
Abstract
Background and Objective
Anaplastic lymphoma kinase gene rearrangements (ALKr) resulting in EML4–ALK proteins occur in a subset of solid tumors and are targeted by ALK inhibitors. Given the development of drug resistance to ALK inhibitors, ALK inhibitors with different kinase selectivity are required.
Methods
This phase I, non-randomized, open-label study evaluated the dose-limiting toxicity (DLT), safety, pharmacokinetics, and antitumor activity of ASP3026, a second-generation ALK inhibitor, in Japanese patients with solid tumors. Between 1 June 2011 and 20 January 2014, 29 patients received different daily doses of ASP3026 in the escalation (25 mg, n = 3; 50 mg, n = 3; 75 mg, n = 3; 125 mg, n = 4; 200 mg, n = 3; or 325 mg, n = 7) and expansion (200 mg, n = 6) cohorts.
Results
Three patients had DLTs at the 325-mg dose: cataract exacerbation, increased aspartate transaminase and alanine transaminase, and impaired hepatic function (all Grade 3 severity). Thus, the maximum tolerated dose was 200 mg. The treatment-emergent adverse event incidence was 100%; the most common events were nausea (n = 8, 27.6%), decreased appetite (n = 10, 34.5%), and fatigue (n = 9, 31.0%) of mild or moderate severity. Six patients were positive for ALK protein and three had ALKr. Two patients achieved partial responses: one with Ewing sarcoma (75-mg dose group) and one with an ALKr-positive inflammatory myofibroblastic tumor (125-mg dose group).
Conclusion
ASP3026 at a 200-mg dose may provide therapeutic benefit for patients with solid tumors, with a tolerable safety profile.
Clinical Trial registration
This study is registered at ClinicalTrials.gov under the identifier NCT01401504 on July 25, 2011
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The maximum tolerated dose (MTD) of the ALK inhibitor ASP3026 in Japanese tumor patients is 200 mg/day. |
This is below the 525 mg/day MTD in US patients. |
ASP3026 has antitumor action on Ewing sarcoma and inflammatory myofibroblastic tumor with ALKr. |
1 Introduction
Anaplastic lymphoma kinase (ALK) gene rearrangements (ALKr) are implicated in non-small cell lung cancer (NSCLC) and other solid tumors [1]. ‘ALKoma’ is the collective term proposed for tumors carrying abnormal ALK as the essential growth driver [2]. Fusion of the echinoderm microtubule-associated protein-like (EML4) and ALK genes results in the expression of EML4–ALK, which promotes cell malignancy [1]. This occurs in approximately 5% of NSCLC cases, and does not coexist with KRAS proto-oncogene, GTPase (KRAS) or epidermal growth factor receptor (EGFR) mutations [3, 4]. In preclinical models, EML4–ALK-dependent lung cancer was observed to respond to ALK inhibitors [3, 5]. For this reason, ALKr have become promising molecular targets for tumors overexpressing EML4–ALK, such as non-Hodgkin lymphoma, melanoma, thyroid carcinoma, esophageal carcinoma, and breast carcinoma, as well as NSCLC [6, 7].
Crizotinib, a first-generation ALK tyrosine kinase inhibitor (TKI), was found to be highly effective for NSCLC with ALKr [8,9,10]. Thus, it is currently the standard first-line treatment for advanced ALKr-positive NSCLC recommended by the National Comprehensive Cancer Network guideline [11]. However, some patients develop resistance to crizotinib after 8–12 months of treatment. Common mechanisms of resistance are acquired ALK tyrosine kinase domain mutations, amplification of the oncogenic fusion gene, activation of alternative signaling pathways, and progression in the central nervous system [12,13,14]. The next-generation ALK TKIs, ceritinib [15] and alectinib [16], have shown activity in patients with crizotinib-naïve NSCLC with ALKr and crizotinib-resistant patients.
