
Abstract
Introduction
Ibuprofen and acetaminophen provide analgesia via different mechanisms of action and do not exhibit drug–drug interactions; therefore, combining low doses of each may provide greater efficacy without compromising safety.
Objectives
The present study assessed the analgesic efficacy of fixed-dose combinations (FDCs) of ibuprofen/acetaminophen (IBU/APAP) compared with ibuprofen 400 mg and placebo.
Methods
This 12-h, double-blind, proof-of-concept study compared three FDCs of IBU/APAP (200 mg/500 mg, 250 mg/500 mg, and 300 mg/500 mg) with ibuprofen 400 mg and placebo in patients with moderate-to-severe pain following third molar extraction. The primary endpoint was the time-weighted sum of pain relief and pain intensity difference scores from 0 to 8 h after dosing (SPRID[4]0–8). Time to meaningful pain relief (TMPR), duration of pain relief, and adverse events (AEs) were also assessed.
Results
In total, 394 patients were randomized. All active treatments were superior to placebo for SPRID[4]0–8 (all p < 0.001) but not significantly different from ibuprofen 400 mg. Median TMPR with FDCs and ibuprofen (44.5–54.1 and 56.2 min, respectively) was faster than with placebo (> 720 min; all p < 0.001 vs. placebo). Duration of pain relief was similar with the FDCs and ibuprofen 400 mg (9.7 –11.1 h) and longer than with placebo (1.6 h; all p < 0.001). AE incidence was comparable with all treatments.
Conclusion
Each IBU/APAP FDC provided analgesic efficacy comparable to that with ibuprofen 400 mg and superior to that with placebo. Each FDC provided MPR in < 1 h, duration of pain relief > 9 h, and tolerability similar to that with ibuprofen and placebo.
ClinicalTrials.gov Registration
NCT01559259
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Combining analgesic treatments with different mechanisms of action may be advantageous for pain control compared with single-agent monotherapy; this study compared three different fixed-dose combinations (FDCs) of ibuprofen and acetaminophen to assess pain relief versus placebo and ibuprofen 400 mg. |
Each FDC ibuprofen/acetaminophen provided analgesia that was superior to that with placebo and similar to that with ibuprofen 400 mg and that occurred at lower exposures than if ibuprofen and acetaminophen were administered at their maximal single-dose over-the-counter strengths (i.e., 400 and 1000 mg, respectively); a trend toward a faster onset of analgesia with the FDC versus with ibuprofen 400 mg was observed. |
Each FDC ibuprofen/acetaminophen combination tested was generally well-tolerated, with an adverse event profile similar to that of both placebo and ibuprofen 400 mg. |
1 Introduction
Ibuprofen and acetaminophen are among the most widely used analgesics in the USA and globally. Their efficacy and safety as prescription and nonprescription treatments for mild-to-moderate pain and fever are well-established [1]. However, for many patients who take over-the-counter (OTC) analgesics, complete relief from pain may be difficult to achieve with a single analgesic agent. Unfortunately, further single-dose increases above 400 mg for ibuprofen and 1000 mg for acetaminophen (i.e., the maximum recommended single-dose OTC strength) provide little if any additional analgesic benefits but may result in a higher risk of adverse events (AEs) [2, 3].
The concept of multimodal analgesia, a process by which different procedures, techniques, and/or medications with differing mechanisms of action are used to achieve adequate pain control, was initially brought forth in the 1990s to improve recovery after surgery [4]. Over time, this strategy has become more commonplace and has been associated with improved patient satisfaction and reduced length of hospital stay [5].
