (PRN) use [105].
A phase I study, RT234-CL101, has been completed for RT234 [106], which compared the pharmacokinetic profiles of RT234 and oral vardenafil in healthy volunteers. The maximum concentration of vardenafil was reached rapidly (time to Cmax [Tmax] = 2 min), there were no serious treatment-emergent AEs, and the drug was well tolerated.
The phase IIb study VIPAH-PRN 2B (NCT04266197) included in our research is currently recruiting. It is a clinical trial designed to evaluate the safety and efficacy of inhaled vardenafil on exercise capacity in 86 patients with PAH. By administering vardenafil via inhalation, the study aims to optimize drug delivery directly to the pulmonary vasculature, where it can exert its vasodilatory effects and improve exercise performance. Indeed, the primary objective of this study is to assess the impact of a single inhaled dose of vardenafil on key measures of cardiorespiratory fitness during exercise. Furthermore, the study will closely monitor the safety profile of inhaled vardenafil in PAH patients [107].
6 Aberrant Epigenetic Changes
6.1 CS1
The balance between acetylation/deacetylation of histone controls the transcription of genes, regulating the structure of chromatin. In particular, histone acetyltransferases add acetyl groups from histones, relaxing chromatin structure and increasing accessibility to transcription enzymes; in contrast, histone deacetylases (HDACs) remove acetyl groups from histones inducing condensation and transcriptional repression [108].
HDACs are involved in many different disease such as diabetes, Parkinson disease, and in cancer progression [109, 110]. Recent studies have identified an over-expression of HDCA1 and HDCA5 in human lung from IPAH patients and the role of HDACs in RV failure across various models of PH. This suggests that HDACs may serve as a promising therapeutic target for PAH [111, 112].
Valproic acid (VPA), used as an antiepileptic agent, is shown to inhibit class I HDAC activity. VPA has been investigated for its benefits in a hypoxia-induced pulmonary hypertension rat model [111].
CS1 is a novel controlled-release formulation of valproic acid that possesses anti-thrombotic, anti-inflammatory, and anti-fibrotic properties. It has shown efficacy in reducing pulmonary pressure and vascular remodeling in patients with PAH [113]. A phase II trial (NCT05224531) is currently underway to evaluate the safety and tolerability of three different doses of CS1 for up to 22 weeks; 30 patients will be enrolled and will be treated with 480 mg, 960 mg, or 1920 mg of CS1 twice daily, with one third of the dose in the morning and two thirds in the evening. The trial will utilize the CardioMEMS HF System to obtain repeated measurements of hemodynamic parameters, providing valuable insights into the drug's effects [114].
7 IL-6 Receptor Modulator
7.1 Satralizumab
The mechanism of action of satralizumab for PAH involves targeting the interleukin-6 (IL-6) receptor, a pro-inflammatory cytokine implicated in various inflammatory and autoimmune pathways such as pulmonary vascular remodeling in PAH. By blocking the IL-6 receptor, satralizumab interferes with IL-6 signaling and can reduce inflammation and immune activation that contribute to the progression of PAH. This may potentially lead to improvements in symptoms and associated pathology in PAH patients [115].
An ongoing phase II randomized controlled trial, SATISFY-JP (NCT05679570), is currently investigating the efficacy of satralizumab in patients with PAH who exhibit an immune-responsive phenotype characterized by serum IL-6 levels ≥ 2.73 pg/mL. The study aims to enroll 24 patients who have shown an inadequate response to currently available medications. The primary outcome measure of the trial will be the percentage change in total PVR from baseline to the end of the 24-week treatment period [116].
8 Smurf-1 Inhibitor
8.1 LTP001
LTP001 is a Smurf-1 inhibitor under study [117]. Smad ubiquitination regulatory factor 1 (Smurf-1) is a E3 ubiquitin ligase involved in degradation of many regulatory proteins. In particular, Smurf-1 marks BMPRs I and II, smad 1 and 5 and its elevated levels have been observed in the chronic hypoxia and monocrotaline preclinical in vivo models of PAH. These levels result in a down-regulation of BMPR-I and -II, suggesting a potential role of Smurf-1 in vascular cell proliferation and remodeling in PAH. Thus, blocking could represent a treatment strategy for the disease [118].