ASP3026 is a novel, selective, orally available and ATP-competitive second-generation ALK TKI, with expected antitumor activity against various solid tumors including NSCLC, based on preclinical data of xenograft tumor-bearing mouse models expressing EML4–ALK [17]. ASP3026 has inhibitory effects on multiple kinases (ALK, ROS proto-oncogene 1, receptor tyrosine kinase, tyrosine kinase non-receptor 1, tyrosine kinase non-receptor 2, receptor tyrosine kinase family, Src family tyrosine kinases, discoidin domain receptor tyrosine kinase 1, and fyn-related Src family tyrosine kinase), which differs from the kinase inhibition profile of crizotinib [17]. ASP3026 can also inhibit cell division and survival in cancer-derived cell lines (EML4–ALK transfected 3T3 cells, NSCLC cell lines expressing EML4–ALK or solute carrier family 34 member 2-ROS and others) and has antitumor activity, including inhibition of growth and tumor regression, against cells expressing EML4–ALK with the L1196M mutation [17]. These preclinical results suggested that patients with NSCLC, other solid tumors, and hematological malignancies expressing ALK and/or ROS fusion kinases, including EML4–ALK, could benefit from treatment with ASP3026, and provided the rationale for the clinical development of ASP3026.
A first-in-human, phase I, dose-escalation study evaluated the safety, pharmacokinetics (PK), and antitumor activity of ASP3026 for advanced solid malignancies in the US. Half of the patients with ALK-positive tumors, especially NSCLC, safely achieved a partial response (PR) with a maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of 525 mg daily [18]. This study aimed to evaluate the safety, PK, and antitumor activity of ASP3026 as a single agent at multiple oral doses in Japanese patients with solid tumors.
2 Materials and Methods
2.1 Study Design
This was a phase I, non-randomized, open-label study of the repeated oral administration of ASP3026 in patients with solid tumors that consisted of two parts, dose escalation and expansion cohorts. This study was conducted in three medical institutions in Japan between 1 June 2011 and 20 January 2014. It was approved by the ethical review board of the participating institutions and posted on ClinicalTrials.gov under the identifier NCT01401504. All study procedures were conducted in accordance with the principles of the Declaration of Helsinki, and all patients provided informed consent to participate in the study.
2.2 Patients
Patients were eligible for this study if they met the following criteria: provision of informed consent; age ≥ 20 years; Eastern Cooperative Oncology Group performance status (ECOG PS) ≤ 2; histologically or cytologically confirmed diagnosis of a relapsed/refractory solid tumor; life expectancy > 12 weeks; ability to take oral medications; either present disease progression despite standard therapies, unavailability of standard therapies or such therapies were not anticipated to result in a durable response, or standard therapies were considered unsuitable or were refused; and met laboratory test requirements. Additionally, patients included in the dose expansion cohort had confirmed ALK-positive tumors and progressive disease (PD) after previous treatment with an ALK TKI. Detailed exclusion criteria are listed in Online Resource 1 (see electronic supplementary material [ESM]).
Patients were withdrawn from treatment for any of the following reasons: PD, dose-limiting toxicity (DLT) during Cycle 1, unacceptable study drug-related toxicity or serious adverse event requiring discontinuation of treatment, protocol violation, or loss to follow-up. Patients were assigned to each dose level of ASP3026 sequentially in the order of registration.
2.3 Endpoints
2.3.1 Primary
The primary outcomes were the assessment of DLT and the safety profile of ASP3026 based on treatment-emergent adverse events (TEAEs; coded according to the Common Terminology Criteria for Adverse Events version 4.0 [CTCAE version 4.0—Japan Clinical Oncology Group]). The DLTs were assessed only during Cycle 1 and were defined as the following study drug-related adverse events: grade 4 neutropenia (absolute neutrophil count [ANC] < 500/mm3) lasting ≥ 7 days; febrile neutropenia defined as an ANC of < 1000/mm3 with a single temperature > 38.3 °C, or a sustained temperature ≥ 38 °C for 1 hour or longer; Grade 3 thrombocytopenia (platelet count ≥ 25,000/mm3 and < 50,000/mm3) with bleeding that required platelet transfusion or Grade 4 thrombocytopenia (platelet count < 25,000/mm3); nausea, vomiting, or diarrhea that was classified as Grade 3 toxicity or worse lasting ≥ 3 days or classified as Grade 4 toxicity despite optimal symptomatic treatment; any other Grade ≥ 3 non-hematologic toxicity (excluding transient electrolyte imbalances); any toxicity resulting in study treatment delay of ≥ 15 days, and any toxicity requiring discontinuation of the study drug. Toxicities were assessed according to the Japan Clinical Oncology Group (JCOG) Japanese translation of the CTCAE version 4.0.
