
Abstract
Background
Hepatic impairment can impact apixaban pharmacokinetics and pharmacodynamics by decreasing cytochrome P450-mediated metabolism and factor X production.
Objective
This study evaluated the effect of mild or moderate (Child–Pugh A and B) hepatic impairment on apixaban pharmacokinetics, pharmacodynamics, and safety.
Methods
This open-label, parallel-group, single-dose study included eight mildly and eight moderately hepatically impaired subjects, and 16 healthy subjects. Subjects received a single oral apixaban 5-mg dose (day 1). Pharmacokinetic, pharmacodynamic, and safety assessments were completed at prespecified time points. Apixaban maximum plasma concentration and area under the concentration–time curve to infinity were compared between subjects with hepatic impairment and healthy subjects.
Results
Apixaban area under the concentration–time curve to infinity point estimates and 90% confidence intervals were 1.03 (0.80–1.32) and 1.09 (0.85–1.41) for subjects with mild and moderate hepatic impairment vs healthy subjects. Maximum plasma concentration results were similar. Mean (standard deviation) apixaban unbound fraction was 6.8% (1.4), 7.9% (1.8), and 7.1% (1.3) in subjects with mild or moderate hepatic impairment and in healthy subjects. Mean change from baseline in international normalized ratio (3 h post-dose) was 14.7%, 12.7%, and 10.7% for subjects with mild or moderate hepatic impairment and healthy subjects, respectively. A direct relationship was observed between apixaban anti-factor Xa activity and plasma concentration across groups. No serious adverse events or discontinuations due to adverse events occurred.
Conclusions
Mild or moderate hepatic impairment had no clinically relevant impact on apixaban pharmacokinetic or pharmacodynamic measures, suggesting that dose adjustment may not be required.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hepatic impairment has the potential to alter a drug’s pharmacokinetic (PK) and pharmacodynamic (PD) profile. |
A single dose of apixaban was well tolerated and the PK/PD profile of apixaban was comparable between healthy subjects and subjects with mild and moderate hepatic impairment. |
These PK and PD data may support administration of apixaban without dose adjustment in patients with mild or moderate hepatic impairment; however, the results should be interpreted cautiously, especially in the presence of more severe hepatic impairment or other risk factors for bleeding. |
1 Introduction
Apixaban is a selective, direct, factor Xa inhibitor approved for prevention of venous thromboembolism following elective hip or knee replacement surgery, treatment of venous thromboembolism, and prevention of stroke and systemic embolism due to nonvalvular atrial fibrillation [1,2,3,4,5,6,7,8]. Apixaban has an oral bioavailability of approximately 50% and its absorption is not affected by food or pH modulators [9, 10]. Elimination of apixaban occurs through multiple pathways, including renal, biliary, and intestinal excretion of the unchanged drug, as well as metabolism to inactive metabolites [11, 12]. In vitro studies indicate that metabolism of apixaban occurs by demethylation, predominantly catalyzed by cytochrome P450 (CYP) 3A4 along with minor contributions from several other CYP enzymes [13, 14].
The effect of hepatic impairment on pharmacokinetics is well described and can be multifaceted, impacting not only the quantity and activity of hepatic enzymes [15,16,17], but also hepatic blood flow, protein binding, absorption, and renal elimination. In addition, the production of clotting factors can also be reduced in patients with hepatic impairment, which could increase the pharmacodynamic (PD) effects of anticoagulant therapy [17,18,19]. This study was conducted to assess the effect of mild (Child–Pugh A) and moderate (Child–Pugh B) hepatic impairment on apixaban pharmacokinetics, protein binding, pharmacodynamics, safety, and tolerability.
2 Methods
2.1 Study Design
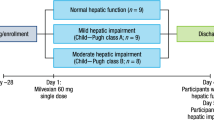
This open-label, non-randomized, parallel-group, single-dose study enrolled hepatically impaired and healthy subjects at three sites, and was approved by the institutional review boards of the respective sites. The study was conducted in accordance with US laws on research in human subjects, Good Clinical Practice, and the principles of the Declaration of Helsinki and its amendments. All subjects provided written informed consent prior to the initiation of study procedures.