NCT05135000 [119] is an ongoing trial designed to evaluate the efficacy and safety of orally administered LTP001 over a duration of approximately 24 weeks. This trial involves 44 participants who have been diagnosed with PAH. The primary outcome being assessed is the change in PVR, measured through right heart catheterization, from baseline to week 25.
9 Conclusions
Over the past three decades, advanced treatment approaches have emerged as the next frontier in addressing PAH, driven by a more precise diagnosis and an enhanced understanding of the molecular pathogenesis. The recent development of novel molecules targeting specific pathobiological PAH pathways has revolutionized the treatment landscape. This pipeline offers an overview of innovative PAH therapies derived from RCTs. Certain included drugs operate through innovative pathways or formulations, amplifying their effectiveness and suggesting a potential synergy that could significantly impact PAH treatment. The inhalation route of PAH therapy holds promise, facilitating a gradual drug delivery to the lungs and potentially allowing less frequent dosing per day, thereby improving the side-effect profile. As ongoing trials are completed, new data will emerge to inform clinical decision making. Although currently available drugs have led to significant progress in slowing disease progression and improving quality of life, further clinical trials are needed to interrupt the progression and potentially cure this disease. Notably, PAH remains an incurable condition to date, with existing treatments failing to confer significant benefits to patients, and the long-term survival rate remains unsatisfactory.
The mortality rate persists unchanged despite intensive treatments and drug combinations. Looking ahead, we hope that these findings in the pipeline, once approved and marketed, will ensure more effective and safe treatments for patients with PAH, improving the prescribed choices of clinicians.
References
Thenappan T, Prins K. Who Group I Pulmonary Hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(3):363–74. https://doi.org/10.1016/J.Ccl.2016.04.001.
Thenappan T, Ormiston M, Ryan J, Archer S. pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018. https://doi.org/10.1136/Bmj.J5492.
Orphanet. Procedural document on the orphanet nomenclature and classification of rare diseases. Orphanet J Rare Dis. 2020;02:1–7.
Task A, Members F, Humbert M, Germany Mmh, Berger Rmf, Denmark Jc, Germany Em, Germany Bn, Germany Kmo, Kingdom Jpu, Quint Jk, Kingdom U, France Gs, France Os, Toshner M, Kingdom U. 2022 Esc/Ers Guidelines For The Diagnosis And Treatment Of Pulmonary Hypertension Developed By The Task Force For The Diagnosis And Treatment Of (Esc) And The European Respiratory Society (ERS). Endorsed By The International Society For Heart And Lu. Eur Heart J. 2022;43(38):3618–3731. https://doi.org/10.1093/Eurheartj/Ehac237(Erratum In: Eur Heart J. 2023 Feb 23).
Zelt JGE, Sugarman J, Weatherald J, Partridge ACR, Liang JC, Swiston J, Brunner N, Chandy G, Stewart DJ, Contreras-Dominguez V, Thakrar M, Helmersen D, Varughese R, Hirani N, Umar F, Dunne R, Doyle-Cox C, Foxall J, Mielniczuk L. Mortality trends in pulmonary arterial hypertension in Canada: a temporal analysis of survival per ESC/ERS guideline era. Eur Respir J. 2022;59(6):2101552. https://doi.org/10.1183/13993003.01552-2021.
Evans JDW, Girerd B, Montani D, Wang X, Galiè N, Austin E, Elliott G, Asano K, Eichstaedt C, Hinderhofer K, Wolf M, Rosenzweig E, Chung W, Soubrier F, Simonneau G, Sitbon O, Gräf S, Kaptoge S, Angelantonio E, Humbert M, Morrell N, Pierre U. Bmpr2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. 2016;4(2):129–37. https://doi.org/10.1016/S2213-2600(15)00544-5.
Condon DF, Agarwal S, Chakraborty A, Auer N, Vazquez R, Patel H, Zamanian RT, de Jesus Perez VA. Novel mechanisms targeted by drug trials in pulmonary arterial hypertension. Chest. 2022;161(4):1060–72. https://doi.org/10.1016/j.chest.2021.10.010.
Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S-24S. https://doi.org/10.1016/j.jacc.2004.02.029.
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–5.
Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–32.
Klinger JR, Abman SH, Gladwin MT. Nitric oxide defciency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;188(6):639–46.
Wharton J, Strange JW, Moller GM, Growcott EJ, Ren X, Franklyn AP, et al. Antiproliferative efects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. 2005;172(1):105–13.
Humbert M, Sitbon O, Simonneau G. Treatment of pulmo-nary arterial hypertension. N Engl J Med. 2004;351(14):1425–36.
Izbicki G, Rosengarten D, Picard E. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2006;354(10):1091–3.
Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafl citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–57.
McLaughlin V, Channick RN, Ghofrani HA, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J. 2015;46:405–13.
Iversen K, Jensen AS, Jensen TV, et al. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebo-controlled, double-blinded trial. Eur Heart J. 2010;31:1124–31.
Vizza CD, Jansa P, Teal S, et al. Sildenafil dosed concomitantly with bosentan for adult pulmonary arterial hypertension in a randomized controlled trial. BMC Cardiovasc Disord. 2017;17:239.
Ling-Yun AI, Chen GUO, Yan DING, et al. CliIIical observation of Bosentan combined with Sildenafil in the treatment of congenital heart disease with severe pulmonary arterial hypertension. China Mod Med. 2016;23:112–4.
Jian-Yong ZHU, Yu-Qin ZENG, Qing HU, et al. Clinical effect of sildenafil combined with Bosentan in treatment of connective tissue disease associated moderate-severe pulmonary arterial hypertension. Chin J Respir Crit Care Med. 2018;17:369–72.
Bermejo J, Yotti R, Garcia-Orta R, Sanchez-Fernandez PL, Castano M, Segovia-Cubero J, Sildenafil for Improving Outcomes after VAlvular Correction (SIOVAC) investigators et al. (2018) Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, double-blind, randomized clinical trial. Eur Heart J. 39:1255–64.
White RJ, Vonk-Noordegraaf A, Rosenkranz S, et al. Clinical outcomes stratified by baseline functional class after initial combination therapy for pulmonary arterial hypertension. Respir Res. 2019;20:208.
Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–44.
Kuwana M, Blair C, Takahashi T, et al. Initial combination therapy of ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) in the modified intentionto-treat population of the AMBITION study: post hoc analysis. Ann Rheum Dis. 2020;79:626–34.
Jian-zhou GUO, Yan-yan MA, Zhi-wei WANG. Clinical effect and changes of cardiac function in patients with pulmonary hypertension after operation of congenital heart disease applicating tadalafil combined with Bosentan. South China J Cardiovasc Dis. 2018;24:40.
De-Zhen Z, An-Meng WEI. Effect of Bosentan combined with tadalafil on activity endurance and cardiac function in patients with pulmonary hypertension. Chin J Clinicians. 2020;48:566–9.
Zhuang Y, Jiang B, Gao H, Zhao W. Randomized study of adding tadalafil to existing ambrisentan in pulmonary arterial hypertension. Hypertens Res. 2014;37:507–12.
Barst RJ, Oudiz RJ, Beardsworth A, Brundage BH, Simonneau G, Ghofrani HA, Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group, et al. Tadalafil monotherapy and as add-on to background bosentan in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2011;30:632–43.
Baughman RP, Culver DA, Cordova FC, et al. Bosentan for sarcoidosis-associated pulmonary hypertension: a double-blind placebo controlled randomized trial. Chest. 2014;145:810–7.
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903.
Gali e N, Humbert M, Vachiery J-L, Vizza C, Kneussl M, Manes A, Arterial Pulmonary Hypertension and Beraprost European (ALPHABET) Study Group, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39:1496–502.
Badesch D, Bodin F, Channick R, et al. Complete results of the first randomized, placebo-controlled study of bosentan, a dual endothelin receptor antagonist, in pulmonary arterial hypertension. Curr Ther Res. 2002;63:227–46.