This same concept, i.e., combining analgesic medications with different mechanisms of action, such as ibuprofen and acetaminophen, may be advantageous for the treatment of acute pain in the OTC setting. Ibuprofen is a nonsteroidal anti-inflammatory drug (NSAID) that inhibits cyclooxygenase (COX)-1 and COX-2 isoenzymes, blocking the subsequent synthesis of proinflammatory prostanoids; acetaminophen is believed to act through inhibition of a subclass of COX enzyme isoforms in the central nervous system [6]. Other proposed mechanisms of action for acetaminophen include activation of central serotonergic pain-inhibitory pathways and inhibition of the l-arginine nitric oxide pathway [7]. Ibuprofen and acetaminophen do not share metabolic pathways, which diminishes the likelihood of drug–drug interactions [8, 9]. Pharmacokinetic studies have demonstrated a lack of drug–drug interactions between ibuprofen and acetaminophen [6, 10, 11]. Ibuprofen and acetaminophen also have different side-effect profiles. At higher doses and longer durations of treatment, ibuprofen and other NSAIDs are associated with the potential for gastrointestinal, cardiovascular, and renal side effects [12], whereas acetaminophen overdose is associated with hepatotoxicity [13]. Therefore, a combination product containing ibuprofen and acetaminophen might allow for the use of lower doses of both agents, reducing safety concerns associated with higher doses of either drug and satisfying the unmet need for a more effective nonprescription analgesic with a favorable safety profile [3]. Previous studies on a fixed-dose combination (FDC) of ibuprofen 200 mg with acetaminophen 500 mg in postsurgical dental pain found that the FDC provided significantly better analgesia than acetaminophen 1000 mg alone and numerically better analgesia than ibuprofen 400 mg alone, without emergent safety concerns [14, 15].
Here, we provide results from a pilot study conducted to determine the overall analgesic efficacy and tolerability of three different FDCs of ibuprofen and acetaminophen, each with a different, lower amount of ibuprofen, compared with ibuprofen 400 mg with a view toward selecting one formulation for further development.
2 Materials and Methods
2.1 Study Design
This was a phase II, 12-h, five-arm, randomized, double-blind, parallel-group, in-patient, placebo-controlled study designed to determine the overall analgesic efficacy of three different FDCs of ibuprofen/acetaminophen (IBU/APAP) compared with ibuprofen 400 mg and placebo (ClinicalTrials.gov identifier: NCT01559259). The study was conducted at a single center (JBR Clinical Research, Salt Lake City, UT, USA) between 10 April 2012 and 13 September 2012. All study procedures were in compliance with the ethical principles originating in or derived from the Declaration of Helsinki, all International Council for Harmonization Good Clinical Practice guidelines, and local regulatory requirements. All patients provided written informed consent. If a patient was underage, a parent or legal guardian signed the informed consent form and the underage patient signed an assent form. The study was managed by Pfizer Inc. and performed by the investigator. Data management and analysis were conducted by Pfizer Inc., and the investigator had full access to and control over data interpretation for this manuscript.
Eligible patients were healthy males or females aged 16–40 years, inclusive, who had undergone extraction of three or more third molar teeth (with at least two having been partial or complete bony mandibular impactions) and had developed at least moderate pain within 5 h of the oral surgery. Other entry criteria included the use of only the following preoperative medications/anesthetics: topical benzocaine, a short-acting parenteral local anesthetic (mepivacaine or lidocaine) with or without a vasoconstrictor and/or nitrous oxide; no contraindications to the study or rescue medications; and being sufficiently reliable, cooperative, and intelligent to record the requested information on the analgesic questionnaire form. Male and female patients of childbearing potential must have agreed to use a highly effective method of contraception throughout the study and for at least 14 days after the last dose of treatment. Key exclusion criteria included the presence or history of significant medical history or laboratory abnormalities determined by the investigator to have placed the patient at increased risk, including the presence or history within the past 2 years of gastrointestinal ulcer or gastrointestinal bleeding, paralytic ileus or other gastrointestinal obstructive disorders, or bleeding disorders. Patients were also excluded for any of the following reasons: at risk for excessive bleeding (such as those on anticoagulant therapy); presence of acute localized dental alveolar infection that could confound postsurgical evaluation; hypersensitivity to ibuprofen, aspirin, or any other NSAID, acetaminophen, tramadol, or other opioids; use of prescription or OTC drugs that are contraindicated for use with ibuprofen or other NSAIDs, acetaminophen, codeine, tramadol, or any other opioid; use of antihistamines within 24 h; use of bisphosphonates within the past 5 years; use of any analgesic or NSAID within five half-lives before taking study medication; current use of monoamine oxidase inhibitors, antipsychotics, or any other neuroleptic; use of any selective serotonin reuptake inhibitor, serotonin norepinephrine reuptake inhibitor, or tricyclic antidepressant for which the dose had not been stable for ≥ 30 days; history of regular alcohol consumption; consumption of caffeine-containing beverages, chocolate, or alcohol within 4 h prior to taking study medication; or habituation to analgesic medications (i.e., routine use of oral analgesics five or more times/week).