Subjects were screened for eligibility within 21 days prior to treatment. Eligible subjects were admitted to the site 1 day before study drug administration for baseline assessments and remained in the clinic until study procedures were completed on day 5. On day 1, after fasting for ≥ 8 h, subjects received a single oral 5-mg dose of apixaban. Serial blood and urine samples were collected for up to 96 h after drug administration for pharmacokinetic (PK), protein binding, and PD analyses.
2.2 Study Population
The study population comprised hepatically impaired and healthy male and female subjects, aged 18–70 years, with a body mass index up to 35 kg/m2. Hepatically impaired subjects were defined as those with stable hepatic insufficiency conforming to Child–Pugh A (mild) or B (moderate) classification [20] based on relevant medical history, physical examination, and one of the following modalities: liver biopsy, computerized tomography, magnetic resonance imaging, radionuclide liver/spleen scan, abdominal laparoscopy, or appropriate serological markers. Eight subjects were enrolled into each hepatic impairment group. Healthy subjects were those with no clinically significant deviation from normal in medical history or routine medical assessments, including physical examination, 12-lead electrocardiogram (ECG), vital signs, and clinical laboratory testing. Sixteen healthy subjects were enrolled and matched 1:1 with hepatically impaired subjects based on age (± 10 years), weight (± 20%), sex, and smoking status (non-smoker/former smoker/current smoker). Women of child-bearing potential who were not nursing or pregnant and using an acceptable method of contraception could be included in the study.
Subjects were excluded from the study if they presented with: any significant history of acute medical illness within the past 2 months, evidence of significant (Child–Pugh C) hepatic impairment or complications due to hepatic impairment (e.g., greater than Stage 1 encephalopathy, greater than moderate ascites, hepatorenal syndrome), presence of cholestatic or autoimmune liver disease, evidence of clinically relevant bleeding (e.g., variceal bleeding), significant coagulopathy or bleeding risks (e.g., varices, recent head trauma), a history of hereditary bleeding or coagulation disorders, vitamin deficiency requiring medical treatment within the past year, or major surgery within 4 weeks prior to the study, or were anticipated to have surgery within 2 weeks of study completion. Medications with the potential to alter apixaban pharmacokinetics or increase the risk of bleeding were prohibited, except for low-dose aspirin (i.e., ≤ 165 mg daily). Hepatically impaired subjects were able to take stable long-term concomitant medications 2–4 h before or after the dose of apixaban as the specific medical condition permitted. Additionally, healthy subjects were excluded if they had any other any evidence of illness, organ dysfunction, or any clinically significant abnormality based on medical history, physical examination, vital signs, ECG, and/or clinical laboratory determinations.
2.3 Bioanalytical Methods
2.3.1 Measurement of Apixaban in Plasma and Urine
Blood samples (2.7 mL per sample) were collected from an indwelling catheter or by direct venipuncture into tubes containing 3.2% sodium citrate at 0 h (pre-dose) and at 1, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 16, 24, 30, 36, 48, 60, 72, and 96 h post-dose. Blood samples were inverted several times for mixing with the anticoagulant then placed on chipped ice. Within 15 min, each sample was centrifuged for 15 min at 1500×g to separate plasma.
Urine was collected pre-dose and during the following post-dose intervals: 0–12, 12–24, 24–48, 48–72, and 72–96 h. Additionally, the 0–12 and 12–24 h samples were used to determine 24-h creatinine clearance. Urine was collected in chilled urine collection jugs and refrigerated during the collection period. The final plasma and urine (15-mL aliquots) samples were stored at or below − 20 °C and until shipped for analysis.
