
Abstract
Background
Individuals with Down syndrome are likely to develop clinical and neuropathological brain changes resembling Alzheimer’s disease dementia by the ages of 35–40 years. Intranasal insulin is a potential treatment for neurodegenerative disease that has been shown to reduce amyloid plaque burden and improve verbal memory performance in normal as well as memory-impaired adults. Investigations have shown that rapid-acting insulins may result in superior cognitive benefits compared with regular insulin.
Objectives
The primary objective of this study was to measure the safety and feasibility of intranasal rapid-acting glulisine in subjects with Down syndrome. Secondarily, we estimated the effects of intranasal glulisine on cognition and memory in Down syndrome.
Methods
A single-center, single-dose, randomized, double-blind, placebo-controlled, cross-over pilot study was performed to test the safety of intranasal glulisine vs placebo in 12 subjects with Down syndrome aged ≥ 35 years. Intranasal administration utilized the Impel NeuroPharma I109 Precision Olfactory Delivery (POD®) device. The primary outcomes were the occurrence of any or related adverse and serious adverse events. Secondary post-treatment cognitive outcome measures included performance on the Fuld Object-Memory Evaluation and Rivermead Behavioral Memory Test.
Results
Intranasal glulisine was safe and well tolerated in the Down syndrome population. No adverse or serious adverse events were observed.
Conclusions
Further investigations are necessary to better evaluate the potential cognitive-enhancing role of intranasal insulin in the Down syndrome population.
ClinicalTrials.gov ID
NCT02432716.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study represents the first study of rapid-acting intranasal insulin in the Down syndrome population |
Intranasal insulin was safe and well tolerated in Down syndrome |
Additional studies are necessary to evaluate the efficacy of rapid-acting insulin in Down syndrome |
1 Introduction
Down syndrome (DS) is the most common chromosomal anomaly recognized at birth, with an incidence of about 1 in 1000 births in the USA [1]. Down syndrome is caused by the presence of all or part of an extra copy of chromosome 21, which can lead to developmental delay along with cognitive impairment [2].
The majority of individuals with DS are likely to develop clinical and neuropathological brain changes resembling Alzheimer’s dementia (AD) by the ages of 35–40 years, which include deposits of extracellular amyloid-beta oligomers, intracellular neurofibrillary tangles, and cerebral glucose hypometabolism [3,4,5]. Whereas cholinesterase inhibitors and memantine are US Food and Drug Administration-approved medications for AD, there are no existing treatment options for cognitive impairment in DS.
Insulin regulates the pathological hallmark proteins associated with both AD and DS, including neurofibrillary tangles and amyloid plaques [6,7,8]. Increasing central insulin levels may decrease neurofibrillary tangle formation through inhibition of tau phosphorylation by maintaining the phosphorylation equilibrium between kinase and phosphatase activity [9]. Insulin reduces amyloid plaque burden through the stimulation of the insulin degrading enzyme [10]. Finally, insulin receptor signaling increases synaptic density, which may counteract the characteristic loss of synapses occurring in AD and DS [11]. The numerous neuropathological similarities that exist in AD and DS characterize the insulin signaling pathway as a promising treatment approach in DS.
Intranasal (IN) insulin has been studied in mild cognitive impairment and AD, showing benefits on verbal retrieval performance in as little as 15 min of a single administration [7] as well as lasting for as long as 4 months [12]. One potential mechanism for the rapid effects on cognition may relate to the effect of insulin on cerebral glucose metabolism; studies have shown that low doses of insulin increase cerebral glucose metabolism in the cortex [7, 13]. Furthermore, studies have suggested that rapid-acting (RA) insulins may have greater effects on memory than regular insulin [14].
For this investigation, we used glulisine, an RA insulin analog lacking the zinc-containing compounds typically found in other insulin formulations that have the potential to cause olfactory neuron toxicity [15, 16]. Furthermore, we employed a novel nasal delivery device known as the Impel NeuroPharma I109 Precision Olfactory Delivery (POD®) device.
