
Abstract
Background and Objective
Tucatinib is a selective tyrosine kinase inhibitor of the human epidermal growth factor receptor 2 (HER2) approved to treat metastatic HER2-positive breast and colorectal cancers. The International Council for Harmonisation of Technical Requirements for Human Use (ICH) E14 guideline mandates that new drugs are assessed for potential effects on cardiac repolarization through electrocardiogram (ECG) evaluation in a QT/corrected QT (TQT) study.
Methods
We evaluated the effect of tucatinib on cardiac repolarization in healthy volunteers in a phase I, randomized, partially double-blind, placebo-and positive-controlled three-period crossover study. The primary endpoint was the placebo-corrected change from baseline in QT interval values, corrected for heart rate using Fridericia’s method (ΔΔQTcF).
Results
After achieving steady-state tucatinib exposures with 300 mg twice daily, the observed ΔΔQTcF ranged from −2.9 msec at 2 hours post-dose to 0 msec at 4 hours post-dose. The upper bound of the 90% confidence interval (CI) was below 5 ms at all post-dose timepoints. Assay sensitivity was confirmed as the lower bound of the 90% CI and was >5 ms following moxifloxacin dosing. As the mean ΔΔQTcF of tucatinib was predicted to be − 1.80 ms (90% CI − 3.90, 0.30) at clinically relevant tucatinib concentrations (511 ng/mL), an effect of tucatinib on QTcF exceeding 10 ms was excluded within observed ranges of tucatinib (up to ~1000 ng/mL). Tucatinib had no clinically relevant effect on heart rate or cardiac conduction. The safety profile of tucatinib was manageable after multiple doses.
Conclusion
Tucatinib had no clinically relevant effects on studied ECG parameters. This study constitutes a clearly negative TQT study per ICH E14 guidance.
Clinical Trial Registration
This trial (NCT03777761) was registered on 17 December 2018.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Steady-state therapeutic exposures of tucatinib had no clinically relevant effect on cardiac repolarization in healthy volunteers. |
Tucatinib had a manageable safety profile throughout the study. |
1 Introduction
Human epidermal growth factor receptor 2 (HER2) gene amplification or overexpression is an oncogenic driver in various solid tumors, including breast, gastric, colon, and lung cancers. Consequently, it has become a well-established therapeutic target in several tumors, including breast, colorectal, and gastric cancers [2].
Tucatinib is a highly selective tyrosine kinase inhibitor of the HER2 receptor [3]. Tucatinib is approved in multiple countries in combination with trastuzumab and capecitabine for adult patients with advanced, unresectable, or metastatic HER2-positive breast cancer who have received prior anti-HER2 therapy [4, 5]. In the international, randomized, double-blind, phase II pivotal HER2CLIMB trial, tucatinib (approved dose of 300 mg twice daily) added to trastuzumab and capecitabine significantly prolonged progression-free survival (including in patients with brain metastases) and overall survival in patients who had previously received trastuzumab, pertuzumab, and trastuzumab emtansine [6, 7].
Tucatinib is predominantly eliminated by the hepatobiliary route and in vitro studies show that it is metabolized primarily by cytochrome P450 (CYP) 2C8 and, to a lesser extent, by CYP3A4 and CYP3A5. In human plasma, tucatinib is the most abundant component (75.6%) followed by its primary metabolite ONT-993 (9.2%), which is responsible for <10% of total pharmacological activity of tucatinib in humans and is formed through oxidation by CYP2C8 [8]. A drug–drug interaction study showed that tucatinib inhibits CYP3A4, CYP2C8, and P-glycoprotein (P-gp)-mediated elimination, and is affected by both CYP3A4 and CYP2C8 inhibition and induction [9].
Tucatinib is also approved in the United States (US) in combination with trastuzumab for the treatment of adult patients with RAS wild-type HER2-positive unresectable or metastatic colorectal cancer that has progressed following treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy [5]. In the international, open-label, phase II MOUNTAINEER trial, tucatinib (approved dose of 300 mg twice daily) added to trastuzumab resulted in a clinically meaningful overall response rate among patients with RAS wild-type HER2-positive unresectable or metastatic colorectal cancer who had previously received a fluoropyrimidine, oxaliplatin, irinotecan, and an antivascular endothelial growth factor monoclonal antibody [10].
The International Council for Harmonisation of Technical Requirements for Human Use (ICH) E14 guideline mandates that most new drugs are assessed for potential effects on cardiac repolarization through electrocardiogram (ECG) evaluation in a QT/corrected QT (TQT study) [1, 11]. Delayed cardiac repolarization, which is represented by a prolonged QT interval, can be induced by medication and predispose patients to developing torsades de pointes, a potentially life-threatening ventricular arrythmia [12].