Galie N, Rubin LJ, Hoeper MM, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371:2093–100.
Ni AI, Wa ER. To investigate the clinical effect of Bosentan in the treatment of patients with congenital heart disease complicated with pulmonary hypertension. China Health Care Nutr. 2018;28:187–96.
McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:1257–63.
Hoeper MM, Leuchte H, Halank M, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2006;28:691–4.
Wilkins MR, Paul GA, Strange JW, Tunariu N, Gin-Sing W, Banya WA, Westwood MA, Stefanidis A, Ng LL, Pennell DJ, Mohiaddin RH, Nihoyannopoulos P, Gibbs JS. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171(11):1292–7. https://doi.org/10.1164/rccm.200410-1411OC.
Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension. Circulation. 2008;117:3010–9.
Gatzoulis MA, Landzberg M, Beghetti M, Berger RM, Efficace M, Gesang S, MAESTRO Study Investigators, et al. Evaluation of macitentan in patients with eisenmenger syndrome. Circulation. 2019;139:51–63.
Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18.
Sitbon O, Bosch J, Cottreel E, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med. 2019;7:594–604.
Jansa P, Pulido T. Macitentan in pulmonary arterial hypertension: a focus on combination therapy in the seraphin trial. Am J Cardiovasc Drugs. 2018;18:1–11.
Galie N, Muller K, Scalise A-V, Grünig E. PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J. 2015;45:1314–22.
Rubin LJ, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. 2015;45:1303–13.
Rosenkranz S, Ghofrani H-A, Beghetti M, et al. Riociguat for pulmonary arterial hypertension associated with congenital heart disease. Heart. 2015;101:1792–9.
Humbert M, Coghlan JG, Ghofrani H-A, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis. 2017;76:422–6.
Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–4.
Badesch DB. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;6:425–34.
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301.
Barst RJ. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin. Heart. 1997;4:299–301.
Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24:353–9.
McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915–22.
Jing ZC, Parikh K, Pulido T, Jerjes-Sanchez C, White RJ, Allen R, Torbicki A, Xu KF, Yehle D, Laliberte K, Arneson C, Rubin LJ. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–33. https://doi.org/10.1161/CIRCULATIONAHA.112.124388.
Hiremath J, Thanikachalam S, Parikh K, Shanmugasundaram S, Bangera S, Shapiro L, TRUST Study Group, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant. 2010;29:137–49.
Ronald JO. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;2:420–7.
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142:1383–90.
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40:874–80.
Coghlan JG, Channick R, Chin K, et al. Targeting the prostacyclin pathway with selexipag in patients with pulmonary arterial hypertension receiving double combination therapy: insights from the randomized controlled GRIPHON study. Am J Cardiovasc Drugs. 2018;18:37–47.
Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522–33.
Goumans MJ, Liu Z, Ten DP. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–27.
Kurakula K, Goumans MJ, Ten DP. Regulatory RNAs controlling vascular (dys)function by affecting TGF-β family signalling. EXCLI J. 2015;14:832–50.
Atkinson C, Stewart S, Upton PD, Machado RD, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8.
Chen N-Y, Collum SD, Luo F, Weng T, Le T-T, Hernandez AM, Philip K, Molina JG, Garcia-Morales LJ, Cao Y, et al. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and Group III pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;311:L238–54.
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Investig. 2012;122:4306–13.
Yang X, Long L, Reynolds P, Morrell N. Expression of mutant Bmpr-Ii in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm Circ 2011;1(1):103–10. https://doi.org/10.4103/2045-8932.78100.
Ormiston M, Upton P, Li W, Morrell N. The promise of recombinant bmp ligands and other approaches targeting Bmpr-Ii in the treatment of pulmonary arterial hypertension. Glob Cardiol Sci Pract. 2015;2015(4):47. https://doi.org/10.5339/Gcsp.2015.47.
Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman AB, Ghofrani HA, Escribano Subias P, Feldman J, Meyer G, Montani D, Olsson KM, Manimaran S, de Oliveira PJ, Badesch DB. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J. 2023;61(1):2201347. https://doi.org/10.1183/13993003.01347-2022.
Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg-Maitland M, McLaughlin VV, Preston IR, Souza R, Waxman AB, Grünig E, Kopeć G, Meyer G, Olsson KM, Rosenkranz S, Xu Y, Miller B, Fowler M, Butler J, Koglin J, de Oliveira Pena J, Humbert M, STELLAR Trial Investigators. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. 2023;388(16):1478–1490. https://doi.org/10.1056/NEJMoa2213558.
Study of sotatercept in newly diagnosed intermediate- and high-risk pah participants (Mk-7962-005/A011-13) (Hyperion). https://Clinicaltrials.Gov/Ct2/Show/Nct04811092. Accessed 09 Jan 2023.
A long-term follow-up study of sotatercept for Pah treatment (Mk-7962-004) (Soteria). https://Clinicaltrials.Gov/Ct2/Show/Nct04796337. Accessed 09 Jan 2023.
A study of sotatercept in participants with Pah who Fc Iii or Fc Iv at high risk of mortality (Mk-7962-006/Zenith) (Zenith). https://Clinicaltrials.Gov/Ct2/Show/Nct04896008. Accessed 09 Jan 2023.
Midgett C, Stitham J, Martin KA, Hwa J. Prostacyclin receptor regulation—from transcription to trafficking. Curr Mol Med. 2011;11(7):517–28. https://doi.org/10.2174/156652411800615144.
Torres F, et al. Efficacy and safety of ralinepag, a novel oral IP agonist, in PAH patients on mono or dual background therapy: results from a phase 2 randomised, parallel group, placebo-controlled trial. Eur Respir J. 2019;54(4).
A study evaluating the efficacy and safety of ralinepag to improve treatment outcomes in PAH patients, NCT03626688. https://clinicaltrials.gov/ct2/show/NCT03626688. Accessed 19 Apr 2023.
A study of ralinepag to evaluate effects on exercise capacity by CPET in subjects with WHO Group 1 PH (CAPACITY), NCT04084678. https://clinicaltrials.gov/ct2/show/NCT04084678. Accessed 19 Apr 2023.
A study evaluating the long-term efficacy and safety of ralinepag in subjects with PAH via an open-label extension. https://clinicaltrials.gov/ct2/show/NCT03683186. Accessed 19 Apr 2023.
LeVarge BL, Channick RN. Inhaled treprostinil for the treatment of pulmonary arterial hypertension. Expert Rev Respir Med. 2012;6:255–65. https://doi.org/10.1586/ers.12.23.
Leifer FG, Konicek DM, Chen KJ, et al. Inhaled treprostinil-prodrug lipid nanoparticle formulations provide long-acting pulmonary vasodilation. Drug Res (Stuttg). 2018;68:605–14. https://doi.org/10.1055/s-0044-100374.
Ismat FA, Usansky HH, Villa R, Zou J, Teper A. Safety, tolerability, and pharmacokinetics of treprostinil palmitil inhalation powder for pulmonary hypertension: a phase 1, randomized, double-blind, single- and multiple-dose study. Adv Ther. 2022;39(11):5144–57. https://doi.org/10.1007/s12325-022-02296-x.
A study of treprostinil palmitil inhalation powder (TPIP) in pulmonary arterial hypertension (PAH)—NCT04791514. https://clinicaltrials.gov/ct2/show/NCT04791514. Accessed 19 Apr 2023.
A study to evaluate the safety and tolerability of treprostinil palmitil inhalation powder in participants with pulmonary hypertension associated with interstitial lung disease—NCT05147805. https://clinicaltrials.gov/ct2/show/NCT05147805. Accessed 19 Apr 2023.
A phase 3 study to evaluate the safety and tolerability of L606 in subjects with Pah. https://Clinicaltrials.Gov/Ct2/Show/Nct04691154?Id=Nct04691154&Draw=2&Rank=1&Load=Cart. Accessed 10 Jan 2023.