The MTD was defined as the highest dose of ASP3026 on a dosing schedule at which < 33% of patients experienced a DLT. The MTD or lower dose, as determined by the Data and Safety Monitoring Board, was the RP2D.
2.3.2 Secondary
The pharmacokinetic parameters for ASP3026 were assessed based on the maximum plasma concentration (Cmax), area under the plasma concentration-time curve from the time of dosing to 24 h (AUC24h), time to maximal plasma concentration (tmax), apparent total clearance (CL/F), and urinary excretion. Pharmacokinetic formulas used were the same as those described by Li et al. [18]. Plasma concentrations of ASP3026 were collected at baseline (pre-dose), Day 1 (post-dose at 30 min, 1 h, 2 h, 3 h, 4 h, 8 h, 12 h, and 24 h), Day 8 (pre-dose), Day 15 (pre-dose), Day 22 (pre-dose), and Day 28 (pre-dose and post-dose at 30 min, 1 h, 2 h, 3 h, 4 h, 8 h, 12 h, and 24 h). Urine samples were collected at baseline (12 h pre-dose), Day 1 to Day 2 (4 h post-dose, 4–8 h post-dose, 8–12 h post-dose, and 12–24 h post-dose), and Day 28 to Day 29 (4 h post-dose, 4–8 h post-dose, 8–12 h post-dose, and 12–24 h post-dose). Plasma and urine samples were stored at ≤ 20 °C and protected from the light until they were analyzed using a validated liquid chromatography tandem-mass spectrometry method in a bioanalytical laboratory.
Antitumor activity of ASP3026 was measured by the overall response rate at each time point and the best overall response rate by Response Evaluation Criteria in Solid Tumors v1.1 based on the results of imaging examinations such as radiography, computed tomography, or magnetic resonance imaging performed on baseline, Day 28 of Cycles 1–4 and then every two cycles thereafter.
2.4 Treatment, Dose Escalation, and Expansion Cohorts
In the dose escalation cohorts, the dose was escalated sequentially from a low to a high level with a 3 + 3 dose escalation scheme based on the modified Fibonacci method. At least three patients were enrolled in each dose escalation cohort. Proceeding to the next cohort (dose escalation) was decided after assessment of DLTs of ASP3026 administered as multiple oral doses once daily in Cycle 1 (every 28 days after the start of the study treatment was defined as one cycle). When one of three patients experienced a DLT in a cohort, three additional patients were enrolled in the same cohort to assess the dose level in a total of six patients. The starting dose level was 25 mg administered orally once daily. Dose escalation proceeded as follows: 25 mg, 50 mg, 75 mg, 125 mg, 200 mg, 325 mg, 525 mg, 800 mg, and 1200 mg.
In the dose escalation cohorts, treatment cycles lasted 28 days. At the end of each cycle, DLTs and other safety variables were evaluated. If patients met the criteria to enter the next cycle, treatment with ASP3026 was continued for subsequent cycles. Treatment was delayed for up to 14 days for patients who did not meet the criteria for the next cycle; if longer delays were required, patients were withdrawn from the study. The RP2D of ASP3026 was employed as a daily dose in the expansion cohort of the study, and safety and tolerability were evaluated.
ASP3026 was available in tablets of 10 mg, 25 mg, or 100 mg, and it was orally administered once daily in the morning, without ingestion of food for at least 2 h prior and 1 h after dosing. ALK protein and ALKr and ROS1 gene rearrangement (ROS1r) status in the tumor cells were examined centrally by immunohistochemistry (IHC) and chromogenic in situ hybridization (CISH) using formalin-fixed paraffin-embedded slides.