The effect of tucatinib on cardiac parameters has been assessed preclinically. Inhibition of the human ether-à-go-go-related gene channel, which plays a critical role in cardiac repolarization [13], produced a half-maximal inhibitory concentration (IC50) of 13.5 µM in vitro, which is more than tenfold higher than peak drug levels measured in patients receiving the maximum tolerated dose of tucatinib [8]. Moreover, no significant effects were observed in blood pressure, heart rate, ECG waveforms, or in QT and corrected QT (QTc) measurements when tucatinib was administered orally at 0, 2.5, 10, or 20 mg/kg twice daily in cynomolgus monkeys [8].
This study evaluated the QT effects of tucatinib at steady-state therapeutic exposures in healthy volunteers.
2 Methods
This was a phase I, randomized, partially double-blind, placebo- and positive-controlled three-period crossover study (NCT03777761) conducted at one site in the US (Covance Clinical Research Unit, Inc., Dallas, TX, USA), with approximately 60 participants planned for recruitment.
2.1 Study Participants
Male and female participants aged 18–65 years with a body mass index (BMI) of 18.0–32.0 kg/m2 and total body weight of 50–100 kg were eligible for inclusion. Participants were enrolled after they were confirmed to be in good general health, based on their medical history, physical examination, hematology, clinical chemistry, negative human immunodeficiency viruses and hepatitis B and C serology, urinalysis, 12-lead ECG, and vital signs. Female volunteers were required to be of nonchildbearing potential, while male volunteers were required to be surgically sterile for at least 90 days or agreed to use contraception if sexually active. Participants were required to have no history or evidence of long QT syndrome and a QTcF ≤450 ms for males or ≤470 ms for females at baseline.
Participants were to refrain from using drugs or substances known to alter drug absorption, metabolism, or elimination processes, including St John’s wort, and known strong inhibitors or inducers of CYP3A4 or 2C8 within 30 days prior to study initiation and throughout the study, unless deemed acceptable by the investigator. Use of tobacco or nicotine-containing products within 3 months prior to screening and throughout the study was prohibited. Participants were also excluded if they consumed foods or beverages containing poppy seeds, grapefruit, Seville oranges, cranberry juice, or ginger within 7 days prior to study initiation and throughout the study. Consumption of alcohol or caffeine-containing foods or beverages within 48 h prior to study initiation and throughout the study was not permitted.
2.2 Study Design
There were three treatment periods (Treatment Period 1 [Days 1–10], 2 [Days 11–20], and 3 [Days 21–26]) (electronic supplementary material [ESM] Fig. S1). Participants were randomized to one of six treatment sequences defined by the two-Williams-squares design: ABC, CAB, and BCA (Square 1); and CBA, ACB, and BAC (Square 2), where Treatment A was tucatinib (Almac, Souderton, PA, USA, and Seagen Inc., Bothell, WA, USA), Treatment B was matching placebo for tucatinib (Almac and Seagen Inc.), and Treatment C was moxifloxacin (Bayer, Pittsburgh, PA, USA, and McKesson Medical Surgical, Richmond, VA, USA).
For each treatment period, tucatinib 300 mg or matching placebo were administered twice daily (approximately 12 h apart under fed conditions, within 10 min after completion of standardized meals) on 4 consecutive days, with a single dose of tucatinib 300 mg or matching placebo administered within 10 min after completion of breakfast on the fifth day. Moxifloxacin was administered as a single dose of 400 mg after at least 8 h fasting on the morning of the fifth day. Moxifloxacin was used as a positive control to determine the assay sensitivity of this study, with an expected peak of QT effect of 10–15 ms [1]. Each dose of tucatinib, placebo, and moxifloxacin was administered orally with approximately 240 mL of room-temperature water. There was a 5-day washout between study treatment administrations (Days 6–10 of Treatment Period 1, and Days 16–20 of Treatment Period 2).
After the screening period (up to 28 days), participants were admitted to the clinical research center on the day before initiating treatment. Participants were discharged on Day 27 upon completion of all pharmacokinetic (PK) and safety assessments, and attended a follow-up visit approximately 7 days after the last dose of study treatment.
2.3 PK Assessments
Blood samples were collected for PK analysis pre-dose, and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, and 36 h post-dose on Days 5, 15, and 25. Sampling windows for blood samples were within 30 min prior to dosing for the pre-dose sample timepoint; ±5 min for sampling timepoints within the first 12 h post-dose; and ±15 min for sampling timepoints at 24 and 36 h post-dose. A single blood sample was collected within 30 min pre-dose on Days 1, 11, and 21.
