
Abstract
Expedited development programs for biological products to be used in the treatment of serious conditions bring about challenges because of the compressed clinical development timeframes. As expedited development does not lessen the quality expectations, one challenge is providing adequate chemistry, manufacturing, and control (CMC) information required to support approval of a biological product. In particular, the analytical comparability and, in some cases, pharmacokinetic comparability studies needed to bridge the clinical material to the commercial material could delay submission of applications for life-saving medicines. While there is the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Topic Q5E guidance on assessing comparability of biological products before and after manufacturing changes, specific guidance on the emerging issue of conducting comparability exercises in the face of expedited drug development is lacking. In July 2019, clinical pharmacologists and product quality chemists from the US FDA and industry representatives convened an FDA workshop for a scientific exchange about considerations and challenges around conducting comparability exercises for expedited programs for biological products. This article highlights discussions from the workshop.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
For biological products with expedited clinical development programs, the reduction in the duration of clinical trial(s) often places strain on the timelines for chemistry, manufacturing, and control activities, including comparability assessments. |
Comparability assessments should be designed using a risk-based approach; there is no one-size-fits-all approach. |
Despite challenges, “non-traditional” clinical pharmacology approaches show promise as a viable means of streamlining pharmacokinetic comparability assessments. |
Expedited development programs could benefit from a framework for bringing machine learning and artificial intelligence into manufacturing. |
1 Introduction
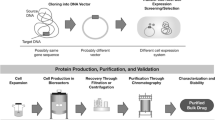
The number of biological product development and approvals rises each year, along with the number of breakthrough therapy and fast-track designations [1]. For expedited clinical development programs, the clinical material used in trial(s) to support approval is often manufactured using a process that might differ from the commercial manufacturing process. In these situations, timelines for chemistry, manufacturing, and control (CMC) activities, including comparability exercises, may need to be compressed [2]. Planning and executing comparability studies under compressed timelines introduces new challenges into product and process development and life cycle management. For example, the application of comparability assessments historically used in non-expedited development programs, which we refer to as “traditional” approaches, may not meet the needs of expedited programs and could delay the delivery of urgently needed therapies to patients. Therefore, the development of a comparability strategy for expedited development programs should consider the type of molecule, type and extent of changes in the manufacturing process, potential impact of the changes on pharmacokinetics, pharmacodynamics, safety and efficacy, and stage of clinical development, among others (Fig. 1). A clear understanding of the mechanism of action (MOA) of the molecule, critical product attributes, and impact of process steps facilitates development of comparability strategies by informing the likelihood of process changes having an impact on biological activity. In this article, we discuss challenges and opportunities in designing comparability assessments for expedited development programs for biological products.

Factors to consider when developing a comparability strategy for therapeutic biologics. CMC chemistry, manufacturing and control, MOA mechanism of action
2 Chemistry, Manufacturing, and Controls: Considerations and Challenges
Biological products undergo manufacturing changes throughout development, which require assessment of comparability between the pre-change and post-change materials. Assessing the comparability of products within expedited development programs requires considered designs and streamlined execution. Thus, prioritizing certain CMC activities, such as identification of critical quality attributes, development of reference standards, and development and validation of assays is essential to enable a successful comparability strategy. In this workshop, Dr. Hanson and Dr. Maggio explored opportunities in the implementation of strategies to support expedited product and manufacturing process development programs.
Dr. Hanson described a comparability philosophy where “the process defines the product” in which in-process controls (IPCs) are used to confirm that the process is running as intended. This strategy leverages modeling from qualified laboratory scale models to establish process and IPC ranges that yield a robust process capable of delivering a safe and efficacious product. If the process ranges are maintained and the IPC acceptance criteria are met, the drug substance and the drug product will meet the release criteria.
As process and product knowledge grows, developers need systematic approaches to update their control strategies. Dr. Hanson proposed the use of modeling approaches to support manufacturing activities (e.g., expand an IPC range). Workshop speakers and panelists were receptive to the idea of modeling approaches but noted that the models must be validated to ensure accurate predictions and that care should be taken to not extrapolate too much from the data. Multivariate analyses to “fingerprint” a process that assesses product performance and updates IPC limits is another approach that could be considered for expedited programs. Dr. Hanson also discussed the potential use of artificial intelligence/machine learning (AI/ML) to reduce the time for understanding how process variability affects product quality. A “define, measure, analyze, implement, control” framework was proposed to define the process of validation of an AI/ML approach based on patient data [3]. While a regulatory framework is available for bringing AI/ML to devices [4], Dr. Hanson suggested that a framework specific to manufacturing will be necessary to bring regulatory consensus with the application of such an approach.
