
Abstract
Background and Introduction
SHR6390 is a new developed highly effective and selective small-molecule oral CDK4/6 inhibitor. We aimed to evaluate the effect of food on the pharmacokinetics of SHR6390 tablets.
Methods
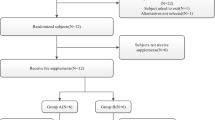
In an open-label two-way crossover study, 24 healthy Chinese volunteers were randomly divided into Group A and Group B, and 12 volunteers in each group received a single oral dose of a SHR6390 150-mg tablet under fasting and high-fat conditions. Blood samples were collected and determined for pharmacokinetic analyses. A liquid chromatography-tandem mass spectrometry method was developed and validated for determining the SHR6390 concentration.
Results
The time to maximum plasma concentration was not significantly affected by a high-fat diet. Compared with the fasting group, maximum plasma concentration, i.e., the area under the concentration–time curve (AUC0–t and AUC0–∞) was altered significantly, as evidenced by an increase of 56.9%, 38.6%, and 37.5% respectively. We identified seven metabolites of SHR6390 from the plasma samples, and we found no sex differences in metabolic pathways. All treatment-emergent adverse events were Grade 1 or 2.
Conclusions
Food intake increased the maximum plasma concentration, AUC0–t, and AUC0–∞ significantly compared with the fasting condition. Meanwhile, single-dose SHR6390 for two treatment cycles is safe. SHR6390 was administered in a fasting status in the pivotal phase III study (NCT03927456) and chosen for the final drug label.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Food intake increased the maximum plasma concentration, AUC0–t, and AUC0–∞ of SHR6390 tablets significantly compared with the fasting condition. |
Single-dose SHR6390 (150 mg) for two treatment cycles is safe. |
1 Introduction
Cyclin-dependent kinase (CDK) is a type of serine/threonine kinase that forms dimers with the corresponding cyclin, and then phosphorylates the downstream protein molecules, promoting the orderly progression of each phase of the cell cycle and achieving cell growth and proliferation. The CDK kinase family includes CDK1–9 and multiple other members, among which CDK4/6 is highly homologous. Through interaction with cyclin D, CDK4/6 phosphorylates Rb protein, dissociates Rb-E2F complex, releases free E2F into the nucleus, regulates protein transcription, and plays a decisive role in the progression of cells from G1 phase to S phase [1].
Disorderly and excessive cell proliferation is an important feature of tumor cells, which is often closely related to the uncontrolled regulation of the G1/S cell cycle. In a small number of tumors (such as retinoblastoma and small-cell lung cancer), uncontrolled regulation of G1/S in the cell cycle is caused by mutations in the Rb gene [2, 3]; and, in most tumors, various genetic or epigenetic changes cause high CDK4/6 activity, which leads to phosphorylation and inhibition of Rb proteins, and ultimately to disorderly cell proliferation [4]. CDK4/6 is therefore an important molecular target for tumor therapy.
In human breast cancer, the incidence of CCND1 gene (encoding cyclin D1) amplification is 15–20%, and the overexpression rate can be as high as 50% [5,6,7]; deletion or mutation of the CDKN2A gene (encoding p16INK4A, which blocks the formation of the CDK4/6-cyclin D dimer) is common [8]. Therefore, CDK4/6 can be used as an important target for the treatment of breast cancer. Palbociclib, a CDK4/6 inhibitor developed by Pfizer, was approved by the US Food and Drug Administration in 2015 and 2016, respectively, in combination with letrozole or fulvestrant in the treatment of advanced HR+/HER2− breast cancer, and was the first CDK4/6 inhibitor on the market. In March 2017, ribociclib (Novartis) was also approved by the Food and Drug Administration in combination with aromatase inhibitors in the treatment of advanced or metastatic HR+/HER2− breast cancer. In September 2017, abemaciclib (Eli Lilly & Company) was approved for the treatment of patients with advanced or metastatic HR+/HER2− breast cancer after endocrine therapy, either alone or in combination with fluviride.