Plasma concentrations of tucatinib and its predominant metabolite, ONT-993, were determined using validated analytical procedures, performed by Covance (Madison, WI, USA) using high-performance liquid chromatography (HPLC) with tandem mass spectrometry (LC-MS/MS), as previously described [14].
PK parameters were determined from plasma concentrations of tucatinib and ONT-993 on Days 5, 15, and 25 using noncompartmental methods and included area under the plasma concentration-time curve over dosing interval (AUC0–τ); maximum observed plasma concentration (Cmax); time of maximum observed plasma concentration (Tmax); apparent plasma terminal elimination half-life (t½); and metabolic ratio based on AUC0–τ (MRAUC; ONT-993 only).
2.4 ECG Assessments
All continuous 12-lead ECG data (Holter) were recorded using a Mortara Surveyor Central System. On Days 1, 11, and 21, continuous ECG recordings were started approximately 1.5 h pre-dose and 12-lead ECGs were extracted at 30, 55, and 80 min pre-dose to establish baseline. On Days 5, 15, and 25, continuous ECG recordings were started approximately 45 min pre-dose relative to administration of study treatment, and replicate 12-lead ECGs were extracted at approximately 30 min pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, and 36 h post-dose relative to administration of study treatment. Sampling windows for ECG extractions were ±5 min for sampling timepoints pre-dose and within the first 12 h post-dose, and ±15 min for sampling timepoints at 24 and 36 h post-dose. Participants were resting in the supine position in an undisturbed environment for at least 10 min prior to and 5 min after each nominal time.
The ECG parameters determined comprised heart rate, PR interval, QRS interval, QT interval, and QTcF. The QT interval was corrected for heart rate using the Fridericia formula (\(QTc =\frac{QT}{RR}\) 1/3) [15]. The primary endpoint of the study was the placebo-corrected change from baseline in QTcF (ΔΔQTcF). Changes from baseline in heart rate (ΔHR), QTcF (ΔQTcF), PR (ΔPR), and QRS (ΔQRS) were evaluated as secondary ECG endpoints.
2.5 Safety
Safety was assessed based on the frequency and severity of treatment-emergent adverse events (TEAEs), clinical laboratory parameters, vital signs, 12-lead ECG, and physical examination. TEAEs were assessed and graded applying the Common Terminology Criteria for Adverse Events version 5.0. TEAEs were summarized by preferred terms and system organ class (coded using the Medical Dictionary for Regulatory Activities [MedDRA] version 21.1).
2.6 Statistical Analysis
Descriptive statistics were applied to summarize demographics, safety, and PK parameters. No formal statistical analyses were planned for PK data.
The ECG data were collected for analysis using the Expert-Precision QT technique, performed using Comprehensive Analysis of Repolarization Signal (The University of Rochester Medical Center, Rochester, NY, USA, and iCardiac Technologies, Rochester, NY, USA) software. The analyses were performed in accordance with the ICH E14 guideline [1] and respective quality assurance documents.
The baseline for all ECG parameters from each treatment period was the average of the three pre-dose timepoints on Days 1, 11, and 21. Analysis of the primary endpoint, ΔΔQTcF, was a by-timepoint analysis based on a linear mixed-effects model with ΔQTcF as the dependent variable; period, sequence, time (categorical), study treatment (tucatinib, moxifloxacin, and placebo), and time-by-treatment interaction as fixed effects; and baseline QTcF as a covariate. From this analysis, the least squares (LS) mean and two-sided 90% confidence intervals (CIs) were calculated for tucatinib versus placebo at each post-dose timepoint. The analysis of the secondary ECG parameters ΔHR, ΔPR, and ΔQRS used the same model as described for ΔΔQTcF.
The relationships between tucatinib and ONT-993 plasma concentrations and ΔΔQTcF were quantified using a linear mixed-effects modeling approach with a full model including ΔΔQTcF as the dependent variable, time-matched plasma concentrations of tucatinib and ONT-993 as the covariates, centered baseline QTcF as an additional covariate, and random intercept and slopes per participant. Model comparison of the full model and reduced models with either tucatinib or ONT-993 only were conducted using Akaike information criterion (AIC). Among these models, the model with the smallest AIC was selected as primary. If the primary model included both analytes (tucatinib and ONT-993), both were used to predict the QT effect. The geometric mean of the individual Cmax values of tucatinib were determined together with the geometric mean concentration of ONT-993 at the Tmax of tucatinib. Similarly, the geometric mean of the individual Cmax of ONT-993 was determined together with the geometric mean concentration of tucatinib at Tmax of ONT-993. The predicted effect using the exposure-response model and its two-sided 90% CI for ΔΔQTcF at these pairs of concentrations were obtained for the model with both analytes (tucatinib and ONT-993). Based on by-timepoint analysis, if the upper bound of the 90% CI was below 10 ms for all post-dose timepoints, it was concluded that tucatinib did not have a significant effect on QT interval prolongation.