2.5 Statistical Analysis/Assessment
The planned sample size was up to 54 patients (up to six patients enrolled in each of the nine cohorts, depending on toxicity findings) in the dose escalation cohort, and up to 15 patients in the expansion cohort. The full analysis set (FAS) comprised patients with solid tumors that received at least one dose of ASP3026, and they were evaluated for at least one efficacy endpoint. The safety analysis set comprised patients who received at least one dose of ASP3026.
Descriptive statistics were used for all evaluated parameters, and 95% confidence intervals (CI) were calculated as appropriate. MedDRA Ver. 13.1 (MedDRA MSSO, McLean, VA, USA) was used to summarize adverse events by system organ class and preferred term. All statistical analyses were performed using SAS Drug Development (ver. 3.4; SAS Institute Inc., Cary, NC, USA) and PC-SAS (ver. 9.1.3; SAS Institute Inc.).
3 Results
3.1 Patient Disposition and Baseline Characteristics
Patients were enrolled between May 2011 and January 2014. A total of 33 patients were enrolled; four patients discontinued the study prior to registration. Twenty-nine patients were registered and assigned to the different daily dose levels of ASP3026 in the escalation cohorts (25 mg, n = 3; 50 mg, n = 3; 75 mg, n = 3; 125 mg, n = 4; 200 mg, n = 3; or 325 mg, n = 7) and the expansion cohort (200 mg, n = 6) (Fig. 1). Of the 29 patients, all took at least one dose of the study drug and were included in the FAS, safety analysis set, and PK analysis set. General treatment compliance was fairly good in this study, and the median overall compliance rate was > 98% in each dose group.

Patient disposition
Baseline characteristics of patients are presented in Table 1. Overall, patients had a median age of 61.0 (range 33–71) years, 55.2% (16/29) were men, and had a mean body mass index of 22.5 (SD 4.64) kg/m2. Most patients (62.1%, 18/29) were former tobacco smokers and most (72.4%, 21/29) had an ECOG PS of 1. None had an ECOG PS of 2, and the ratio of ECOG PS 1 to 0 was almost 3:1. Fifteen (51.7%) of the 29 patients assigned to treatment had hepatic metastasis. In the escalation cohort, five patients had previously been treated with crizotinib, including two patients in the 50-mg dose group, two patients in the 125-mg group, and one patient in the 200-mg dose group. In the expansion cohort, all six patients had been previously treated using an ALK inhibitor, including crizotinib (n = 3), crizotinib and ceritinib (n = 1), ceritinib and alectinib (n = 1), and alectinib (n = 1).
The most frequent reason for discontinuation was disease progression, and the incidence was 89.7% (26/29). Two patients (6.9%) discontinued the study because of adverse events (one patient for Grade 2 pyrexia and one patient for Grade 3 aspartate transaminase [AST]/alanine transaminase [ALT] increase).
Because pre-analytical data were not available for all cases, precut slides for 20 cases were used to evaluate ALK, ALKr, and ROS1r status. Of these, 18 of 20 cases were examined by ALK IHC and six (33%) were determined to be positive for ALK protein. Fourteen samples underwent ALK CISH and three (21%) were found to have an ALKr. ROS1 CISH was performed on 10 samples, but none were found to have ROS1r. There was a 93% concordance between ALK positivity and ALKr status. One patient showed discordance, being positive for ALK protein but negative for ALKr. Finally, EGFR gene analysis of all 29 patients showed that 11 patients were characterized as wild type while four patients were positive for an EGFR mutation and the remaining 14 patients were of unknown status. Two of the four patients with EGFR mutations had been treated with an EGFR-TKI, and in both cases, EGFR-TKI treatment was followed by other treatments. One patient was a 57-year-old woman with lung cancer (adenocarcinoma) who had never smoked tobacco and had an ECOG PS of 1. She received regimens of cisplatin/pemetrexed triweekly for 2.5 months, followed by gefitinib 250 mg/day orally for approximately 2.5 months, erlotinib 150 mg/day orally for approximately 1.5 months, and docetaxel triweekly for about 1 month. The other patient was a 67-year-old woman with lung cancer (adenocarcinoma) who had never smoked tobacco and had an ECOG PS of 1. She received an initial regimen consisting of cisplatin/gemcitabine hydrochloride for approximately 2.5 months, followed by gefitinib 250 mg/day orally for almost 2 years. Then she received one cycle of pemetrexed, followed by an investigational drug (ARQ197) for 2.5 months, erlotinib 150 mg/day orally for roughly 2.5 months, then one cycle of paclitaxel/bevacizumab/carboplatin, and finally, docetaxel for approximately 8 months.