In contrast, Dr. Maggio described a philosophy where “the product defines the process”. He suggested that current analytical tools allow a more thorough understanding of the criticality of specific attributes and the levels that may affect safety or efficacy. Therefore, manufacturing process ranges and IPCs are based on understanding of attributes and their relationship to safety/efficacy, potentially justifying a greater operating space. Dr. Maggio discussed streamlining process and product characterization activities by developing a database of CMC/process information from similar molecules and processes to leverage prior knowledge and that the risk assessment that leverages this prior information would be included in the control strategy section of the biologics license application (BLA) submission. A regulatory consideration for this approach is the justification of the relevance of the prior knowledge and its applicability to the current program. To expedite CMC activities required for licensure, the commercial site could be used to manufacture clinical lots. If the manufacturing process was locked down before the manufacture of registrational trial clinical material, and the clinical material was manufactured at the commercial scale, then developers could leverage stability data from the clinical lots to support the shelf life, and limited to no comparability would be needed before commercialization. However, a plan for CMC activities that may need to continue or be deferred post approval should be discussed with regulators before the BLA submission. Close collaboration with regulators to align on opportunities for expedited approval is considered crucial to successfully implementing novel science-based approaches for timeline acceleration of CMC development.
3 Clinical Pharmacology Considerations and Challenges
Dr. Putnam described how “traditional” clinical pharmacology comparability approaches are used in product development. For major changes made during or after registrational trial(s), the “traditional” approach to demonstrate pharmacokinetic comparability between pre- and post-change materials is a dedicated, powered, head-to-head bioequivalence study. As this approach may lead to significant delays in regulatory submissions [5], speakers from industry presented ideas focused around “non-traditional” approaches to assess pharmacokinetic comparability.
The population pharmacokinetic (popPK) modeling approach, particularly with the evolution of model-informed drug development [6], is a new research area that could be explored in pharmacokinetic comparability exercises. While popPK analyses have been included in pharmacokinetic comparability exercises to support product approvals [5, 7], popPK analysis alone is not yet considered sufficient by regulators to demonstrate pharmacokinetic comparability. Dr. Zhuang shared dinutuximab [5, 7, 8] as this type of case example, where—in addition to a popPK analysis—a non-compartmental analysis (NCA) was used to support the pharmacokinetic comparability assessment. In this example, a change in manufacturer occurred after completion of a phase III trial in a pediatric high-risk neuroblastoma population, pre-approval. The pharmacokinetic comparability study was conducted in 28 pediatric patients (aged ≤ 8 years) with high-risk neuroblastoma. Patients were randomly assigned to one of two treatment sequences, such that all patients received one product during cycles 1 and 2 and the other product during cycles 3–5. Pharmacokinetic sampling was obtained at 22 time points over the course of the study. Pharmacokinetic data from an independent study in a pediatric high-risk neuroblastoma population (n = 9) using dinutuximab from the original manufacturer was used to develop a structural pharmacokinetic model. The popPK model predicted that pharmacokinetic parameters between the products from the two manufacturers were comparable [5, 7, 8]. The NCA results also showed that the 90% confidence intervals of ratios of dose-normalized area under the plasma concentration–time curve (AUC)last and AUC0–216 h were between 0.8 and 1.25, concluding pharmacokinetic comparability [7]. Furthermore, comparability exercises solely applying a popPK approach present additional challenges in new indications/patient populations. If the registrational trial(s) have already been completed in one indication/patient population, collecting sparse pharmacokinetics with the post-change material in a new indication for assessing comparability may be difficult to interpret as any observed differences in pharmacokinetics may be a result of the material and/or the population. Additional considerations include whether the product MOA is similar in both indications and the overall safety/efficacy profiles. Despite challenges, workshop speakers and panelists were receptive to further exploration of the use of popPK/modeling approaches to better understand the advantages and disadvantages, including the value of these approaches in comparability exercises.
Dr. Martin shared an overview of the risk-based approach to comparability assessments that Pfizer currently uses in decision making to support manufacturing changes:
-
Step 1 involves estimating the product risk level, which includes various factors such as those described in Fig. 1.
-
Step 2 involves categorizing the type of CMC change(s) (e.g., minor, moderate, or major categories).