Assay sensitivity was also evaluated by analyzing the effect of moxifloxacin on ΔΔQTcF using the same model as the primary analysis. For the timepoints at 1, 2, and 3 h post-dose, the contrast in treatment ΔΔQTcF = “moxifloxacin versus placebo” was tested against the one-sided null hypothesis ΔΔQTcF ≤5 ms on the significance level of 5%. Multiplicity was controlled using the Hochberg procedure. If ΔΔQTcF was significantly larger than 5 ms for at least one timepoint, assay sensitivity was considered shown. Two-sided 90% CIs were obtained for the contrast at all timepoints.
Statistical analyses were performed using SAS® version 9.4 or a later version (SAS Institute Inc., Cary, NC, USA) or WinNonlin Version 8.1 (Certara L.P., Princeton, NJ, USA).
Approximately 60 participants were to be enrolled to ensure that 48 participants completed the study. Assuming a one-sided 5% significance level and a within-participant standard deviation of 7 ms for ΔQTcF, as well as a true mean difference of 3 ms in ΔQTcF between tucatinib and placebo, a sample size of 48 evaluable participants was expected to provide 99.4% power to demonstrate that the upper bounds of all the two-sided 90% CIs on ΔΔQTcF fell below 10 ms for up to 11 timepoints. Furthermore, to demonstrate assay sensitivity, this sample size would provide a power of 90.5% to exclude a mean difference of 5 ms in ΔQTcF between the moxifloxacin and placebo groups from the lower bound of the two-sided 90% CI for at least one of the three prespecified timepoints.
The safety population consisted of all participants who received at least one dose of study treatment and had at least one post-dose safety assessment. The PK population consisted of all participants who received at least one dose of study treatment and had at least one evaluable post-dose plasma assessment for tucatinib or ONT-993. A participant may have been excluded from the PK summary statistics and statistical analysis if they vomited at or within two times of the median Tmax. The QT/QTc population consisted of all participants in the safety population with measurements at baseline as well as on-treatment with at least one post-dose timepoint with a valid ΔQTcF value. The QT/QTc population was used for the by-timepoint and categorical analyses of the cardiodynamic ECG parameters. The PK/QTc population consisted of all participants who were in both the QT/QTc and PK populations with at least one pair of post-dose PK and QTcF data from the same timepoint. The PK/QTc population was used for the exposure-response analysis.
3 Results
3.1 Study Participants
A total of 55 participants were enrolled in the study, of whom 53 received at least one dose of study treatment and were included in one of six treatment sequences: ABC (n = 9), CAB (n = 8), BCA (n = 9), CBA (n = 9), ACB (n = 9), and BAC (n = 9). Fifty-one participants completed the study. Two participants withdrew from the study prior to receiving any study treatment and were excluded from the safety and PK populations (in the CAB sequence, one participant withdrew consent and another participant was withdrawn by physician decision due to out-of-range vital signs on Day 5 prior to dosing). Two participants in the BCA sequence who withdrew from the study prior to receiving tucatinib were excluded from the PK population (one participant withdrew consent on Day 15 and another participant was withdrawn on Day 2 by physician decision due to urinary incontinence [part of the participant’s medical history]).
Overall, participants had a mean age of 42 years (Table 1). The majority of participants were male (84.9%) and Black or African American (52.8%). Participants had a mean weight of 81.5 kg, mean height of 173.2 cm, and a mean BMI of 27.1 kg/m2. Baseline ECG parameters were within expectations for a healthy population. Respective mean heart rate, QTcF, PR, and QRS were between 57.6 and 58.2 beats per min, 400.8 and 401.8 ms, 154.5 and 158.1 ms, and 106.4 and 106.8 ms, respectively.