SHR6390 is a highly effective and selective, small-molecule oral CDK4/6 inhibitor developed by Jiangsu Hengrui Pharmaceuticals Co., Ltd. In vitro studies showed that SHR6390 selectively inhibited CDK4/6 with half maximal inhibitory concentration values of 10–12 nM, which were comparable to that of PD-0332991 (palbociclib [Pfizer Inc.]). Furthermore, SHR6390 showed an effect similar to PD-0332991 in inhibiting the CDK4/6-Rb signal pathway, inducing G1 phase arrest, and selectively inhibiting the proliferation of tumor cells retaining Rb expression. In multiple xenograft models, SHR6390 significantly inhibited tumor growth (including human colon cancer, ovarian cancer, and breast cancer) with tumor inhibition rates close to or higher than that of PD-0332991.
A new Class 1.1 chemical drug, SHR6390, successfully obtained clinical trial approval by the China National Medical Products Administration in October 2015, with the approval numbers 2015L02257, 2015L02258, and 2015L02259. At present, SHR6390 has entered the phase I clinical study, and it is believed that SHR6390 will provide more treatment options for patients with tumors. However, at the present time, there are no studies of the food effect on the bioavailability of SHR6390 tablets. For an oral medicine, it is necessary to understand the effect of food on the drug. To develop a more appropriate administration regimen and obtain preliminary data on human metabolism, we conducted this study to investigate the effect of food on SHR6390.
2 Materials and Methods
2.1 Ethics Approval
The protocol of this clinical study was approved and authorized by the Ethics Committee of the Clinical Research Institute of the Qingdao University Hospital (Qingdao, China). All clinical procedures were carried out in the Phase I Clinical Trial Unit of the Qingdao University Hospital. In addition, this study was conducted according to the Declaration of Helsinki and the Guidelines for Good Clinical Practice. The clinical registration number is CTR20191262 (http://www.chinadrugtrials.org.cn/). All recruited volunteers agreed and provided written informed consent before the study was initiated.
2.2 Subjects
Male and female healthy subjects aged 18 years and above and with a body weight index of 19–26 kg/m2 were enrolled in the study. The major exclusion criteria were: (1) clear history of cardiovascular, central nervous system, liver, kidney, or other organ system diseases; (2) abnormal 12-lead electrocardiogram; (3) chronic or active gastrointestinal diseases such as esophagitis, gastritis, gastric ulcer, enteritis, gastrointestinal bleeding, gastroesophageal reflux disease, gastrointestinal obstruction, esophageal varices, or gastrointestinal surgery (part of the digestive tract resection, cholecystectomy), and the investigator believes is still clinically significant; (4) smoked five of more cigarettes/day before dosing; (5) those who were allergic to the active ingredients of the preparation and related compounds and any excipients; (6) drank or ate any caffeine-containing beverage or food within 48 hours of study initiation; and (7) regular drinkers within 6 months before the test, i.e., drinking more than 14 units of alcohol per week (1 unit = 360 mL of beer, or 45 mL of spirits, or 150 mL of wine), any alcohol-containing products cannot be stopped during the study and those who were positive for an alcohol breath test.
2.3 Study Design and Administration
This was a phase I, randomized, open-label, two-period, crossover study conducted in healthy subjects. The study consisted of two groups (A and B). The effect of food on the pharmacokinetics of SHR6390 was determined. The study was designed as an open-label, randomized, two-treatment-period crossover study. Briefly, 24 healthy volunteers took a single oral dose of SHR6390 (150 mg:125 mg × 1, 25 mg × 1) and were randomized (12:12) into either a fed condition (high-calorie intake, approximately 800–1000 calories; high-fat intake consisting of approximately 50% of the total caloric content of the meal, 30 min before dosing) and a fasting condition (no food for 10 h). After a 14-day washout period, study participants received the same amount of SHR6390 in the conditions of the other group. A mouth check was implemented to assess whether the drug was taken.
2.4 Tolerability Measurements
Treatment-emergent adverse events (TEAEs) were defined according to the Common Terminology Criteria for the Classification of Adverse Events of the National Cancer Institute (version 4.03). The following parameters were applied for TEAE measurements: mild, moderate, or severe intensity; duration; clinical outcome; and severity. The specific items of this study included vital signs (sitting blood pressure, body temperature, and heart rate), physical examinations, electrocardiograms, as well as clinical laboratory tests (hematology tests, biochemistry tests, urinalysis, and coagulation indexes).