3.2 Primary Endpoints (DLT, TEAEs, and MTD)
DLTs were assessed only during Cycle 1. No DLT was observed among patients receiving a daily dose of ASP3026 from 25 mg to 200 mg. In the escalation cohort receiving a daily dose of 325 mg, three out of the six patients who were evaluated presented with DLT. A 61-year-old male patient had exacerbation of a cataract (Grade 3) that was possibly drug-related and required treatment. He did not recover during the study period; however, the dose was not changed. A 62-year-old male presented with AST increased and ALT increased (Grade 3) that did not recover spontaneously, led to treatment discontinuation, and was probably drug-related. A 54-year-old male patient presented Grade 3 hepatic function abnormalities/hepatic dysfunction that did not recover spontaneously, led to treatment discontinuation, and was probably drug-related. Both of these patients with liver-related DLTs had hepatic metastasis, which became aggravated and led to treatment discontinuation in both cases. After conducting the dose escalation assessments (25–325 mg) and based on the DLTs found, the MTD and RP2D of ASP3026 for Japanese patients was set at 200 mg.
The incidence of TEAEs was 100% (29/29) in all treatment groups (Table 2). The overall incidence of drug-related AEs was 82.8% (24/29). The detailed incidences of drug-related AEs in the dose escalation cohorts are shown in Table 3.
In the 125-mg cohort, a 53-year-old woman experienced two Grade 5 TEAEs (lymphangiosis carcinomatosa and disseminated intravascular coagulation), but neither of these events was considered to be related to exposure to ASP3026. Her case accounted for the one death that occurred during the study. Another patient experienced a Grade 4 TEAE (metastases to meninges). The total incidence for each grade was 3.4% (1/29). The total incidence of Grade 1, Grade 2, and Grade 3 TEAEs in all cohorts was 20.7% (6/29), 27.6% (8/29), and 44.8% (13/29), respectively (Online Resource 2, see ESM).
TEAEs occurring in three or more patients who received at least one dose of the study drug are shown in Online Resource 3 (see ESM). Most events were of mild or moderate severity (Grades 1–3). The most commonly reported TEAEs, regardless of relatedness to the study drug, were as follows (n, %): decreased appetite (10, 34.5), fatigue (9, 31.0), nausea (8, 27.6), hepatic function abnormal (7, 24.1), vomiting (7, 24.1), and diarrhea (7, 24.1).
Two cases of prolongation of the QT interval were observed: one was a Grade 1 adverse event that occurred in the 50-mg cohort and the other was a Grade 2 adverse event that occurred in the 325-mg cohort. The former was not considered related to the study drug, but the latter was considered to be possibly related to the study drug. However, this event resolved without a change in treatment. One patient in the 325-mg dose group presented with both Grade 3 ALT and AST increases and another presented with Grade 3 hepatic function abnormalities/hepatic dysfunction. These hepatic function abnormalities were considered as probably related to the study drug. Of note, two patients who were confirmed to have EGFR mutations had previously been treated with EGFR-TKIs, but neither of them presented hepatic function abnormalities/hepatic dysfunction during treatment with ASP3026.
Ophthalmological examination was limited to the 325-mg dose group in the dose escalation cohort and the 200-mg dose group in the expansion cohort. A 61-year-old woman had a cataract at baseline that worsened to Grade 3 in Cycle 1 and was considered a DLT. No new cases of cataracts were reported in this study.