Apixaban was quantitatively determined in plasma and urine using validated high-performance liquid chromatography tandem mass spectrometry methods as described previously [21]. The lower limit of quantification was 1.0 ng/mL for both plasma and urine assays. The between-run and within-run variability, coefficient of variation (CV), for apixaban in plasma quality-control samples was ≤ 12.5% and ≤ 15.2%, respectively, with deviations from the nominal concentration of no more than ± 3.52%. The between-run and within-run variability, CV, for apixaban in urine quality-control samples was ≤ 4.52% and ≤ 2.44%, respectively, with deviations from the nominal concentration of no more than ± 6.89%. Stability of apixaban in plasma and urine was established at − 20 °C and all samples were analyzed within the period of analyte stability.
2.3.2 Determination of Apixaban Protein Binding
An additional 5-mL blood sample for the assessment of apixaban protein binding was collected 3 h after apixaban administration into a tube containing no anticoagulant and centrifuged (15 min at 1000×g) to obtain serum. Ex vivo protein binding of apixaban in human serum was determined by equilibrium dialysis using the HTD96b dialysis device and HTD96a/b dialysis membrane strips (HTDialysis, LLC, Gales Ferry, CT, USA). Prior to the assay, dialysis membranes were pre-conditioned with de-ionized water followed by 0.133 mol/L of potassium phosphate buffer (pH 7.4) for 15 min. A 0.15-mL aliquot of serum was added to one side of the dialysis cell, and an equal volume of the phosphate buffer was added to the opposite side of the cell. The plate was sealed with adhesive tape and incubated in a VWR™ waterjacketed CO2 incubator (Sheldon Manufacturing Inc., Cornelius, OR, USA) at 37 °C with gentle orbital shaking for 4 h. After incubation, a 75-µL aliquot was collected from the buffer side of each dialysis cell and mixed with an equal volume of blank serum. A 75-µL aliquot from the serum side of each dialysis cell was also collected and mixed with an equal volume of the phosphate buffer. The original serum sample from each subject was placed at 4 °C for 4 h, and then a 75-µL aliquot of sample was mixed with an equal volume of the phosphate buffer. Experiments were performed in triplicate. Apixaban concentration was determined via a validated liquid chromatography with tandem mass spectrometry method with a dynamic range of 1.00–200 ng/mL using 100 μL of human serum:buffer performed at Alta Analytical Laboratory Inc. (El Dorado Hills, CA, USA). The between-run and within-run variability, CV, for apixaban in serum:buffer quality-control samples was ≤ 2.13% and ≤ 2.81%, respectively, with deviations from the nominal concentration of no more than ± 2.10%.
2.3.3 Apixaban PD Sample Collection and Analysis
Serial blood samples (4.5 mL) for apixaban PD assessments including international normalized ratio (INR), activated partial thromboplastin time (aPTT), and anti-factor Xa activity were collected 0 h (pre-dose) and at 3, 3.5, 6, 12, 24, 48, 72, and 96 h post-dose into tubes containing 3.2% citrate. Immediately upon collection, the tubes were gently mixed by inversion. Platelet-poor plasma was separated by centrifuging at 2500×g for 15 min and retaining the supernatant, which was stored at or below − 20 °C and shipped for analysis.
Analyses of INR and aPTT were performed by Esoterix (Aurora, CO, USA) using a Trinity Biotech MDA180 coagulation analyzer and Thromoborel® S Dade Behring reagent with an international sensitivity index of approximately 1.14–1.16. The aPTT sample analysis was performed using a Trinity Biotech MDA180 coagulation analyzer. Anti-factor Xa activity was determined by a validated method using the Rotachrom® Heparin assay and accompanying reagents on an STA-Compact® analyser (Diagnostica Stago, Inc., Parsippany, NJ, USA) [23].
2.4 Endpoints
2.4.1 PK Analysis
Apixaban parameters were calculated by noncompartmental methods using Kinetica Version 4.4.1 in eToolbox package (EP Version 2.6.1; Thermo Electron Corporation, Philadelphia, PA, USA). Apixaban peak plasma concentration (Cmax) and the time to reach the peak concentration (tmax) were obtained from experimental observations. The slope (λ) of the terminal phase of the plasma concentration–time profile was determined by the method of least squares (log-linear regression of at least three data points). The half-life (t1/2) was estimated as ln2/λ. The area under the curve from time zero to the time of last quantifiable concentration (AUClast) was determined using the log-linear trapezoidal method. The area under the curve from time zero to infinity (AUC∞) was determined by summing AUClast and the extrapolated area calculated by dividing the last measured concentration by the slope of the terminal log-linear phase. Apixaban fraction unbound (fu) was obtained from experimental observations following equilibrium dialysis and calculated as follows: 100 − fraction bound (%) = fu (%). Renal clearance (CLR) was determined by dividing the cumulative amount of apixaban excreted in the urine by the plasma AUC of apixaban over the corresponding time interval.