2.5 Pharmacokinetic Assessment
For the pharmacokinetic assessment, 4 mL of blood samples were collected into heparin lithium anticoagulant tubes at different timepoints: pre-dose, and 0.5, 1, 2, 3, 4, 6, 8, 10, 24 48, 72, 96, 120, and 144 hours after SHR6390 administration. The collected blood samples were centrifuged at 2–8 ℃, 2000 g for 10 minutes, and the plasma was collected separately and stored in a − 70 °C freezer before analysis. Pharmacokinetic parameters of SHR6390 were estimated using non-compartmental methods based on plasma concentrations with WinNonlin® Certara Company (version 7.0). The pharmacokinetic parameters estimated included: time to maximum plasma concentration (Tmax), maximum plasma concentration (Cmax), area under the plasma concentration–time curve (AUC0–t or AUC0–∞), mean retention time, apparent clearance, volume of apparent distribution, and terminal elimination half-life. All collected samples were analyzed at Frontage (Shanghai) Pharmaceutical Technology Co., Ltd (Shanghai, China) using a validated liquid chromatography-tandem mass spectrometry method. Twenty microliters of internal standard (d8-Palbociclib, 20 ng/mL in acetonitrile:deionized water [1:1, v:v]) was added into the 50-μL plasma sample. Then, the sample was subject to protein precipitation with 400 μL of acetonitrile, vortexed and centrifuged, and the resulting supernatant was acidified with 0.1% formic acid. The supernatant was loaded on a Waters Xbridge C18, 3.5-μm column. The mobile phase A was performed with a 0.1% formic acid and ammonium formate 10 mM aqueous solution, while mobile phase B was 10 mM of ammonium formate in water: acetonitrile (90:10, v:v). The calibration range of the SHR6390 was 0.390–390 ng/mL. The lower limit of quantification was 0.390 ng/mL. The accuracy of the assay was − 7.7% to 1.0%, and the precision was within 8.0% of the coefficient of variation.
2.6 Statistical Analysis
Area under the concentration–time curve (AUC0–t and AUC0–∞) and Cmax (systemic exposure parameters) were naturally log-transformed and assayed with a mixed-effects model. The analysis of variance model was applied to analyze the point estimates and the ratios of population geometric means of the AUC and Cmax with 90% confidence intervals for the comparison between the fed and fasting conditions. The index food conditions (either fasting or fed), period, and sequence were considered in the model with the fixed effects of the food state. The factor of subjects nested within a sequence was considered as random effects. A conclusion of no effect of food on the pharmacokinetics of SHR6390 was made if the 90% confidence intervals were within the range of 0.80–1.25 for SHR6390 Cmax and AUC [9]. After data conversion, the Cmax and AUC of SHR6390 were assessed by an analysis of variance to evaluate the sex differences of the parameters under the fasting and fed conditions. The geometric mean ratio of the Cmax and AUC of female and male subjects and their 90% confidence intervals were calculated as well. All statistical data were analyzed with SAS (version 9.4; SAS Institute Inc., Cary, NC, USA).
3 Results
3.1 Demographics of the Fasting and Fed Groups
A total of 24 volunteers were enrolled from 81 candidates to undergo safety analyses until the completion of the study. Baseline characteristics between fasting and fed groups are shown in Table 1.
3.2 Safety
There were no deaths, serious adverse events (AEs), or discontinuations. Treatment-emergent AEs were preliminarily observed in the treatment groups (Table 2). Twelve clinical TEAEs were reported in eight subjects (33.3%, 8/24). All TEAEs were Grade 1 or 2. Twelve TEAEs occurred in six subjects (6/24; 25%) in the fasting condition and two subjects (2/24; 8.3%) in the fed condition. According to the existing data, the incidence of AEs in the fed state is lower than that in the fasting state. Most TEAEs spontaneously disappeared without any specific intervention during the observation.