Upon experiencing at least moderate pain after dental extractions, patients were stratified by sex and baseline pain (i.e., moderate vs. severe) and were randomized 1:3:3:3:3 within a stratum to receive a single oral dose of placebo, ibuprofen 400 mg, FDC IBU/APAP 200 mg/500 mg, FDC IBU/APAP 250 mg/500 mg, or FDC IBU/APAP 300 mg/500 mg. All treatments were manufactured by the sponsor (Pfizer Inc.) and administered as two immediate-release tablets under double-blinded conditions. The PCH Consumer Study Supply group provided randomization codes generated by the PCH Biostatistics Department directly to the individual(s) responsible for dispensing study medication at the site who were not otherwise involved in any part of the trial. Patients were blindfolded during study drug administration.
Patients who did not experience adequate pain relief from study medication were permitted to take rescue medication, which consisted of either immediate-release tramadol 50–100 mg or codeine phosphate 15–60 mg. Patients could receive two additional doses of rescue medication at the study center. Patients were permitted to take rescue medication at any time but were encouraged to wait at least 1 h after taking study medication to allow time for the medication to exert its effect. Patients who took rescue medication remained at the study site for the full duration of the study and continued to perform assessments.
2.2 Study Assessments and Endpoints
At baseline, patients rated their pain severity using a categorical and a numerical pain severity rating (PSR) scale. The categorical PSR was a 4-point scale (PSR4, none, mild, moderate, severe), with each category assigned a value from 0 to 3, respectively. The experience of moderate pain at baseline was confirmed by a score of > 50 mm on a 100-mm visual analog pain scale. Pain was also assessed using an 11-point numerical PSR (PSR11, 0–10; 0 = none to 10 = worst possible pain).
Following the administration of study medication, patients were followed and evaluated on site for 12 h. During that time, study participants provided self-ratings of pain severity (on both the categorical PSR4 and numerical PSR11 scales as described above) and pain relief, using a 5-point categorical pain relief rating (PRR) scale (0 = none, 1 = a little, 2 = some, 3 = a lot, and 4 = complete). The PRR self-assessments took place at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 h post-treatment, after taking study medication, immediately before taking rescue medication, or at time of study withdrawal (if either occurred).
Additional patient assessments included time to first perceptible relief (TFPR) and time to meaningful pain relief (TMPR) using the double-stopwatch method [16], in which the first stopwatch was pressed by the patient when pain relief was first perceived, and the second when that relief became meaningful. Duration of pain relief was measured by the time to treatment failure (i.e., the time of first dose of rescue medication or dropout because of lack of efficacy). At 12 h or immediately before taking rescue medication for the first time, patients also provided a Patient Global Evaluation based on a 6-point categorical scale ranging from 0 (very poor) to 5 (excellent).
The primary efficacy endpoint was the time-weighted sum of pain relief and pain intensity difference (PID) scores from baseline (time 0) through 8 h (SPRID[4]0–8) after dosing, based on PRR and categorical PSR4. Secondary endpoints included SPRID[4]0–2, SPRID[4]0–6, and SPRID[4]0–12; TFPR and TMPR; cumulative proportion of patients attaining FPR and MPR by each time point; time-weighted sum of PRR scores over 0–2 h (TOTPAR0–2), 0–6 h (TOTPAR0–6), 0–8 h (TOTPAR0–8), and 0–12 h (TOTPAR0–12); time-weighted sum of categorical and numerical (PID) scores (based on PSR4 and PSR11) over the same intervals (SPID[4] and SPID[11]); duration of relief (as measured by time to treatment failure or patient discontinuation); cumulative proportion of treatment failures at each time point; and Patient Global Evaluation of study medication.
Patients were monitored for any AEs during the study. All AEs reported through 14 days after administration of study drug were reported regardless of relationship.
2.3 Populations and Statistics
The primary analysis set (full analysis set) comprised all randomized patients who received study medication and who provided a baseline pain assessment. The safety analysis set comprised all patients who received at least one dose of study drug.