3.3 Secondary Endpoints (Pharmacokinetics)
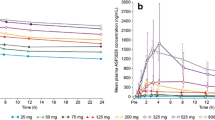
In the escalation cohort, the PK parameters, median Cmax and AUC24h, increased in a dose-dependent manner with increasing dose from 25 mg to 325 mg both on Day 1 (Fig. 2a, Online Resource 4, see ESM) and Day 28 (Fig. 2b, Online Resource 5, see ESM). The urinary excretion was low for ASP3026 at all dose levels. Although wide variability was observed, dose proportionality was suggested. An accumulation in AUC24h (geometric least squares [LS] mean from 1.91 to 2.69) and Cmax (geometric LS mean from 1.33 to 2.05) was observed from Day 1 to Day 28. The mean tmax across the dose groups ranged from 2.12 h to 4.52 h on Day 1 (median 1.97–3.97 h) and from 2.48 h to 3.75 h on Day 28 (median 2.48–3.00 h). The steady state was achieved from Day 8 and on.

a Median (max) plasma concentration of ASP3026 (semi-log scale plot), Day 1 (pharmacokinetic analysis set). b Median (max) plasma concentration of ASP3026, (semi-log scale plot), Day 28 (pharmacokinetic analysis set). These plasma concentrations are approximately the same as those shown by Li et al. [18]
3.4 Antitumor Activity
Across all treatment cycles, PR was observed in two patients, one patient (33.3%, 1/3) in the 75-mg dose group and one (25.0%, 1/4) in the 125-mg dose group in the dose escalation cohort (one patient had Ewing sarcoma, of unknown ALK/ALKr status, and the other had ALKr-positive inflammatory myofibroblastic tumor [IMT]). The patient in the 75-mg dose group completed 28 cycles of ASP3026 treatment and the patient in the 125-mg dose group completed seven cycles. The maximum duration of treatment was 28 cycles in the 75-mg dose group and nine cycles in the 200-mg dose group.
At the RP2D of 200-mg daily dose of ASP3026, none of the patients in either the dose escalation cohort (n = 3) or the expansion cohort (n = 6) consisting of patients with confirmed ALKr and who had PD after previous treatment with an ALK inhibitor (these included crizotinib, ceritinib, and alectinib, and one patient had received two ALK inhibitors previously) achieved PR in Cycle 1. At the RP2D of a 200-mg daily dose of ASP3026, three patients in the escalation cohort (n = 3) and three patients in the expansion cohort (n = 6) showed stable disease in Cycle 1 (Table 4).
4 Discussion
In this phase I study, we investigated for the first time, the safety, PK, and antitumor activity of ASP3026 administered as a single agent, at multiple oral doses in Japanese patients with solid tumors. Based on the DLTs found, the MTD and RP2D of ASP3026 was found to be 200 mg once daily. In comparison, the MTD and RP2D of ASP3026 in the study in the US was found to be 525 mg daily [18]. Considering that the exposure in this study was similar to that in the US study, the lower MTD identified in this study may be associated with a heightened sensitivity of Japanese patients to ASP3026. It is also possible that these results are associated with the small number of patients treated at each dose level.
Overall, the study drug was well tolerated. Although all patients experienced a TEAE (incidence 100%), most TEAEs were of mild or moderate severity. The total incidence of Grade 1, Grade 2, Grade 3, Grade 4, and Grade 5 TEAEs in all cohorts was 20.7% (6/29), 27.6% (8/29), 44.8% (13/29), 3.4% (1/29), and 3.4% (1/29), respectively. The total incidence of drug-related adverse events was 82.8%. One patient died in the study, but this was not considered a drug-related adverse event. These findings are similar to those reported in a similar study of ASP3026 conducted in the US in patients with advanced solid tumors [18] as well as early-phase studies on ceritinib [15] and alectinib [16]. However, in the present study, adverse events were mostly Grade 3 or less, with very few Grade 4 and 5 events (with an incidence of 3% each). The most common adverse events in a phase I study of crizotinib were visual disorders [8]. Notably, in the present study, only four patients reported eye disorders (13.8%, 4/29), all of which were Grade 3 or less in severity. Only one patient had exacerbation of a cataract and none of the patients developed new treatment-related cataracts. The mechanism by which ALK inhibitors may worsen or cause cataracts remains to be clarified.