2.5 Statistical Analysis
Thirty-two subjects (eight hepatically impaired subjects in each of the two Child–Pugh classes [A and B] and 16 healthy subjects) were expected to provide 92% and 96% power to detect 1.5-fold increases in geometric means of Cmax and AUC∞ in subjects with hepatic impairment compared with healthy subjects, respectively. Eight hepatically impaired subjects in each of the Child–Pugh classes would also provide 83% and 88% confidence, respectively, that the estimated impaired:control ratios of Cmax and AUC∞ geometric means were within 20% of true population values for each Child–Pugh class. These calculations assumed that Cmax and AUC∞ were log-normally distributed with inter-subject standard deviations (SDs) of 0.29 and 0.26 for ln(Cmax) and ln(AUC∞), respectively, as estimated from previous data (data on file).
Summary statistics of apixaban PK parameters were presented by group. Geometric means and CV were calculated for Cmax, AUC∞, AUClast, and CLR for each group. Medians, minima, and maxima were provided for tmax. Means and SDs were provided for the remaining PK parameters. One-way analyses of variance were performed on the log-transformed values of Cmax, AUC∞, and AUClast. Group means and differences on the log scale between subjects with mild hepatic impairment and healthy subjects, and subjects with moderate hepatic impairment and healthy subjects, were exponentiated to obtain point estimates and 90% confidence intervals for the ratios of Cmax, AUC∞, and AUClast geometric means (impaired:control).
Summary statistics were tabulated for INR, aPTT, and anti-factor Xa activity along with change from baseline by group for INR and aPTT. Baseline in this study was the day 1 pre-dose value. For each measure, plots of mean percent change from baseline over time and scatter plots against apixaban plasma concentration were generated by group. SAS/STAT® Version 8.2 (SAS Institute, Cary, NC, USA) was used to perform statistical analyses.
2.6 Safety Assessments
Safety assessments were based on medical reviews of adverse event (AE) reports and results of physical examinations, vital sign measurements, ECGs, and clinical laboratory tests. Safety assessments were performed at screening, prior to study drug administration, and at scheduled intervals following drug administration through discharge from the study.
3 Results
3.1 Study Population
Thirty-two subjects were enrolled, treated, and completed the study (16 healthy subjects, eight subjects with mild hepatic impairment and eight subjects with moderate hepatic impairment). Baseline characteristics, including age, weight, smoking status, and 24-h creatinine clearance, and demographics were similar across all treatment groups (Table 1). The median (range) Child–Pugh scores for subjects in the mild and moderate hepatic impairment groups were 5 (5–6) and 7 (7–8), respectively. Chronic hepatitis C infection was the most common cause of hepatic impairment in these subjects. One subject with mild hepatic impairment had low-grade encephalopathy. All subjects with moderate hepatic impairment had low-grade encephalopathy and slight ascites, with the exception of one subject who had moderate ascites. Total bilirubin was less than 2 mg/dL and baseline prothrombin time was no more than 4 s greater than the upper limit of normal in all subjects with hepatic impairment. Albumin was between 2.8 and 3.5 g/dL in two subjects, one in each hepatic impairment group.