3.2 PK of Tucatinib and ONT-993
Figure 1 shows the arithmetic mean plasma concentrations of tucatinib and ONT-993 after the last dose administration following multiple doses of tucatinib (300 mg twice daily for 4 days and a single dose on the fifth day). Steady-state PK parameters for tucatinib and ONT-993 following multiple doses of tucatinib are shown in Table 2. Median Tmax was 3.00 h (range 1.00–4.10 h) for both tucatinib and ONT-993. Steady-state geometric mean (coefficient of variation [CV%]) Cmax was 519 (26.7) ng/mL for tucatinib and 65.0 (40.5) ng/mL for ONT-993. Steady-state plasma exposures based on geometric mean (CV%) AUC0–τ were 3520 (26.0) h*ng/mL for tucatinib and 409 (36.5) h*ng/mL for ONT-993, and the geometric mean (CV%) MRAUC for ONT-993 was 0.116 (35.4).

Arithmetic mean (±SD) plasma concentrations of tucatinib and ONT-993 after last dose administration following multiple oral doses of 300 mg tucatinib (twice daily for 4 days and a single dose on the fifth day). Blue open squares represent tucatinib; red open circles represent ONT-993. SD standard deviation
3.3 Cardiac Parameters
The mean ΔQTcF of tucatinib closely followed the placebo pattern across post-dose timepoints (ESM Fig. S2). Therefore, the mean ΔΔQTcF was very small, ranging from − 2.9 ms at 2 h post-dose to 0 ms at 4 h post-dose (Table 3 and Fig. 2). The upper bound of the 90% CI for ΔΔQTcF of tucatinib was <5 ms at all post-dose timepoints.

ΔΔQTcF across timepoints after 300 mg tucatinib (twice daily for 4 days and a single dose on the fifth day) at steady-state, or 400 mg moxifloxacin (QT/QTc population). Blue open squares represent tucatinib; red open triangles represent moxifloxacin; errors bars represent the 90% CI based on a linear mixed-effects model; gray dashed line represents a value of 10 ms. ΔΔQTcF placebo-corrected change from baseline in QT interval corrected for heart rate using Fridericia’s method, CI confidence interval, QTc corrected QT
After dosing with moxifloxacin, the mean ΔΔQTcF increased and peaked at 16.6 ms (90% CI 14.03, 19.08) at 3 h post-dose (Table 3 and Fig. 2). The lower bound of the 90% CI for ΔΔQTcF of moxifloxacin was >5 ms at all predefined timepoints (1, 2, and 3 h).
After multiple oral 300 mg doses (twice daily for 4 days and a single dose on the fifth day), tucatinib had no clinically relevant effect on heart rate or cardiac conduction, i.e., the PR and QRS intervals (ESM Fig. S3).
3.4 Concentration-QTc Model
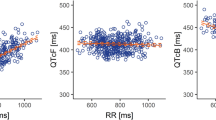
A linear mixed-effects model with tucatinib and ONT-993 as analytes was applied to the data. The relationship between the individually observed tucatinib and ONT-993 plasma concentrations and ΔΔQTcF is shown in Fig. 3a and ESM Fig. S4a. The mean ΔΔQTcF within each tucatinib and ONT-993 concentration decile and the model-predicted mean ΔΔQTcF with 90% CI are shown in Fig. 3b and ESM Fig. S4b, respectively. The estimated population slope of the concentration-QTc relationship was shallow and not statistically significant: − 0.003 ms per ng/mL (90% CI − 0.0090, 0.0036) for tucatinib and 0.04 ms per ng/mL (90% CI − 0.009, 0.085) for ONT-993, with a moderate statistically significant intercept of − 2.9 ms (90% CI − 4.96, 0.80). The model-predicted and estimated ΔΔQTcF at the peak of the geometric mean concentrations of tucatinib and ONT-993 are shown in Fig. 3c and ESM Fig. S4c, respectively. The ΔΔQTcF of tucatinib was predicted to be − 1.80 ms (90% CI − 3.90, 0.30) with a tucatinib plasma concentration of 511 ng/mL and an ONT-993 plasma concentration of 64 ng/mL.