Based on results of a previous similarly designed study [16], it was expected that the difference in SPRID[4]0–8 between the ibuprofen 400 mg and any FDC group would be 5.9 units. Based on this assumption, a sample size of 90 patients per arm would give approximately 80% power to detect this difference in SPRID[4]0–8 between treatments at the 5% level of significance (two-sided). All computations assumed a root mean square of error of 14.0 units (observed in previous study). Thus, a total of 390 patients were required to complete the study. Assuming a 5% dropout rate, a total of 410 patients were to be enrolled. Statistical comparisons between the individual FDCs were not made.
The PID (both scales), PRR, and their sum (PRID) at each time point, as well as the corresponding summary scales, SPID, TOTPAR, and SPRID, were analyzed using an analysis of variance model, with treatment group, sex, baseline categorical PSR4, and treatment-by-baseline categorical PSR4 as interaction terms. For each comparison, the treatment difference based on the least squares mean and the associated p values were computed. The TMPR, TFPR, and time to treatment failure were analyzed using a proportional hazards regression model with terms for treatment, baseline categorical PSR4, and sex. The 95% confidence intervals for each pairwise treatment difference were computed using the hazard ratio and associated 95% Wald confidence interval. The cumulative proportion of patients with MPR, FPR, complete relief, and treatment failure at each specified time point was compared using the Cochran–Mantel–Haenszel row mean score test, controlling for baseline categorical PSR4 and sex, using table scores. Patient Global Evaluation was analyzed using the Cochran–Mantel–Haenszel row mean score test, controlling for baseline categorical PSR4 and sex, using modified ridit scores.
All statistical computation was conducted using SAS version 9.2 (SAS Institute, Cary, NC, USA). Since this was a pilot or proof-of-concept study, statistically significant treatment differences were declared if the probability of random occurrence among or between the treatment groups, p, was ≤ 0.05, and treatment differences were declared marginally significant if 0.05 < p ≤ 0.10. All tests were two-sided, and no adjustments for multiple comparisons/endpoints were performed.
3 Results
3.1 Participants and Baseline Characteristics
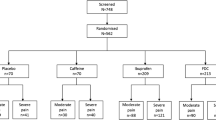
A total of 576 patients were screened, and 394 were randomized (Fig. 1). Baseline characteristics within each of the treatment cohorts were well-balanced between groups (Table 1). Approximately 50% of patients were female, and the majority (> 95%) were White; the mean age was 18.1 years. Overall, 61.9% of patients ranked their pain on study entry as severe on the categorical PSR4 scale.

Patient disposition. APAP acetaminophen, FDC fixed-dose combination, IBU ibuprofen
3.2 Efficacy
Figure 2 illustrates the course of pain relief over time with the FDCs, ibuprofen 400 mg, and placebo. Better pain relief was observed with all FDC formulations versus placebo from the 0.25-h time point through the 10- or 11-h time points, whereas ibuprofen 400 mg was better than placebo from 0.50 h to the end of the study. The FDC formulations provided better pain relief than ibuprofen 400 mg at earlier time points, but ibuprofen 400 mg trended better than some of the FDC formulations at later time points. Mean PSR11 (electronic supplementary material [ESM] 1), PSR4 (ESM 2), PID (ESM 3), and PRID[4] (ESM 4) scores over time reflect similar patterns.

Five-point categorical pain relief rating scores over time. APAP acetaminophen, FDC fixed-dose combination, IBU ibuprofen. *p ≤ 0.001 vs. placebo. †p ≤ 0.01 vs. placebo. ‡p ≤ 0.05 vs. placebo. §p ≤ 0.01 vs. IBU 400 mg. IIp ≤ 0.05 vs. IBU 400 mg. ¶p ≤ 0.05 vs. FDC IBU/APAP 200 mg/500 mg
All active treatments were significantly better than placebo on the primary endpoint of SPRID[4]0–8 (p < 0.001 for all comparisons; Fig. 3). There was no significant difference between any of the FDC formulations and ibuprofen 400 mg for this primary measure.

Mean time-weighted sum of pain relief and pain intensity difference scores. APAP acetaminophen, FDC fixed-dose combination, IBU ibuprofen, SE standard error, SPRID[4] time-weighted sum of pain relief and pain intensity difference scores based on the categorical pain severity rating scale and pain relief scores from 0–2, 0–6, 0–8, and 0–12 h. *p < 0.001 vs. placebo. †p < 0.05 vs. IBU 400 mg
A similar result was seen for the secondary endpoints of SPRID[4]0–2, SPRID[4]0–6, and SPRID[4]0–12 (Fig. 3). However, FDC IBU/APAP 200 mg/500 mg and FDC IBU/APAP 300 mg/500 mg were significant versus ibuprofen 400 mg for SPRID[4]0–2 (p < 0.05). Similar patterns were seen for time-weighted SPID[4] and SPID[11] scores over the same time intervals (data not shown).