3.2 Pharmacokinetics
Apixaban plasma concentration–time profiles were similar for healthy subjects and for those with mild or moderate impairment (Fig. 1). Apixaban Cmax was reached approximately 3 h after administration. Hepatic impairment did not have a significant impact on apixaban Cmax or AUC∞ (Table 2). The apixaban geometric mean Cmax and AUC values in hepatically impaired subjects were within 15% and 9% of the mean values observed for healthy subjects. Apixaban fu was similar between healthy subjects and subjects with mild or moderate hepatic impairment (mean [SD]: 7.1% [1.3], 6.8% [1.4], 7.9% [1.8], respectively). Apixaban CLR was similar in healthy subjects and in subjects with mild and moderate hepatic impairment (geometric mean [%CV]: 0.59 L/h [41], 0.89 L/h [25], 0.56 [49], respectively). Apixaban mean t1/2 was similar in healthy subjects and in subjects with mild hepatic impairment but was approximately 2 h longer in subjects with moderate hepatic impairment (mean [SD]: 14.8 h [10.2], 14.7 h [7.0], 17.1 h [16.8], respectively) (Table 2).

Mean (standard deviation) plasma concentration–time profiles of apixaban after single-dose administration (5 mg) in hepatically impaired and healthy subjects
3.3 Pharmacodynamics
Apixaban pharmacodynamics was similar between healthy subjects and subjects with mild or moderate hepatic impairment. A direct relationship between apixaban plasma concentration and anti-factor Xa activity was observed in healthy subjects and subjects with hepatic impairment (Fig. 2a), and anti-factor Xa profiles were comparable among all three groups (Fig. 2b). Hepatically impaired subjects had a slightly higher baseline INR (mean [SD]: 1.17 [0.19], 1.15 [0.16], and 1.04 [0.08] for mild, moderate, and healthy subjects, respectively) and baseline aPTT (mean [SD]: 32.8 s [7.53], 30.8 s [3.30], and 29.9 s [2.90] for mild, moderate, and healthy subjects, respectively). However, percent changes from baseline appeared comparable to those observed in healthy subjects (Fig. 3). No subject had an INR ≥ 2. A shallow and variable apixaban concentration-related increase in INR and aPTT was observed (data not shown).

a Anti-factor Xa activity vs apixaban plasma concentration and b mean apixaban anti-factor Xa activity over time in healthy subjects and in subjects with mild or moderate hepatic impairment. Anti-factor Xa activity values under the lower limit of quantification are excluded. LMWH low-molecular-weight heparin

Percentage change from baseline for a international normalized ratio (INR) and b activated partial thromboplastin time (aPTT) in healthy subjects and subjects with mild or moderate hepatic impairment
3.4 Safety
There were no deaths or serious AEs. No clinically significant vital sign or ECG-related findings were reported. Few AEs occurred following apixaban administration, with no appreciable difference between hepatically impaired and healthy subjects.
4 Discussion
Apixaban has multiple routes of elimination, including CYP-mediated metabolism, renal excretion, biliary excretion, and direct excretion into the intestine [11, 22]. Although metabolism and biliary excretion play a role in the clearance of apixaban [11, 14], there were no apparent differences in the apixaban PK profiles observed for subjects with mild or moderate hepatic impairment compared with healthy subjects in this study, despite evident differences in CYP activity. Apixaban Cmax and AUC were comparable among subjects with hepatic impairment and healthy subjects. There were no appreciable differences in fu, CLR, or t1/2 across groups. The values for CLR observed across groups in this study are consistent with those observed previously in healthy subjects [23,24,25,26]. Therefore, it is possible that renal elimination of apixaban and direct intestinal excretion helped compensate for potential decreases in apixaban metabolism. However, there was a small numeric trend towards an increase in mean t1/2 with moderate hepatic impairment. This may indicate that apixaban metabolism, and potentially biliary excretion, could be further decreased in patients with more severe forms of hepatic impairment than present in this study. It is possible that the alternate pathways of elimination may not be able to compensate fully for a more significant reduction in hepatic clearance. It should be noted that apixaban is not approved for use in patients with severe hepatic impairment.