-
Step 3 involves understanding the outcome of the analytical comparability exercise, and he suggested a sliding scale for the degree of differences observed between demonstrating and not demonstrating analytical comparability.
-
Steps 4 and 5 are intended to assess the need for and type of animal or human testing for when analytical comparability was demonstrated and for when analytical data show some differences, respectively.
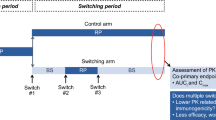
Dr. Martin also provided an overview of a quantitative tool that uses exposure–response data (Fig. 2) to aid in understanding the impact of pharmacokinetic/pharmacodynamic (PK/PD) differences in support of comparability assessments (instructions and code provided in the electronic supplementary material [ESM] S1). Differences in pharmacokinetics may not relate to pharmacodynamics or efficacy differences under many scenarios highlighted by the tool, so lack of pharmacokinetic comparability may be of less risk to patients under these scenarios. He shared that a challenge to applying this type of tool in expedited programs for new molecular entities is that clinical data to support sufficient understanding of exposure–response relationships to help inform the impact of PK/PD differences will likely be limited. However, such tools have utility in informing risk and study design and could be useful for comparability assessments in lifecycle management of molecules with well-characterized exposure–response relationships.

Workflow using R tool to evaluate different case scenerios. Cmax maximum plasma drug concentration, EC50 half maximal effective concentration, PD pharmacodynamics, PK pharmacokinetics
Dr. Zhuang presented that pharmacodynamic biomarker data from pre- and post-change materials may also contribute to the comparability assessment [5, 9], particularly when a biomarker is validated to predict clinical efficacy. Dr. Zhuang shared case examples to describe how biomarkers could provide additional support for the comparability evaluation. One case example was for vedolizumab, where changes in manufacturing process and formulation were made before phase III clinical trials were initiated. As part of the comparability exercise, a PK/PD comparability study was conducted in healthy subjects. The study evaluated ACT-1 and MAdCAM-1-Fc binding to α4β7 as pharmacodynamic biomarkers. Both processes/formulations of vedolizumab included in the study resulted in near maximal inhibition of ACT-1 and MAdCAM-1-Fc binding to α4β7 [7]. Another case example shared was for evolocumab, where changes in presentation (prefilled syringe vs. autoinjector/pen) were made during development. The pharmacodynamic biomarkers evaluated in the healthy subject PK/PD comparability study were low-density lipoprotein cholesterol (LDL-C) and proprotein convertase subtilisin/kexin type 9 (PCSK9). The study showed that reductions over time in LDL-C and PCSK9 were nearly identical between the two presentations [7]. Generally, the use of pharmacodynamic biomarkers is an area of opportunity for developers and could serve an increasing role in comparability assessments.
Dr. Putnam discussed that if developers intend to incorporate post-change material into an ongoing registrational trial and to use clinical experience (e.g., efficacy, safety, PK/PD) to support the filing instead of a dedicated, powered head-to-head pharmacokinetic comparability study, two approaches could be considered: (1) dosing randomized patients into the main trial with the post-change material, typically at the first dose, at steady state, or in an open-label extension, or (2) dosing patients with the post-change material in a separate cohort. The timing of introducing post-change material into an ongoing registrational trial is an important consideration in planning for a comparability assessment. An advantage of introducing post-change material in some patients (i.e., a subset or separate cohort) at the first dose is that patients receive only one material. However, a challenge is that the sample size of the subset may not be considered adequate for the efficacy analyses. If a comparative pharmacokinetic analysis is conducted from patients switching between the pre- and post-change materials, selection of the exposure metric is an important consideration. For example, regulatory panelists suggested that, when safety/efficacy data with the post-change material are limited, comparisons of maximum concentration and AUC over the dosing interval at steady state (AUCtau,ss) may be preferable to peak and/or trough comparisons. Dr. Putnam suggested that this is another opportunity to apply modeling approaches to estimate exposure (e.g., AUCtau,ss), as frequent patient visits for pharmacokinetic sampling in a phase III trial may be logistically difficult.
Dr. Zhuang described how a “totality of evidence” approach may be applied when the post-change material is introduced into a registrational trial. For secukinumab, clinical efficacy data, together with a powered pharmacokinetic comparability study, were used to bridge between a lyophilized and liquid formulation in a pre-filled syringe. However, to support registration of an autoinjector presentation, cross-study evaluations of clinical safety and efficacy data from several phase III trials were conducted [5, 7]. She also presented results from an exposure–response analysis conducted by the FDA review team [5, 7] and noted that a dedicated pharmacokinetic study was not needed to bridge the autoinjector presentation.