In terms of antitumor activity, one patient with Ewing sarcoma receiving ASP3026 75 mg and one with inflammatory myofibroblastic tumor with ALKr receiving 125 mg [19] in the dose escalation cohorts achieved best overall PR across all treatment cycles. Based on each patient’s molecular profiling, we consider that these two patients achieved PR because of the following reasons. The patient with IMT was previously reported as having a RAN binding protein 2 (RANBP2)-ALKr by real-time RT-PCR [19]. In that case, the antitumor activity of ASP3026 is attributed to its effect on the RANBP2-ALKr protein product. Furthermore, ALKr has been documented in approximately 60% of patients with Ewing sarcomas [20]. In the present study, the patient with Ewing sarcoma may have had ALKr. To clarify the activity of ASP3026 on such conditions, it would be ideal to conduct a ‘basket trial’ of ALKr in patients with soft tissue tumors such as IMTs and Ewing sarcoma in the future.
Among the patients with crizotinib-resistant NSCLC (n = 9), there were no PRs. However, two patients had stable disease. Based on the small patient numbers, it was not possible to conclusively evaluate the antitumor activity of ASP3026 in this study. However, the seemingly lower response rate may be attributed to the lower MTD used in the expansion cohort. Nevertheless, together with the data from the study conducted in the US, the data from the present study suggest that ASP3026 is clinically active in crizotinib-resistant patients.
Regarding the PK parameters evaluated, Cmax and AUC increased dose dependently, and a steady state was achieved by Day 8 of treatment. These findings were consistent with the findings of the US ASP3026 trial [18].
5 Conclusions
Our study is the first to investigate the safety, PK, and antitumor activity of ASP3026 in Japanese patients with advanced solid tumors, and we have established the MTD of ASP3026 in Japanese patients with advanced solid tumors. Based on the present results, ASP3026 may be clinically active in patients with Ewing sarcoma and IMT with ALKr, in addition to NSCLC, as shown in the US study. In conclusion, ASP3026 at a dose of 200 mg has a potential for therapeutic benefit for Japanese patients with solid tumors, with a tolerable safety profile.
References
Mano H. Non-solid oncogenes in solid tumors: EML4–ALK fusion genes in lung cancer. Cancer Sci. 2008;99:2349–55.
Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discov. 2012;2:495–502.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6.
Horn L, Pao W. EML4–ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27:4232–5.
Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4–ALK-positive lung cancer. Proc Natl Acad Sci USA. 2008;105:19893–7.
Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420:345–61.
Dirks WG, Fähnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100:49–56.
Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–9.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94.
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–77.
Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, et al. NCCN guidelines insights: non-small cell lung cancer, version 4. 2016. J Natl Compr Canc Netw. 2016;14:255–64.
Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17.
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82.
Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014;12:429–39.
Shaw AT, Kim DW, Mehra R, Tan DSW, Felip E, Chow LQM, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–97.
Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–28.
Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, et al. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol Cancer Ther. 2014;13:329–40.
Li T, LoRusso P, Maitland ML, Ou SH, Bahceci E, Ball HA, Park JW, et al. First-in-human, open-label dose-escalation and dose-expansion study of the safety, pharmacokinetics, and antitumor effects of an oral ALK inhibitor ASP3026 in patients with advanced solid tumors. J Hematol Oncol. 2016;9:23.
Ono A, Murakami H, Serizawa M, Wakuda K, Kenmotsu H, Naito T, et al. Drastic initial response and subsequent response to two ALK inhibitors in a patient with a highly aggressive ALK-rearranged inflammatory myofibroblastic tumor arising in the pleural cavity. Lung Cancer. 2016;99:151–4.
Fleuren ED, Roeffen MH, Leenders WP, Flucke UE, Vlenterie M, Schreuder HW, et al. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int J Cancer. 2013;133:427–36.
Acknowledgements
The authors would like to thank Dr Keyra Martinez Dunn, MD, of Edanz Evidence Generation, on behalf of Springer Healthcare for providing medical writing support, which was funded by Astellas Pharma Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Astellas Pharma, Inc.