3.3 Food Effect on the Pharmacokinetics of SHR6390
The concentration–time profiles of SHR6390 were analyzed under fasting and fed conditions. The median Tmax of the fed group was 5.0 hours, while the fasting group was 6.0 hours (Fig. 1), indicating that the Tmax was not significantly affected by a high-fat diet on the absorption rate of SHR6390. The pharmacokinetic parameters for SHR6390 when administered under fasting conditions or fed conditions are presented in Table 3 and a summary of the high-fat food effect is presented in Table 4. Between the fasting and fed groups, the Cmax and AUC of the fed group were higher than those of the fasting group, indicating that the high-fat meals have a tendency to increase the absorption of SHR6390. In addition, comparing the mean terminal elimination half-life of the fasting group and the high-fat fed group, indicating that the high-fat meals had no significant effect on the elimination of SHR6390. The exposure level of SHR6390 after a high-fat meal increased by 37.5–56.9% compared with that of the fasting condition. These results indicate that the high-fat meals can increase the exposure level of SHR6390. The effect of sex on the pharmacokinetics of SHR6390 was further analyzed. In the fasting group, compared with male subjects, the Cmax, AUC0–t, and AUC0–∞ of female subjects increased by 79.0%, 49.0%, and 43.9%,respectively. However, in the fed group, compared with male subjects, the Cmax, AUC0–t, and AUC0–∞ of female subjects increased by 25.9%, 29.2%, and 26.3%, respectively. The difference in exposure levels of SHR6390 between male and female subjects in the fed group was smaller than that in the fasting group.

The time-concentration profiles of mean SHR6390 plasma concentration
3.4 Metabolic Transformation of SHR6390
Plasma samples were examined to analyze the metabolic transformation by detecting the SHR6390 metabolites with the UPLC-PDA-Q-TOF system in all subjects of fasting and fed groups. Seven metabolites were detected in the plasma of healthy subjects after a single dose of SHR6390 150-mg tablets, as shown in Table 5. From the peak area of liquid chromatography-mass spectrometry detection, the plasma is dominated by the original drug M0, followed by the mono-oxidation and dehydrogenation metabolite M2, the mono-oxidation metabolite M3-1, and the di-oxidation metabolite M5, and the mass spectrum peak areas of all metabolites. All the detected metabolites are less than 10% of the original drug. There is no sex difference in the metabolic pathways of SHR6390 in the plasma of subjects. The proposed metabolic pathway of SHR6390 in the human body is shown in Fig. 2.

The proposed metabolic pathway of SHR6390
4 Discussion
The aim of this open-label phase I trial was to evaluate the effects of food intake on the safety profile and pharmacokinetics of oral SHR6390. This study provides data to support the administration of SHR6390 with food.
The study was carried out on 24 volunteers according to the guidelines “Assessing the Effects of Food on Drugs in INDs and NDAs: Clinical Pharmacology Considerations” from the Food and Drug Administration, in which during the fasting and fed treatment periods, the sponsor should collect samples from the study subjects (e.g., 12–18 samples per subject in each period). Additionally, it was a randomized and crossover design, with no placebo or control group [10].
The results of the pharmacokinetic study showed that after oral administration of SHR6390 150 mg in subjects under fasting and fed states, the terminal elimination half-life of SHR6390 was similar in the fasting and fed groups and the Tmax under the fed condition was 1 hour earlier compared with administration of SHR6390 under fasting conditions, indicating that the Tmax was not significantly affected by a high-fat diet on the absorption rate of SHR6390. A mixed-effects model analysis showed that the ratio of the geometric mean value and its 90% confidence interval between fed and fasting groups for Cmax, AUC0–t, and AUC0–∞ were 156.85% (138.42%, 177.72%), 138.57% (126.01%, 152.39%), and 137.50% (125.54%, 150.59%), respectively, indicating a high-fat diet could increase the exposure of SHR6390 in healthy subjects. At the dose level of 150 mg, Cmax, AUC0–t, and AUC0–∞ were increased by 56.9%, 38.6%, and 37.5% after an oral dose under a fed state as compared with those under a fasting state.