Relationship between tucatinib concentrations and ΔΔQTcF (PK/QTc population). a Scatter plot of observed plasma concentrations for tucatinib (blue squares) and ΔΔQTcF from the full model. Red lines are mean predicted (solid) and 90% CI (dashed). b Model-predicted (mean, 90% CI, black line and shaded area) and observed (mean, 90% CI, red dots and vertical lines) ΔΔQTcF across deciles of tucatinib plasma concentrations from the full model. c Model-predicted (mean, 90% CI, black line and shaded area) and estimated (mean, 90% CI, blue square and shaded area) ΔΔQTcF at geometric mean Cmax (blue square). Gray dashed lines represent a value of 10 ms. CI confidence interval, Cmax maximum observed plasma concentration, ΔΔQTcF placebo-corrected change from baseline in QT interval corrected for heart rate using Fridericia’s method, PK pharmacokinetic, QTc corrected QT
3.5 Safety
Overall, 19 (35.8%) participants reported a total of 58 TEAEs. TEAEs occurred more frequently with tucatinib (12/51 [23.5%] participants) than placebo (7/53 [13.2%] participants) or moxifloxacin (7/52 [13.5%] participants). There were no deaths or serious adverse events. All TEAEs resolved and no participants withdrew from the study due to a TEAE. Fifty-seven TEAEs were considered to be Grade 1 in severity. One TEAE of increased aspartate aminotransferase (AST), which was classified as Grade 3, was reported after the participant had completed treatment during the follow-up visit (Day 32; 27 days after the last dose of tucatinib and 17 days after the last dose of moxifloxacin); this TEAE was considered unrelated to tucatinib or moxifloxacin and had resolved by an unscheduled visit on Day 39. There were no clinically significant findings in hematology, urinalysis, vital signs, or ECGs during the study. Except for the TEAE of increased AST, there were no significant clinical chemistry findings.
4 Discussion
This study evaluated the QT effects of tucatinib in healthy volunteers; it was performed in line with ICH E14 guidance, which mandates that most new drugs are assessed for potential effects on cardiac repolarization in a TQT study [1, 11]. Steady-state tucatinib exposures were achieved after the administration of tucatinib 300 mg (twice daily for 4 days and a single dose on the fifth day), with geometric mean (CV%) tucatinib Cmax and AUC0–τ values of 519 (26.7) ng/mL and 3520 (26.0) h*ng/mL, respectively. This enabled the evaluation of therapeutically relevant tucatinib concentrations on cardiac parameters.
The ICH E14 guideline recommends using a positive control to establish assay sensitivity. Assuming the positive control prolongs the mean QT/QTc interval by approximately 5 ms, it would ensure the study is able to detect such an effect of the study drug [1]. Moxifloxacin is the most frequently used positive control in TQT studies [16]. In this study, moxifloxacin 400 mg was associated with a clear increase in mean ΔΔQTcF, with a peak value of 16.6 ms (90% CI 14.03, 19.08) at 3 h post-dose. In addition, the lower bound of the 90% CI was >5 ms, which confirmed the sensitivity of the assay.
The mean ΔQTcF of tucatinib closely followed the placebo pattern across post-dose timepoints. The mean ΔΔQTcF was very small, ranging from − 2.9 ms at 2 h post-dose to 0.0 ms at 4 h post-dose. In addition, the upper bound of the 90% CI for ΔΔQTcF of tucatinib was below 5 ms at all post-dose timepoints, while the lower bound of the 90% CI for ΔΔQTcF of moxifloxacin was >5 ms at all timepoints. These results indicate that tucatinib had no statistically significant or clinically relevant effect on ΔΔQTcF in a clinical setting within the limits of assay sensitivity. Moreover, tucatinib had no clinically relevant effect on heart rate or cardiac conduction (PR and QRS intervals).
The mean ΔΔQTcF of tucatinib was predicted to be − 1.80 ms (90% CI − 3.90, 0.30) at tucatinib and ONT-993 plasma concentrations of 511 ng/mL and 64 ng/mL, respectively. Based on a concentration-QTc model, a QTcF effect exceeding 10 ms was excluded within the observed ranges of tucatinib (~1000 ng/mL) and ONT-993 plasma concentrations (~150 ng/mL); this was consistent with the results from the by-timepoint analysis. The estimated population slope of the concentration-QTc relationship was also shallow and not statistically significant (−0.003 ms per ng/mL [90% CI − 0.0090, 0.0036] for tucatinib and 0.04 ms per ng/mL [90% CI − 0.009, 0.085] for ONT-993), correlating with a low risk of ventricular arrythmia.
Throughout this study, tucatinib had a manageable safety profile after multiple oral doses (300 mg twice daily for 4 days and a single dose on the fifth day). Nineteen of 53 (35.8%) participants reported a total of 58 TEAEs, all of which resolved. Fifty-seven of 58 (98.3%) TEAEs were Grade 1 in severity. One Grade 3 TEAE of increased AST was recorded at the follow-up visit (27 days after the last dose of tucatinib and 17 days after the last dose of moxifloxacin). This TEAE was considered unrelated to tucatinib or moxifloxacin and had been resolved by an unscheduled visit on Day 39.