Conflict of interest
Akira Ono has received honoraria from AstraZeneca, Ono Pharmaceutical, Chugai Pharmaceutical Co., Ltd., and MSD. Haruyasu Murakami has received honoraria from AstraZeneca and has received research funding from Astellas, AstraZeneca, Eli Lilly, and Taiho Pharmaceutical. Takashi Seto has received honoraria from AstraZeneca, Chugai Pharmaceutical, Eli Lilly Japan, Nippon Boehringer Ingelheim, Ono Pharmaceutical, Pfizer Japan, Taiho Pharmaceutical, and Merck Sharp & Dohme, and has received grants from Astellas Pharma, AstraZeneca, Chugai Pharmaceutical, Daiichi Sankyo, Eli Lilly Japan, Eisai, Kissei Pharmaceutical, Merck Serono, Merck Sharp & Dohme, Nippon Boehringer Ingelheim, Novartis, Pfizer Japan, Verastem, and Yakult Honsha. Toshio Shimizu has received research funds from Bristol-Myers Squibb, Daiichi Sankyo, Takeda-Millennium, Five Prime, PharmaMar, 3D Medicine, and SymBio Pharmaceuticals. Shigeru Takeshita, Kentaro Takeda, and Erkut Bahceci are employees of Astellas Pharma Global Development, Inc. Junko Toyoshima, Itsuro Nagase, and Maiko Morishita are employees of Astellas Pharma Inc. Satoshi Morita has received honoraria from AstraZeneca, Chugai, Bristol-Myers Squibb, and Eli Lilly, and has received research funds from Boehringer Ingelheim. Masahiro Fukuoka has received honoraria from Astellas Pharma Inc. Kazuhiko Nakagawa has received honoraria from Eli Lilly Japan K.K., AstraZeneca K.K., Pfizer Japan Inc., Astellas Pharma Inc., Nippon Boehringer Ingelheim Co., Ltd., MSD K.K., and Ono Pharmaceutical Co., Ltd. and has received research funds from MSD K.K., Quintiles Inc., Eisai Co., Ltd., Daiichi Sankyo Co., Ltd., AbbVie Inc., ICON Japan K.K., Eli Lilly Japan K.K., Bristol-Myers Squibb Company, Ono Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., A2 Healthcare Corp., Astellas Pharma Inc., inVentiv Health Japan, AC Medical Inc., Gritstone Oncology Inc., Nippon Boehringer Ingelheim Co., Ltd., Takeda Pharmaceutical Co., Ltd., and Chugai Pharmaceutical Co., Ltd. Sawori Watanabe declares no conflict of interest. This study was sponsored by Astellas Pharma Inc (APGD Japan).
Ethical approval
This study was approved by the ethical review board of the participating institutions and posted on ClinicalTrials.gov under the identifier NCT01401504. All study procedures were conducted in accordance with the principles of the Declaration of Helsinki.
Consent to participate
All patients provided informed consent to participate in the study.
Consent for publication
Not applicable.
Availability of data and material
Datasets from this study have not been made available in an open data repository.
Authors’ contributions
Akira Ono, Haruyasu Murakami, Toshio Shimizu, Takashi Seto, Sawori Watanabe and Kazuhiko Nakagawa substantially contributed to the acquisition and interpretation of data, as well as drafting and critically revising the manuscript. Masahiro Fukuoka, Satoshi Morita, Shigeru Takeshita, Kentaro Takeda, Junko Toyoshima, Itsuro Nagase, Erkut Bahceci and Maiko Morishita substantially contributed to the study design, and the analysis and interpretation of data, as well as drafting and critically revising the manuscript. All authors approved the final version to be published and agree to be accountable for all aspects of the work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ono, A., Murakami, H., Seto, T. et al. Safety and Antitumor Activity of Repeated ASP3026 Administration in Japanese Patients with Solid Tumors: A Phase I Study. Drugs R D 21, 65–78 (2021). https://doi.org/10.1007/s40268-020-00331-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00331-2