The study was completed in 24 Chinese healthy volunteers receiving a single dose of SHR6390 150 mg separately in two trial cycles. A total of eight subjects (33.3%) experienced at least one TEAE, among whom six subjects (25.0%) experienced TEAEs after administration in a fasting state and two subjects (8.3%) experienced TEAEs after a high-fat diet. A total of seven subjects (29.2%) experienced at least one treatment-related adverse event, among whom five subjects (20.8%) experienced treatment-related adverse events after administration in a fasting state and two subjects (8.3%) experienced TEAEs after a high-fat diet. No subjects experienced a Grade 3 or higher AE, no subjects withdrew from the study because of an AE, and no serious AEs were reported. Our study showed that the incidence of AEs when taking the drug post-prandially is lower than that when taking it pre-prandially, and the situation is similar to that of the same drug palbociclib [11].
5 Conclusions
The results of this study suggest that SHR6390 is metabolized through dehydrogenation, mono-oxidation dehydrogenation, mono-oxidation, mono-oxidation hydrogenation, deoxidation, and mono-oxidation glucuronic acid conjugation. The exposure in terms of Cmax, AUC0–t, and AUC0–∞ is higher in the fed state than that in the fasting state. Meanwhile, single-dose SHR6390 (150 mg) for two treatment cycles is safe. SHR6390 was administered in the fasting state in the pivotal phase III study (NCT03927456) and chosen for the final drug label.
References
Lange CA, Yee D. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr Relat Cancer. 2011;18:C19-24.
Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–6.
Harbour JW, Lai SL, Whang-Peng J, et al. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science. 1988;241:353–7.
Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–83.
Burdette-Radoux S, Tozer RG, Lohmann RC, et al. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest New Drugs. 2004;22(3):315–22.
Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23(18):4215–24.
Santarius T, Shipley J, Brewer D, et al. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10(1):59–64.
Liggett WH Jr, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16(3):1197–206.
US Food and Drug Administration. Guidance for industry: food-effect bioavailability and fed bioequivalence studies (last updated: December 2002). https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070241.pdf. Accessed 27 Aug 2018.
US Food and Drug Administration. Guidance for industry: assessing the effects of food on drugs in INDs and NDAs: clinical pharmacology considerations. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-effects-food-drugs-inds-and-ndas-clinical-pharmacology-considerations. Accessed 28 Apr 2022.
Ruiz-Garcia A, Plotka A, O’Gorman M, et al. Effect of food on the bioavailability of palbociclib. Cancer Chemother Pharmacol. 2017;79(3):527–33.
Acknowledgments
The authors thank the volunteers enrolled in this trial, as well as the staff who contributed to this trial. The authors are grateful to Jiangsu Hengrui Pharmaceuticals Co., Ltd., China, for their support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work was supported by grants from the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2020ZX09201-018) and the Natural Science Foundation of Shandong Province, with the grant number ZR2019MH101.
Conflicts of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Ethics approval
The protocol of this clinical study was approved and authorized by the Ethics Committee of the Clinical Research Institute of the Qingdao University Hospital (Qingdao, China).
Consent to participate
All recruited volunteers agreed and provided written informed consent before the study was initiated.
Consent for publication
All authors approved the publication of this manuscript.
Availability of data and material
The data is unavailable for confidentiality reasons.
Code availability
Not applicable.
Authors’ contributions
The authors are solely responsible for the design and conduct of this study. Yan-ping LIU, Ming-hui HU, Chen-jing Wang and Yu CAO performed a review of the topic, and wrote and revised the manuscript. Ping-ping LIN, Ting LI and Shu-qin LIU took part in analyzing pharmacokinetics data. Yu-ya WANG, Shao-rong LI and Xiang-kun LI took part in the analysis and interpretation of data, and prepared all figures and tables. All the authors approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liu, Yp., Hu, Mh., Lin, Pp. et al. Evaluation of the Effect of Food on the Pharmacokinetics of SHR6390, An Oral CDK4/6 Inhibitor, in Healthy Volunteers. Drugs R D 22, 175–182 (2022). https://doi.org/10.1007/s40268-022-00390-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-022-00390-7