Evaluation of safety in previous studies demonstrated there were no clinically relevant ECG abnormalities when tucatinib was administered to volunteers in the presence of a strong CYP2C8 inhibitor (tucatinib concentrations increased by threefold when tucatinib was concomitantly administered with gemfibrozil) [17], or to volunteers with moderate or severe hepatic impairment (tucatinib concentrations increased by 1.6-fold in volunteers with severe hepatic impairment) [9]. Moreover, no significant cardiac effects were observed in cynomolgus monkeys administered tucatinib 0, 2.5, 10, or 20 mg/kg twice daily [8]; these doses are higher than the approved dose of 300 mg twice daily cited within the tucatinib prescribing information [4, 5].
One limitation of this study was the relatively low number of female participants included. It is hypothesized that female patients may be more likely to experience drug-induced QT prolongation [18, 19], which is important to note as tucatinib in combination with trastuzumab and capecitabine is indicated in adults with advanced, unresectable, or metastatic HER2-positive breast cancer, which predominantly occurs in female patients. Another limitation was that the study was conducted in healthy volunteers; retrospective studies have shown an increased risk of QT prolongation in patients with cancer compared with individuals without cancer, with several potential clinical factors involved [20,21,22].
5 Conclusion
In conclusion, tucatinib after multiple oral doses (300 mg twice daily for 4 days and a single dose on the fifth day) had no clinically relevant effects on the studied ECG parameters. Therefore, this study constitutes a clearly negative TQT study, as defined in the ICH E14 guidance document [1].
References
E14 Implementation Working Group. ICH E14 guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs questions & answers (R3). Available at: https://database.ich.org/sites/default/files/E14_Q%26As_R3_Q%26As.pdf. Accessed 23 Mar 2023.
Siena S, Sartore-Bianchi A, Marsoni S, Hurwitz H, McCall S, Penault-Llorca F, et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann Oncol. 2018;29(5):1108–19. https://doi.org/10.1093/annonc/mdy100.
Kulukian A, Lee P, Taylor J, Rosler R, de Vries P, Watson D, et al. Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol Cancer Ther. 2020;19(4):976–87. https://doi.org/10.1158/1535-7163.Mct-19-0873.
Seagen Inc. Tukysa (tucatinib): European summary of product characteristics. Available at: https://www.ema.europa.eu/en/documents/product-information/tukysa-epar-product-information_en.pdf. Accessed 12 Jan 2023.
US FDA. TUKYSA US Prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/213411s004lbl.pdf. Accessed 6 Mar 2023.
Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382(7):597–609. https://doi.org/10.1056/NEJMoa1914609.
Curigliano G, Mueller V, Borges V, Hamilton E, Hurvitz S, Loi S, et al. Tucatinib versus placebo added to trastuzumab and capecitabine for patients with pretreated HER2+ metastatic breast cancer with and without brain metastases (HER2CLIMB): final overall survival analysis. Ann Oncol. 2022;33(3):321–9. https://doi.org/10.1016/j.annonc.2021.12.005.
US FDA. Multi-disciplinary review and evaluation-NDA 213411 (tucatinib). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213411Orig1s000MultidisciplineR.pdf. Accessed 4 Mar 2023.
Topletz-Erickson AR, Lee AJ, Mayor JG, Sun H, Abdulrasool LI, Rustia EL, et al. The pharmacokinetics and safety of tucatinib in volunteers with hepatic impairment. Clin Pharmacokinet. 2022;61(12):1761–70. https://doi.org/10.1007/s40262-022-01183-6.
Strickler JH, Cercek A, Siena S, Andre T, Ng K, Van Cutsem E, et al. Primary analysis of MOUNTAINEER: A phase 2 study of tucatinib and trastuzumab for HER2 positive mCRC [abstract LBA-2]. European Society of Medical Oncology World Congress on Gastrointestinal Cancer; 2022.
Lester RM, Paglialunga S, Johnson IA. QT assessment in early drug development: the long and the short of it. Int J Mol Sci. 2019;20(6):1324. https://doi.org/10.3390/ijms20061324.
Li M, Ramos LG. Drug-induced QT prolongation and torsades de pointes. P T. 2017;42(7):473–7.
He S, Moutaoufik MT, Islam S, Persad A, Wu A, Aly KA, et al. HERG channel and cancer: a mechanistic review of carcinogenic processes and therapeutic potential. Biochim Biophys Acta Rev Cancer. 2020;1873(2):188355. https://doi.org/10.1016/j.bbcan.2020.188355.