In the current study, we were interested in demonstrating the safety and tolerability of IN RA insulin glulisine in subjects with DS. We had a secondary objective of measuring the effect of IN RA insulin glulisine on cognition and memory.
2 Materials and Methods
This 8-week single-center study (ClinicalTrials.gov ID NCT02432716) enrolled 12 adult subjects with DS aged 35–80 years. All subjects either had prior karyotyping or underwent karyotyping through the study to confirm the diagnosis.
Study eligibility was determined by a neurological evaluation (cranial nerves motor examination, coordination, sensation, reflexes, and gait), medical history, physical examination, electrocardiogram, and laboratory studies. Patients were excluded for any history of additional psychiatric or neurologic diagnoses expected to impact cognition beside DS, history of drug or alcohol abuse, status of residing in a skilled nursing facility, presence of seizure disorder, Hachinski score > 4, history of diabetes mellitus or insulin use, history of insulin intolerance, recent exposure within 30 days to an experimental drug for DS, and inability to comply with the protocol for treatment. Any participants with a neuropsychiatric index-C aggression or agitation subscore > 4 (severity > 2; frequency > 2) were excluded from the trial. If screening laboratory studies showed glycosylated hemoglobin > 6.1%, patients were prohibited from participating in the investigation (refer to Table 1 of the Electronic Supplementary Material [ESM] for full inclusion and exclusion criteria).
The design was a phase II, double-blinded, placebo-controlled, randomized cross-over study. Randomization was stratified by sex a priori by the permuted block method. One clinician at the site was unblinded to randomization and prepared the study dose for each subject. This person did not have responsibility for obtaining study data. The investigators were provided with a sealed envelope containing the true sequence of glulisine and saline for each group if the blind needed to be broken because of unforeseen circumstances.
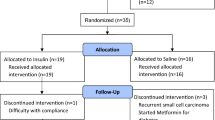
All subjects underwent a screening visit where fasting laboratory samples for basic metabolic panel, glucose, and insulin levels were drawn. During this visit, a test dose of IN saline was delivered to ensure subjects tolerated the nasal delivery device. Subjects were then randomized using a 1:1 allocation ratio to initially receive either IN glulisine 20 IU or placebo (Fig. 1). This dose was based on that used in our prior IN glulisine study in mild-moderate AD [17]. The study dose was administered within the clinical setting by the primary investigator at two separate visits with a 7- to 24-day washout period between visits. Subjects initially assigned to glulisine later received placebo during the subsequent visit at least 1 week later, and vice versa. A total of 0.20 mM of glulisine or placebo was administered using the POD® device to deliver 0.10 mL of agent in each nostril for a total of 20 IU. Subjects were seated during the administration.

Study design
Twenty minutes after receiving the IN treatment, subjects were administered a cognitive battery consisting of the Rivermead Behavioral Memory Test (RBMT) [18] and Fuld Object Memory Evaluation (FOME) [19] under the supervision of a licensed fellowship-trained neuropsychologist. These tests were chosen based on their relevance to cognitive impairment in the DS population and for purposes of minimizing floor effects. This investigation was not powered to detect changes on cognitive assessment and, thus, these batteries were administered for the purpose of feasibility rather than efficacy.
For safety assessment, all subjects underwent completion of vital signs, a physical examination, and a neurological examination at each research visit. A finger-stick glucose level was obtained at the start of each treatment visit and 30 min post-administration. Serum insulin levels, a basic metabolic panel, and glucose levels were obtained during the baseline visit and post-treatment for all subjects. A final safety visit in which subjects completed vital signs, a general/neurological examination, and laboratory studies was scheduled 7–24 days after the second treatment visit.
Baseline patient characteristics across randomized groups were compared using t-tests and Fisher’s exact tests. Treatment effects on both primary and secondary endpoints were assessed using common statistical methodology for AB/BA crossover designed trials. We planned to test the occurrence of adverse events using a mixed-effects Poisson regression model accounting for period effect, the treatment-period interaction, subject age, and sex. Cognitive outcomes from the FOME and RBMT were tested between glulisine and saline periods using two-sided two-sample t-tests. Both treatment and period differences were assessed for these analyses; although period effects were expected to be non-significant owing to the balanced nature of our design. Vitals and laboratory values were summarized by visit and a change in these values were compared for glulisine and saline periods using paired two-sample t-tests. All statistical analyses were conducted in SAS Version 9.4. The study was performed in accordance with the Declaration of Helsinki and the protocol was approved by the Regions Hospital Institutional Review Board under protocol #14-105. All participants and their caregivers provided written informed consent prior to inclusion into the study.