All active treatment groups provided pain relief superior to that with placebo (p < 0.001), as measured by TOTPAR, for all comparisons (Table 2). In addition, FDC IBU/APAP 200 mg/500 mg and FDC IBU/APAP 300 mg/500 mg provided superior pain relief relative to ibuprofen 400 mg for the 0- to 2-h interval (TOTPAR0–2; p = 0.031 and p = 0.011, respectively).
TFPR with the IBU/APAP FDCs ranged from 18.5 to 22.8 min compared with 24.9 min with ibuprofen 400 mg and > 720 min with placebo (Table 3). TFPR was faster (p < 0.001) with all active treatment groups versus placebo. Additionally, FDC IBU/APAP 200 mg/500 mg and FDC IBU/APAP 300 mg/500 mg had faster TFPR than ibuprofen 400 mg (p = 0.012 and p = 0.030, respectively). Higher percentages of patients reported FPR versus placebo from the first evaluation point at 0.25 h (p = 0.031) through the end of the study (p < 0.001 for all time points from 0.5 to 12 h).
Figure 4 presents the Kaplan–Meier curves for TMPR. The median TMPR with the FDC IBU/APAP formulations ranged from 44.5 to 54.1 minutes compared with 56.2 min with ibuprofen 400 mg and > 720 min with placebo (Table 3). All of the active treatment regimens provided faster TMPR than placebo (p < 0.001). FDC IBU/APAP 200 mg/500 mg also had faster median TMPR than ibuprofen 400 mg (44.5 vs. 56.2 min; p = 0.014); the other FDCs did not reach statistical significance for this comparison.

Kaplan–Meier estimate of time to meaningful pain relief. APAP acetaminophen, IBU, ibuprofen. *p ≤ 0.001 vs. placebo. †p ≤ 0.05 vs. IBU 400 mg
Figure 5 shows Kaplan–Meier curves for duration of pain relief as measured by time to treatment failure. All active treatments were significant versus placebo (p < 0.001). The median time to treatment failure with placebo was 1.6 h, whereas the median time to treatment failure with FDC IBU/APAP 200 mg/500 mg, FDC IBU/APAP 250 mg/500 mg, and FDC IBU/APAP 300 mg/500 mg was 9.7, 10.1, and 11.1 h, respectively. Results for the FDCs were not significantly different from ibuprofen 400 mg (10.4 h; Table 3).

Kaplan–Meier estimate of time to treatment failure. APAP acetaminophen, FDC fixed-dose combination, IBU ibuprofen. *p ≤ 0.001 vs. placebo
For Patient Global Evaluation, all of the FDC formulations and ibuprofen 400 mg yielded better scores than placebo (p < 0.001 for all comparisons), but there was no significant difference between any of the FDCs and ibuprofen 400 mg (Table 4).
3.3 Safety
The overall incidence of AEs was comparable among all treatment groups, with no significant differences seen for any system organ class. A total of 256 treatment-emergent AEs were reported by 127 patients (32.2%). The majority of AEs were mild or moderate in severity, and none were determined to be treatment related. There were no serious AEs. Nausea, vomiting, and dizziness were the most commonly reported AEs (Table 5). The incidence of nausea was greater in the placebo group relative to the active treatment groups. No differences were observed between the FDC groups and the ibuprofen 400 mg group. Two patients—one each in the FDC IBU/APAP 200 mg/500 mg and FDC IBU/APAP 250 mg/500 mg groups—discontinued because of vomiting within 1 h of taking study medication; both events were considered related to the surgical procedure rather than treatment.
4 Discussion
This proof-of-concept study demonstrated that each of the tested FDCs of ibuprofen and acetaminophen (200 mg/500 mg, 250 mg/500 mg, and 300 mg/500 mg) provided overall pain relief over 8 h that was superior to that with placebo and comparable to that with ibuprofen 400 mg. No difference was seen between the FDCs and ibuprofen 400 mg in terms of pain relief over 0–6 and 0–12 h. All of the FDC dose strengths evaluated showed better pain relief at 30 min post dose (all p ≤ 0.01) than ibuprofen 400 mg, suggesting a faster onset of action.