Lazarus H, Denning J, Wring S, Palacios M, Hoffman S, Crizer K, Kamau-Kelley W, Symonds W, Feldman J. A trial design to maximize knowledge of the effects of rodatristat ethyl in the treatment of pulmonary arterial hypertension (elevate 2). Pulm Circ. 2022;12(2):E12088. https://doi.org/10.1002/Pul2.12088.
A study of rodatristat ethyl in patients with pulmonary arterial hypertension (Elevate 2). https://Clinicaltrials.Gov/Ct2/Show/Nct04712669. Accessed 10 Jan 2023.
Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, Funk G-C, Hamilton G, Novotny C, Burian B, Block L-H. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–46. https://doi.org/10.1172/Jci17500.
Phase 2 study to assess safety, tolerability and efficacy of once weekly Sc pemziviptadil (Pb1046) in subjects with symptomatic Pah (Vip). https://Clinicaltrials.Gov/Ct2/Show/Nct03556020. Accessed 10 Jan 2023.
Long-term, open label extension study of pemziviptadil (Pb1046) in Pah subjects following completion of study Pb1046-Pt-Cl-0004 (Vip Extend). https://www.Clinicaltrials.Gov/Ct2/Show/Nct03795428. Accessed 15 Jan 2023.
Bajwa E, Cislak D, Palcza J, Feng HP, Messina E, Reynders T, Denef J, Corcea V, Lai E, Stoch S. Effects of an inhaled soluble guanylate cyclase (Sgc) stimulator Mk-5475 in pulmonary arterial hypertension (Pah). Respir Med. 2023;206: 107065. https://doi.org/10.1016/J.Rmed.2022.107065.
A study of the efficacy and safety of Mk-5475 in participants with pulmonary arterial hypertension (Insignia-Pah: Phase 2/3 study of an inhaled Sgc Stimulator In Pah) (Mk-5475-007). https://Clinicaltrials.Gov/Ct2/Show/Nct04732221. Accessed 15 Jan 2023.
Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. https://doi.org/10.1038/nbt.1990.
Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21. https://doi.org/10.1172/JCI24838.
Perros F, Montani D, Dorfmüller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–8. https://doi.org/10.1164/rccm.200707-1037OC.
Ghofrani HA, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182(9):1171–7.
Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez-Sánchez MA, Grimminger F, Grünig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–38. https://doi.org/10.1161/CIRCULATIONAHA.112.000765.
Positioning imatinib for pulmonary arterial hypertension (Pipah). https://Clinicaltrials.Gov/Ct2/Show/Nct04416750. Accessed 15 Jan 2023.
A study of Av-101 (dry powder inhaled imatinib) in patients with pulmonary arterial hypertension (Pah) (Impahct). https://Clinicaltrials.Gov/Ct2/Show/Nct05036135. Accessed 15 Jan 2023.
Inhaled imatinib pulmonary arterial hypertension clinical trial—follow up long term extension (IMPAHCT-FUL)—NCT05557942. https://clinicaltrials.gov/ct2/show/NCT05557942. Accessed 9 Apr 2023.
Galkin A, Sitapara R, Clemons B, Garcia E, Kennedy M, Guimond D, Carter L, Douthitt A, Osterhout R, Gandjeva A, Slee D, Salter-Cid L, Tuder R, Zisman L. Inhaled seralutinib exhibits potent efficacy in models of pulmonary arterial hypertension. Eur Respir J. 2022;60(6):2102356. https://doi.org/10.1183/13993003.02356-2021.
Li J, Yamashita M, Cravets M, et al. Phase 1A randomized double-blind placebo-controlled single-ascending dose and multiple-ascending dose studies of orally inhaled GB002 in healthy adult subjects. Am J Respir Crit Care Med. 2020;201:A2907.
Open-label extension study of Gb002 in adult subjects with pulmonary arterial hypertension (Pah). https://Clinicaltrials.Gov/Ct2/Show/Nct04816604. Accessed 16 Jan 2023.
Mair K, Wright A, Duggan N, Rowlands D, Hussey M, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, Maclean M. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190(4):456–67. https://doi.org/10.1164/Rccm.201403-0483oc.
Tamoxifen therapy to treat pulmonary arterial hypertension (T3pah). https://Clinicaltrials.Gov/Ct2/Show/Nct03528902. Accessed 16 Jan 2023.