3 Results
The participants consisted of six female and six male individuals with a mean age of 42.7 years (range 35–53 years) (Table 1). All subjects were Caucasian. Nine subjects had completed high school (75%), two subjects had completed college (16.7%), and data were missing for a single subject (8.3%). The mean baseline glycosylated hemoglobin was 5.4. The randomized groups had similar baseline characteristics for age, sex, ethnicity, education, and glycosylated hemoglobin.
All subjects tolerated the test IN saline dose. Safety data for vital signs were missing for a single participant in the post-saline group. There were no adverse events or serious adverse events reported by any participating subjects. Given the lack of adverse events, we were unable to perform the planned mixed-effects Poisson regression analysis; however, this outcome indicates the treatment was well tolerated among our sample. Vital signs showed a significant increase in systolic blood pressure compared to baseline with glulisine as compared with placebo (Table 2). Otherwise, there were no significant effects of glulisine relative to placebo on heart rate, respirations, temperature, O2 saturation, or peripheral glucose. There were no significant differences between post-glulisine and post-placebo groups compared to baseline for serum glucose and insulin levels.
On the FOME, there was no significant impact of IN glulisine on learning, immediate recall, delayed recall, memory retention, recognition memory, and retention estimate (Table 3). On the RBMT, there was significantly improved immediate recall in the post-placebo group compared with the post-glulisine group. Furthermore, a trend toward improved performance on memory retention (p = 0.067) was found in the post-glulisine group compared with the post-saline group. Otherwise, no significant difference was found between treatment and placebo groups for delayed recall and memory recognition (refer to Table 2 of the ESM for performance at individual visits).
4 Discussion
We showed that a single dose of IN glulisine was safe and well tolerated in this patient population. Similar to past IN clinical trials, our investigation confirmed the lack of an impact of IN insulin in the central nervous system on peripheral glucose levels, suggesting that this safety outcome may not require close monitoring in future IN insulin clinical trials. Although prior studies have demonstrated that the effects of IN insulin include elevated blood pressures [20], we did not find any evidence for clinically significant hypertension. Systolic blood pressure was increased with glulisine compared with placebo, but there was no elevation of blood pressures into the clinically significant range. There was concern among the investigators that subjects with DS as a result of their developmental delay would be unable to tolerate the insertion of the Impel POD® device into the nares; our experience showed this population was not oppositional to the IN route of drug delivery.
This pilot study was not powered to detect the effect of IN glulisine on cognitive performance in subjects with DS. There was a non-significant trend toward improved memory retention in the post-glulisine group on the RBMT, warranting further investigation of IN glulisine on this outcome variable. Factors to be considered in future studies to determine clinical efficacy should consider the daily dose of IN insulin as well as the longitudinal duration of treatment. The IN glulisine dose used in our single-dose cross-over clinical trial (20 IU) is lower than the daily dose of 40 IU administered in more extensive randomized, double-blind, placebo-controlled studies in mild cognitive impairment and AD [12]. The FOME and RBMS were appropriate cognitive batteries to assess outcomes in this patient population, lacking either a ceiling or floor effect.
The strengths of this study include the fact that all subjects with DS were confirmed using chromosomal analysis. The investigation used an IN device designed specifically to target the upper one-third of the nasal sinus for brain-nose delivery. Finally, this study is the first to our knowledge to explore the role of insulin for cognitive impairment in patients with DS.
Limitations of this study include the relatively small number of participating subjects, which impacted the ability to demonstrate clinical efficacy with the employed neuropsychometric measures. In addition, this trial evaluated outcomes in the setting of a single dose, and whether longitudinal administration of IN RA insulin is safe and well tolerated remains in question. Finally, the study was limited by a lack of diversity with a cohort that was 100% Caucasian.