TMPR and duration of relief are other important metrics for acute analgesics. The time to onset of pain relief was faster with the FDCs than with placebo. There was also a trend toward faster TMPR relative to ibuprofen 400 mg alone, but this reached statistical significance only with FDC IBU/APAP 200 mg/500 mg (44.5 vs. 56.2 min; p = 0.014). This faster TMPR seen with the FDC is consistent with that seen in previous dental pain studies in which FDC IBU/APAP 200 mg/500 mg was compared with ibuprofen 400 mg [14, 15]. The difference in pain relief between FDC IBU/APAP 200 mg/500 mg and ibuprofen 400 mg, and between FDC IBU/APAP 300 mg/500 mg and ibuprofen 400 mg, for the TOTPAR0–2 interval further supported the faster onset of action; FDC IBU/APAP 250 mg/500 mg did not reach statistical significance versus ibuprofen 400 mg but showed a similar trend. This is perhaps to be expected, as an acetaminophen tablet has a faster time to maximum concentration than an ibuprofen tablet [6]. TFPR followed a similar pattern.
The duration of relief, as measured by time to treatment failure, was longer with the FDCs than with the placebo. There was no significant difference between the FDCs and ibuprofen 400 mg; however, the duration of action increased directionally with the amount of ibuprofen in the combination. After 8 h, ibuprofen 400 mg and FDCs with ibuprofen 250 mg or 300 mg performed better than FDC IBU/APAP 200 mg/500 mg, indicating that duration of pain relief is driven by ibuprofen, with higher doses providing longer relief.
The most frequent AEs were nausea, vomiting, and dizziness. These AEs are commonly encountered following removal of wisdom teeth and may be related to the surgical procedure and/or use of opioid rescue medication. These AEs occurred most frequently in the placebo group and may have been due to greater pain and greater use of opioid rescue medications in this group. Overall, the AE profile of the FDCs was comparable to those with ibuprofen 400 mg and placebo. There were no unexpected AEs, and the safety profile of the FDCs was consistent with that seen in previous studies [14, 15, 17].
The study is limited in that no protection for multiple comparisons was implemented; however, this decision was made a priori because of the proof-of-concept nature of the study conducted.
This study demonstrates that each of the three FDCs of IBU/APAP tested had an efficacy and safety profile comparable to that of ibuprofen 400 mg. Additionally, there was some suggestion of a more rapid onset of action with the FDCs than with ibuprofen alone. This efficacy occurred at lower exposures than when ibuprofen and acetaminophen are administered at their maximum single-dose OTC strengths (i.e., 400 and 1000 mg, respectively) per dose. Since multiple studies have demonstrated that ibuprofen 400 mg is superior to acetaminophen 1000 mg [18,19,20], and two previous studies demonstrated that FDC IBU/APAP 200 mg/500 mg was superior to acetaminophen 1000 mg [14, 15], all of the FDCs evaluated herein would also be expected to be superior to the maximal dose of acetaminophen.
Data Sharing Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer. On 1 August 2019, Pfizer Consumer Healthcare became part of GSK Consumer Healthcare.
References
Moore RA, Wiffen PJ, Derry S, Maguire T, Roy YM, Tyrrell L. Non-prescription (OTC) oral analgesics for acute pain—an overview of Cochrane reviews. Cochrane Database Syst Rev. 2015;11:CD010794.
Becker DE. Pain management: part 1: managing acute and postoperative dental pain. Anesth Prog. 2010;57:67–78 (quiz 9–80).
Mehlisch DR. The efficacy of combination analgesic therapy in relieving dental pain. J Am Dent Assoc. 2002;133:861–71.
Kehlet H. Multimodal approach to control postoperative pathophysiology and rehabilitation. Br J Anaesth. 1997;78:606–17.
Starkweather APM. Enhanced recovery programs and pain management. Top Pain Manag. 2017;32:1–12.
Tanner T, Aspley S, Munn A, Thomas T. The pharmacokinetic profile of a novel fixed-dose combination tablet of ibuprofen and paracetamol. BMC Clin Pharmacol. 2010;10:10.