Meyer JA, DeChenne S, Foerder CA, Hengel SM. Bioanalysis of tucatinib and metabolite, and a five-way cross-validation to support clinical pharmacokinetic analysis. Bioanalysis. 2023;14(22):1443–52. https://doi.org/10.4155/bio-2022-0199.
Fridericia LS. The duration of systole in an electrocardiogram in normal humans and in patients with heart disease. Ann Noninvasive Electrocardiol. 2003;8(4):343–51. https://doi.org/10.1046/j.1542-474x.2003.08413.x.
Yan LK, Zhang J, Ng MJ, Dang Q. Statistical characteristics of moxifloxacin-induced QTc effect. J Biopharm Stat. 2010;20(3):497–507. https://doi.org/10.1080/10543400903581945.
Topletz-Erickson A, Lee A, Rustia EL, Sun H, Mayor JG, Abdulrasool LI, et al. Evaluation of safety and clinically relevant drug-drug interactions with tucatinib in healthy volunteers. Clin Pharmacokinet. 2022;61(10):1417–26. https://doi.org/10.1007/s40262-022-01144-z.
Huo J, Wei F, Cai C, Lyn-Cook B, Pang L. Sex-related differences in drug-induced QT prolongation and Torsades de Pointes: a new model system with human iPSC-CMs. Toxicol Sci. 2019;167(2):360–74. https://doi.org/10.1093/toxsci/kfy239.
Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89(11):1363–72. https://doi.org/10.1136/heart.89.11.1363.
Kim P, Masha L, Olson A, Iliescu C, Karimzad K, Hassan S, et al. QT prolongation in cancer patients. Front Cardiovasc Med. 2021;8:613625. https://doi.org/10.3389/fcvm.2021.613625.
Lin Y, Yu H, Liu F, Chen C, Zhang Y, Wang B, et al. Hospitalized cancer patients with acquired long QT syndrome-a matched case-control study. Cardiooncology. 2020;6:3. https://doi.org/10.1186/s40959-020-0057-2.
Hardy JR, Bundock D, Cross J, Gibbons K, Pinkerton R, Kindl K, et al. Prevalence of QTc prolongation in patients with advanced cancer receiving palliative care-a cause for concern? J Pain Symptom Manag. 2020;59(4):856–63. https://doi.org/10.1016/j.jpainsymman.2019.12.356.
Acknowledgments
The authors would like to thank all study participants, as well as the investigators Jennifer Zon and Alexander King from Covance, Anthony Lee from Seagen for his contribution to the study oversight, and Borje Darpo from IRT for conducting the PK-QT related analyses. Medical writing support was provided by Charlotte Simpson, PhD, and editorial support, including formatting, proofreading, and submission, was provided by Travis Taylor, BA, both of Scion, London, UK, supported by Seagen according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M22-1460). Some of these results were presented at the 2021 American College of Clinical Pharmacology annual meeting.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for this publication was provided by Seagen Inc., Bothell, WA, USA.
Conflicts of Interest
The authors wrote this article with the assistance of a medical writer funded by the sponsor. All authors had full access to the relevant data, vouch for the completeness and accuracy of the data and for adherence of the trial to the protocol, and assume final responsibility for the content of the article and the decision to submit the article for publication. Ariel R. Topletz-Erickson, JoAl G. Mayor, Hsu-Tai Liu, Layth I. Abdulrasool, and Christopher J. Endres are employees of Seagen and hold stocks and shares in Seagen.
Availability of Data and Material
Qualified researchers may request access to certain data and related study documents consistent with the Principles for Responsible Clinical Trial Data Sharing. Interested researchers can use http://www.clinicalstudydatarequest.com to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care.
Ethics Approval
Before the start of this study, the study protocol and informed consent form were approved by a central Institutional Review Board (Midlands Independent Review Board, Overland Park, KS, USA). All amendments to the protocol were also approved by the Institutional Review Board. The study was designed and implemented in accordance with the International Council for Harmonisation of Technical Requirements for Human Use (ICH) E14 guidance on The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs [1]. The study was also conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable regional and local legislation.
Consent to Participate
All participants provided written consent prior to study enrolment.
Author Contributions
All authors contributed to the study design; analysis and interpretation of the data; and critical revisions of the article. All authors provided final approval for publication.
Consent for publication
All participants provided written consent prior to study enrolment.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Topletz-Erickson, A.R., Mayor, J.G., Liu, HT. et al. Effect of Tucatinib on Cardiac Repolarization in Healthy Volunteers. Drugs R D 23, 411–419 (2023). https://doi.org/10.1007/s40268-023-00440-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-023-00440-8