5 Conclusions
Intranasal RA insulin was safe and well tolerated in the DS population. Considering the lack of available therapeutic interventions for cognitive decline in DS, as well as the potential impact on cerebral amyloid plaques, further efficacy trials of IN insulin in DS are warranted to better understand the relationship between IN insulin and cognition in this population.
References
Shin M, Besser LM, Kucik JE, et al. Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics. 2009;124(6):1565–71.
Mohan M, Carpenter PK, Bennett C. Donepezil for dementia in people with Down syndrome. Cochrane Database Syst Rev. 2009;1:CD007178.
Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–90.
Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17(3):278–82.
Lao PJ, Handen BL, Betthauser TJ, et al. Alzheimer-like pattern of hypometabolism emerges with elevated amyloid-beta burden in Down syndrome. J Alzheimers Dis. 2018;61(2):631–44.
Westerman MA, Cooper-Blacketer D, Mariash A, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22(5):1858–67.
Reger MA, Watson GS, Frey WH 2nd, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27(3):451–8.
Reger MA, Craft S. Intranasal insulin administration: a method for dissociating central and peripheral effects of insulin. Drugs Today (Barc). 2006;42(11):729–39.
Ke YD, Delerue F, Gladbach A, et al. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4(11):e7917.
Zhang Q, Jiang X, Jiang W, et al. Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int J Pharm. 2004;275(1–2):85–96.
Chiu SL, Chen CM, Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008;58(5):708–19.
Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38.
Bingham EM, Hopkins D, Smith D. The role of insulin in human brain glucose metabolism: an 18fluorodeoxyglucose positron emission tomography study. Diabetes. 2002;51(12):3384–90
Benedict C, Hallschmid M, Schmitz K, et al. Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology. 2007;32(1):239–43.
Jafek BW, Linschoten MR, Murrow BW. Anosmia after intranasal zinc gluconate use. Am J Rhinol. 2004;18(3):137–41.
Davidson TM, Smith WM. The Bradford Hill criteria and zinc-induced anosmia: a causality analysis. Arch Otolaryngol Head Neck Surg. 2010;136(7):673–6.
Rosenbloom MH, Barclay TR, Pyle M, et al. A single-dose pilot trial of intranasal rapid-acting insulin in apolipoprotein E4 carriers with mild-moderate Alzheimer’s disease. CNS Drugs. 2014;28(12):1185–9.
Aldrich FK, Wilson B. Rivermead Behavioural Memory Test for Children (RBMT-C): a preliminary evaluation. Br J Clin Psychol. 1991;30(Pt 2):161–8.
La Rue A, D'Elia LF, Clark EO, et al. Clinical tests of memory in dementia, depression, and healthy aging. Psychol Aging. 1986;1(1):69–77.
Benedict C, Dodt C, Hallschmid M, et al. Immediate but not long-term intranasal administration of insulin raises blood pressure in human beings. Metabolism. 2005;54(10):1356–61.
Acknowledgements
We thank Jim and Dorothy Nelson for their generous gift. We also thank the Nelsons and Al Fletcher for their encouragement and sharing their knowledge.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by philanthropic dollars raised by the Regions Hospital Foundation.
Conflict of interest
Michael Rosenbloom, Terry Barclay, Justin Johnsen, Lauren Erickson, Aleta Svitak, Maria Pyle, William Frey, and Leah R. Hanson have no conflicts of interest that are directly relevant to the content of this study.
Ethics approval
This study was approved by the HealthPartners Institutional Review Board.
Consent to participate
Written informed consent was obtained from the caregiver and subject (or subject’s legally authorized representative) prior to any study-related procedures.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rosenbloom, M., Barclay, T., Johnsen, J. et al. Double-Blind Placebo-Controlled Pilot Investigation of the Safety of a Single Dose of Rapid-Acting Intranasal Insulin in Down Syndrome. Drugs R D 20, 11–15 (2020). https://doi.org/10.1007/s40268-020-00296-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00296-2