Wang Z, Jiang X, Zhang X, Tian G, Yang R, Wu J, Zou X, Liu Z, Yang X, Wu C, Shi J, Li J, Suo J, Wang Y, Zhang R, Xu Z, Gong X, He Y, Zhu W, Aisa H, Jiang H, Xu Y, Shen J. Pharmacokinetics-driven optimization of 4(3 H)-pyrimidinones as phosphodiesterase type 5 inhibitors leading to Tpn171, a clinical candidate for the treatment of pulmonary arterial hypertension. J Med Chem. 2019;62(10):4979–90. https://doi.org/10.1021/Acs.Jmedchem.9b00123.
Acute haemodynamic study of Tpn171h in patients with pulmonary arterial hypertension. https://Clinicaltrials.Gov/Ct2/Show/Nct04483115. Accessed 20 Jan 2023.
Jing Z, Yu Z, Shen J, Wu B, Xu K, Zhu X, Pan L, Zhang Z, Liu X, Zhang Y, Jiang X, Galiè N. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2011;183(12):1723–9. https://doi.org/10.1164/Rccm.201101-0093oc.
Eldon MA, Parsley EL, Maurer M, Tarara TE, Okikawa J, Weers JG. Safety, tolerability, and pharmacokinetics of RT234 (vardenafil inhalation powder): a first-in-human, ascending single- and multiple-dose study in healthy subjects. J Aerosol Med Pulm Drug Deliv. 2021;34:251–61. https://doi.org/10.1089/jamp.2020.1651.
Vardenafil inhaled for pulmonary arterial hypertension Prn phase 2b study (Vipah-Prn 2b). https://Clinicaltrials.Gov/Ct2/Show/Nct04266197. Accessed 20 Jan 2023.
Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100.
Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–39.
Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease. Pharmacol Ther. 2013;140:34–52.
Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126(4):455–67. https://doi.org/10.1161/CIRCULATIONAHA.112.103176.
Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI, Ma JS, Kook H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J. 2010;74(4):760–70. https://doi.org/10.1253/circj.CJ-09-0580.
Cereno. 2022. https://Cerenoscientific.Com/Pipeline/Cs1/.
Effect of Cs1 in subjects with pulmonary arterial hypertension. https://Clinicaltrials.Gov/Ct2/Show/Nct05224531. Accessed 20 Jan 2023.
Fric J, Zelante T, Wong AY, Mertes A, Yu HB, Ricciardi-Castagnoli P. NFAT control of innate immunity. Blood. 2012;120:1380–9. https://doi.org/10.1182/blood2012-02-404475.
Satralizumab in the treatment of pulmonary arterial hypertension (SATISFY-JP Trial), NCT05679570. https://clinicaltrials.gov/ct2/show/NCT05679570. Accessed 9 April 2023.
https://synapse.patsnap.com/drug/04efca99118e43d8adc38cd21d9c28ef.
Murakami K, Mathew R, Huang J, Farahani R, Peng H, Olson SC, Etlinger JD. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp Biol Med (Maywood). 2010;235(7):805–13. https://doi.org/10.1258/ebm.2010.009383.
Study of efficacy and safety of LTP001 in pulmonary arterial hypertension, NCT05135000. https://clinicaltrials.gov/ct2/show/NCT05135000. Accessed 10 Apr 2023.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Italian Ministry of Health, Rome, Italy (Ricerca Corrente).
Author contributions
AP: conceptualization and review of the paper. MEN and EDM: methodology. AC and AP: validation, supervision, and project administration. MEN and EDM: investigation, formal analysis and data curation. MEN and EDM: writing—original draft preparation. BS, MN, AC, VP and AP: writing, review and editing. All authors have read and agreed to the published version of the manuscript.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Novara, M.E., Di Martino, E., Stephens, B. et al. Future Perspectives of Pulmonary Arterial Hypertension: A Review of Novel Pipeline Treatments and Indications. Drugs R D 24, 13–28 (2024). https://doi.org/10.1007/s40268-024-00453-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-024-00453-x