Hepatic impairment not only has the potential to alter a drug’s pharmacokinetics, but in the case of apixaban and other anticoagulants, hepatic impairment has the potential to also impact pharmacodynamics. This is because the liver is responsible for the synthesis of vitamin K-dependent clotting factors and other components of the hemostatic system and can contribute to the development of sequelae, such as gastric and esophageal varices, which may increase the risk of bleeding. Several studies have shown that the extent of coagulopathy in this patient population is directly proportional to the degree of hepatic impairment [15, 18, 19]. In this study, higher baseline INR and aPTT values were apparent in hepatically impaired subjects, which likely reflects decreases in the synthesis of coagulation factors secondary to liver dysfunction [15]. However, the pharmacodynamics of apixaban appeared to be similar between healthy subjects and subjects with mild or moderate hepatic impairment. The INR and aPTT changes from baseline observed in these hepatically impaired subjects appeared comparable to that observed in healthy subjects. The concentration–response relationships between apixaban and INR, and between apixaban and aPTT, were shallow and variable across groups, consistent with results from previous studies with apixaban [9, 11]. A close linear relationship between apixaban plasma concentration and anti-factor Xa activity was observed in both healthy subjects and in those with mild or moderate hepatic impairment. Anti-factor Xa activity profiles were also comparable among all groups. However, this may be related to the method of measurement rather than reflecting true similarities in endogenous factor X or Xa across the groups. These data provide further evidence that traditional clotting time tests, such as INR and aPTT, may not be suitable for assessing exposure to direct factor Xa inhibitors. In contrast, the anti-factor Xa activity assay exhibited a much clearer association with apixaban plasma concentration.
Notably, this study did not include subjects with severe hepatic impairment (Child–Pugh C), and the subjects with moderate hepatic impairment in this study had a median (range) Child–Pugh score of 7 (7–8), which is at the [27] lower end of the full Child–Pugh Class B score range, 7–9. Subjects with more severe hepatic impairment (i.e., higher Child–Pugh scores) might have increases in apixaban exposure due to more significantly reduced CYP-mediated metabolism, despite the presence of other elimination pathways. It may be challenging to predict the PD effect at a given exposure owing to a decrease in the clotting factor synthesis and potential dysfunction in the coagulation cascade in patients with more severe hepatic impairment. It is also possible that the risk of bleeding may be increased in patients with established varices or other bleeding risk factors not present in this study, even if apixaban exposure is not increased. As noted in the product label, [28] limited clinical experience precludes dosing recommendations in patients with moderate hepatic impairment and apixaban is not recommended in patients with severe hepatic impairment.
In this study, a single dose of apixaban was well tolerated and the PK and PD profiles of apixaban were comparable between healthy subjects and subjects with mild and moderate hepatic impairment. While these PK and PD data may support administration of apixaban without dose adjustment in patients with mild or moderate hepatic impairment, these results should be interpreted with caution, especially in the presence of more severe hepatic impairment or other risk factors for bleeding.
References
Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369:799–808.
Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, et al. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368:699–708.
Buller H, Deitchman D, Prins M, Segers A. Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis: the Botticelli DVT dose-ranging study. J Thromb Haemost. 2008;6:1313–8.
Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364:806–17.
Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek E, Hanna M, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981–92.
Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM, et al. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med. 2010;363:2487–98.
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P, et al. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375:807–15.
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361:594–604.
Frost C, Wang J, Nepal S, Schuster A, Barrett YC, Mosqueda-Garcia R, et al. Apixaban, an oral, direct factor Xa inhibitor: single-dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75:476–87.
Upreti VV, Song Y, Wang J, Byon W, Boyd RA, Pursley JM, et al. Effect of famotidine on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Clin Pharmacol. 2013;5:59–66.
Raghavan N, Frost CE, Yu Z, He K, Zhang H, Humphreys WG, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37:74–81.
Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD, et al. Comparative metabolism of 14C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos. 2009;37:1738–48.
Wang L, Raghavan N, He K, Luettgen JM, Humphreys WG, Knabb RM, et al. Sulfation of o-demethyl apixaban: enzyme identification and species comparison. Drug Metab Dispos. 2009;37:802–8.
Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38:448–58.
Morgan DJ, McLean AJ. Clinical pharmacokinetic and pharmacodynamic considerations in patients with liver disease: an update. Clin Pharmacokinet. 1995;29:370–91.
Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147–61.