Finally, applying a risk-based approach to the evaluation of clinical immunogenicity [2, 5, 9] was also discussed. In principle, clinical trials in target disease populations with repeated dosing are considered the most relevant for immunogenicity assessments. In addition to the reporting of the immunogenicity incidence, the impact of immunogenicity on pharmacokinetics, pharmacodynamics, efficacy, and safety should also be analyzed and reported. Dr. Putnam noted that, for expedited programs, the small patient sample sizes may limit the incidence reporting for immunogenicity with post-change material. However, in a scenario where the incidence of immunogenicity with the pre-change material is low, and more importantly, with no observed impact on pharmacokinetics, pharmacodynamics, efficacy, or safety, one would not expect a post-change material with minimal changes to have differences in immunogenicity. Dr. Martin also agreed that obtaining clinical data with post-change material, in conjunction with impact analyses, may be sufficient to mitigate immunogenicity concerns.
4 Conclusion
Carefully designed, risk-based comparability strategies are critical for expedited product development programs. To support manufacturing activities, modeling approaches that use laboratory-scale models, extensive product and process characterization with state-of-the-art analytical tools, and prior knowledge may be leveraged to expedite development. Expedited development programs could also benefit from a framework for bringing AI/ML into manufacturing. Despite challenges, “non-traditional” clinical pharmacology approaches also show promise as a viable means of streamlining comparability assessments. Early discussions with regulators during development can provide necessary agreements on expedited development approaches before filing to minimize risk. The ultimate goal is to provide patients with earlier access to safe and effective life-saving medicines.
References
US Food and Drug Administration. IND Activity. https://www.fda.gov/drugs/drug-and-biologic-approval-and-ind-activity-reports/ind-activity. Accessed Sept 2019.
Dye E, et al. Examining manufacturing readiness for breakthrough drug development. AAPS PharmSciTech. 2016;17:529–38.
Xu S, et al. Skin sensors are the future of health care. Nature. 2019;571:319–21.
Proposed regulatory framework for modifications to artificial intelligence/machine learning (AI/ML)-based software as a medical device (SaMD). April 2, 2019. https://www.regulations.gov/document?D=FDA-2019-N-1185-0001. Accessed Sept 2019.
Zhuang Y, et al. Risk-based comparability assessment for monoclonal antibodies during drug development: a clinical pharmacology perspective. AAPS J. 2018;20:109.
Zhu H, et al. Model-informed drug development: a regulatory perspective on progress. Clin Pharmacol Ther. 2019;106:91–3.
US Food and Drug Administration. Drug information (labels and reviews) from Drugs@FDA. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. Accessed Sept 2019.
Marachelian A, et al. Comparative pharmacokinetics, safety, and tolerability of two sources of ch14.18 in pediatric patients with high-risk neuroblastoma following myeloablative therapy. Cancer Chemother Pharmacol. 2016;77:405–12.
Putnam WS, et al. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28:509–16.
Acknowledgements
The authors thank both the attendees of the 2 July 2019 FDA workshop and the FDA panelists who facilitated the discussion: Hong Zhao, Yow-Ming Wang, Susan Kirshner, Qing Zhou, Ramesh Potla, and Christopher Downey.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This paper reports discussions from a workshop hosted by the US FDA.
Conflict of interest
SJS, ECYC, JC, and LARR have no conflicts of interest that are directly relevant to the content of this article. WSP is an employee of Genentech, Inc., and a Roche shareholder. YZ is an employee of Janssen Research & Development, LLC, and a Johnson & Johnson shareholder. SWM is an employee of Pfizer, Inc., and a Pfizer and Amgen shareholder. FM is an employee of Amgen, Inc., and an Amgen shareholder. PH is an employee of Takeda Pharmaceuticals, Inc., and a Takeda shareholder.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
SJS, WSP, ECYC, JC, YZ, SWM, PH, FM, and LARR wrote the manuscript.
Additional information
The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the FDA.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schrieber, S.J., Putnam, W.S., Chow, E.C.Y. et al. Comparability Considerations and Challenges for Expedited Development Programs for Biological Products. Drugs R D 20, 301–306 (2020). https://doi.org/10.1007/s40268-020-00321-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00321-4