Anderson BJ. Paracetamol (acetaminophen): mechanisms of action. Paediatr Anaesth. 2008;18:915–21.
Davies NM. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin Pharmacokinet. 1998;34:101–54.
Athersuch TJ, Antoine DJ, Boobis AR, et al. Paracetamol metabolism, hepatotoxicity, biomarkers and therapeutic interventions: a perspective. Toxicol Res (Camb). 2018;7:347–57.
Wright CE 3rd, Antal EJ, Gillespie WR, Albert KS. Ibuprofen and acetaminophen kinetics when taken concurrently. Clin Pharmacol Ther. 1983;34:707–10.
Tarabar S, Kelsh D, Vince B, et al. Phase I pharmacokinetic study of fixed-dose combinations of ibuprofen and acetaminophen in healthy adult and adolescent populations. Drugs R&D. 2020;20:23–37.
Moore N, Pollack C, Butkerait P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther Clin Risk Manag. 2015;11:1061–75.
Jaeschke H. Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig Dis. 2015;33:464–71.
Mehlisch DR, Aspley S, Daniels SE, Bandy DP. Comparison of the analgesic efficacy of concurrent ibuprofen and paracetamol with ibuprofen or paracetamol alone in the management of moderate to severe acute postoperative dental pain in adolescents and adults: a randomized, double-blind, placebo-controlled, parallel-group, single-dose, two-center, modified factorial study. Clin Ther. 2010;32:882–95.
Mehlisch DR, Aspley S, Daniels SE, Southerden KA, Christensen KS. A single-tablet fixed-dose combination of racemic ibuprofen/paracetamol in the management of moderate to severe postoperative dental pain in adult and adolescent patients: a multicenter, two-stage, randomized, double-blind, parallel-group, placebo-controlled, factorial study. Clin Ther. 2010;32:1033–49.
Brain P, Leyva R, Doyle G, Kellstein D. Onset of analgesia and efficacy of ibuprofen sodium in postsurgical dental pain: a randomized, placebo-controlled study versus standard ibuprofen. Clin J Pain. 2015;31:444–50.
Doherty M, Hawkey C, Goulder M, et al. A randomised controlled trial of ibuprofen, paracetamol or a combination tablet of ibuprofen/paracetamol in community-derived people with knee pain. Ann Rheum Dis. 2011;70:1534–41.
Cooper SA, Schachtel BP, Goldman E, Gelb S, Cohn P. Ibuprofen and acetaminophen in the relief of acute pain: a randomized, double-blind, placebo-controlled study. J Clin Pharmacol. 1989;29:1026–30.
Hersh EV, Levin LM, Cooper SA, et al. Ibuprofen liquigel for oral surgery pain. Clin Ther. 2000;22:1306–18.
Packman B, Packman E, Doyle G, et al. Solubilized ibuprofen: evaluation of onset, relief, and safety of a novel formulation in the treatment of episodic tension-type headache. Headache. 2000;40:561–7.
Acknowledgements
The authors thank Zhongwei Zhou for extensive statistical programming work.
Funding
This study was sponsored by Pfizer Consumer Healthcare. Medical writing support was provided by John H. Simmons, MD, of Peloton Advantage, LLC, an OPEN Health company, and was funded by Pfizer.
Author information
Authors and Affiliations
Contributions
Study design: DK, RL. Collection and assembly of data: RL. Data analysis: RL (statistician). Data interpretation: DK, RL. Manuscript preparation: DK, RL. Manuscript review and revisions: DK, RL. Final approval of manuscript: DK, RL.
Corresponding author
Ethics declarations
Conflict of interest
David Kellstein is a former employee of Pfizer Consumer Healthcare. Rina Leyva is a current employee of GSK Consumer Healthcare. On 1 August 2019, Pfizer Consumer Healthcare became part of GSK Consumer Healthcare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Sterling Institutional Review Board, Atlanta, GA, USA) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kellstein, D., Leyva, R. Evaluation of Fixed-Dose Combinations of Ibuprofen and Acetaminophen in the Treatment of Postsurgical Dental Pain: A Pilot, Dose-Ranging, Randomized Study. Drugs R D 20, 237–247 (2020). https://doi.org/10.1007/s40268-020-00310-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00310-7