Williams RL, Mamelok RD. Hepatic disease and drug pharmacokinetics. Clin Pharmacokinet. 1980;5:528–47.
Verbeeck RK, Horsmans Y. Effect of hepatic insufficiency on pharmacokinetics and drug dosing. Pharm World Sci. 1998;20:183–92.
Wahlander K, Eriksson-Lepkowska M, Frison L, Fager G, Eriksson UG. No influence of mild-to-moderate hepatic impairment on the pharmacokinetics and pharmacodynamics of ximelagatran, an oral direct thrombin inhibitor. Clin Pharmacokinet. 2003;42:755–64.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9.
Pursley J, Shen JX, Schuster A, Dang OT, Lehman J, Buonarati MH, et al. LC-MS/MS determination of apixaban (BMS-562247) and its major metabolite in human plasma: an application of polarity switching and monolithic HPLC column. Bioanalysis. 2014;6:2071–82.
Zhang D, Frost CE, He K, Rodrigues AD, Wang X, Wang L, et al. Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: administration of activated charcoal to bile duct-cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab Dispos. 2013;41:906–15.
Cui Y, Song Y, Wang J, Yu Z, Schuster A, Barrett YC, et al. Single- and multiple-dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin Pharmacol. 2013;5:177–84.
Frost C, Nepal S, Wang J, Schuster A, Byon W, Boyd RA, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76:776–86.
Frost CE, Song Y, Shenker A, Wang J, Barrett YC, Schuster A, et al. Effects of age and sex on the single-dose pharmacokinetics and pharmacodynamics of apixaban. Clin Pharmacokinet. 2015;54:651–62.
Upreti VV, Wang J, Barrett YC, Byon W, Boyd RA, Pursley J, et al. Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br J Clin Pharmacol. 2013;76:908–16.
Tsoris A, Marlar CA. Use of the Child Pugh score in liver disease. Treasure Island: StatPearls; 2020.
Bristol-Myers Squibb. Eliquis® (apixaban tablets). Summary of product characteristics. November 2019. https://www.ema.europa.eu/en/documents/product-information/eliquis-epar-product-information_en.pdf. Accessed 26 Dec 2019.
Acknowledgements
The authors gratefully acknowledge the contributions of Andrew Shenker, Alexander Bragat, Xioali Wang, and the staff of the New Orleans Center for Clinical Research Knoxville and the Center for Clinical Trials Research, Gainesville, FL, USA. The authors also acknowledge Dr. Alan Schuster, Dr. Zhigang Yu, and Dr. Jessie Wang, who are all previous employees of Bristol Myers Squibb, who were involved with the interpretation of the data but do not qualify for full authorship. This study was sponsored by Bristol Myers Squibb and Pfizer. Editorial and writing support was provided by Dana Fox, PhD, CMPP, at Caudex and Nicole Draghi, PhD, CMPP formerly of Caudex, and was funded by Bristol Myers Squibb and Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Bristol Myers Squibb and Pfizer.
Conflicts of Interest/Competing Interests
All authors are employees and shareholders of Bristol Myers Squibb, Princeton, NJ, USA.
Ethics approval
This study was approved by the institutional review boards of the respective sites and was conducted in accordance with US laws on research in human subjects, Good Clinical Practice, and the principles of the Declaration of Helsinki and its amendments.
Consent to participate
All subjects provided written informed consent prior to the initiation of study procedures.
Consent for publication
Not applicable.
Availability of data and material
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html.
Code availability
Not applicable.
Authors’ contributions
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. All authors made a substantial contribution to the conception and design, acquisition of data OR analysis and interpretation of data. All authors were involved in writing and critically revising the manuscript for important intellectual content. All authors approved the final version of the submitted for publication and agree to be accountable for all aspects of the work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Frost, C.E., Ly, V. & Garonzik, S.M. Apixaban Pharmacokinetics and Pharmacodynamics in Subjects with Mild or Moderate Hepatic Impairment. Drugs R D 21, 375–384 (2021). https://doi.org/10.1007/s40268-021-00359-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-021-00359-y