
Abstract
Background and Objective
Given that a high intrapatient variability (IPV) of tacrolimus whole blood concentration increases the risk for a poor kidney transplant outcome, some experts advocate routine IPV monitoring for detection of high-risk patients. However, attempts to estimate the variance of tacrolimus trough concentrations (TTC) are limited by the need for patients to receive a fixed dose over time and/or the use of linear statistical models. A goal of this study is to overcome the current limitations through the novel application of statistical methodology generalizing the relationship between TTC and dose through the use of nonparametric functional regression modeling.
Methods
With TTC as a response and dose as a covariate, the model employs an unknown bivariate function, allowing for the potentially complex, nonlinear relationship between the two parameters. A dose-adjusted variance of TTC is then derived based on standard functional principal component analysis (FPCA). To assess the model, it was compared against an FPCA-based model and linear mixed-effects models using prediction error, bias, and coverage probabilities for simulated data as well as phase III data from the Astellas new drug application studies for extended-release tacrolimus.
Results
Our numerical investigation indicates that the new model better predicts dose-adjusted TTCs compared with the prediction of linear mixed effects models. Estimated coverage probabilities also indicate that the new model accurately accounts for the variance of TTC during the periods of large fluctuation in dose, whereas the linear mixed effects model consistently underestimates the coverage probabilities because of the inaccurate characterization of TTC fluctuation.
Conclusion
This is the first known application of a functional regression model to assess complex relationships between TTC and dose in a real clinical setting. This new method has applicability in future clinical trials including real-world data sets due to flexibility of the nonparametric modeling approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tacrolimus is widely accepted as a profoundly effective immunosuppressive drug. However, shortly after its introduction in the 1990s, strategies to minimize tacrolimus dosage were sought as a potential means of improving long-term outcomes [1] based on previous reports connecting cyclosporine, another member of the calcineurin inhibitor (CNI) class, with the appearance of progressive fibrosis on biopsy [2]. Nevertheless, as more studies became available [1, 3, 4], it appeared that the lesions commonly associated with CNI nephrotoxicity were in fact more frequent in patients who were either nonadherent to tacrolimus therapy or who had high intrapatient variability (IPV) in tacrolimus levels [5, 6], implicating fibrosis as a final common pathway resulting from both over-as well as under-immunosuppression.
Further studies have suggested an association between intragraft inflammation and the development of interstitial fibrosis and/or loss of the allograft [7,8,9,10,11,12]. Indeed, subclinical inflammation appearing early after transplantation may represent a failure of conventional immunosuppression to prevent a resurgence of alloimmunity [3]. In such cases, optimizing (rather than minimizing) a patient’s exposure to drugs such as tacrolimus may be prudent [1] in order to balance the risk of adverse effects with adequate protection against the alloimmune response. However, in the case of tacrolimus, this could be complicated by intrapatient fluctuations in therapeutic tacrolimus blood concentrations [6], potentially exposing patients to periods of over- as well as under-immunosuppression [13,14,15], as discussed above. Such IPV, which can originate from a number of biological sources or from medication nonadherence [6, 16,17,18], has been shown to predict poor transplant outcomes. Associations with chronic allograft nephropathy and donor-specific antibody formation in renal transplant recipients are well-documented. High IPV has also been linked to allograft dysfunction, decreased patient and graft survival, and acute rejection in both renal and nonrenal allografts [6, 13, 16, 19,20,21,22,23,24,25].
Although the relationship between tacrolimus IPV and transplant-related outcomes is being increasingly recognized, IPV calculation has, to date, relied on fixed-dose assumptions and linear modeling [6, 25]. While such an approach may be useful for research purposes, it cannot be used with predictive intent due to real-world dosing requirements, the nonlinear complexity of biological systems, and because such models fail to fully account for the changing variability of the tacrolimus trough concentration (TTC) over time. A method that accounts for higher levels of complexity is therefore needed to achieve true clinical utility.
As such, we adopted a flexible class of functional regression models [26,27,28,29,30,31,32,33,34,35] to account for the complex nonlinear relationship between TTC and patient dose over time. Performance of the model was then assessed under a variety of conditions using simulated data, as well as data from previously conducted phase III clinical trials. Simulations were designed to validate the hypothesis that, compared with conventional approaches (e.g. prediction based on linear mixed-effects models [LME]), this type of model can better capture the true dynamics of TTC when the knowledge of patients’ past dosing and TTC observations are available. The ultimate goal of this effort was to generate a robust model for predicting dose-adjusted TTC that could be further utilized to predict the variance of TTC during treatment periods.
2 Methods
2.1 Study Model
The methodology applied in this study assumed that TTC variation can be formulated as an unknown function with respect to time, with such variances considered as a series of covariances defined for each time point. Given that TTC fluctuation is high during the early phase after transplantation, we propose that because this methodology accounts for changing IPV and dosing over time, it will allow interpretation of TTC fluctuation patterns in a more clinically meaningful way.
A flexible class of functional regression models [26,27,28,29,30,31,32,33,34,35] was utilized to allow for the correct quantification of the complex nonlinear relationship between TTC and patient dose over time. The observed data were denoted as \(\{ (D_{ij} , t_{ij} ):j = 1, \ldots ,m_{i} \}\) and \(\{ (Y_{ij} , t_{ij} ):j = 1, \ldots ,m_{i} \}\), where \(D_{ij}\) and \(Y_{ij}\) are tacrolimus dose and TTC, respectively, for the \(i\)th patient (\(i = 1, \ldots , n\)) observed at time points \(\{ t_{i1} , \ldots t_{{im_{i} }} \}\).
Due to the inherent variation in how individual patients are monitored, any model must account for the fact that the number of observations may be different for each patient, and may also be irregularly spaced. Therefore, \(Y_{ij} = Y_{i} (t_{ij} )\) was used to express the dependence of TTC on the observed time, where \(Y_{i} ( \cdot )\) is a smooth random curve defined over a closed and bounded interval, \(T\). In many observational studies, covariates can be contaminated with noise as patient-exposure information may be measured with error or under conditions of nonadherence. To accommodate such realistic scenarios, it was further assumed that \(D_{ij} = X_{i} (t_{ij} ) + \delta_{ij}\), where \(X_{i} ( \cdot )\) is a true, latent curve with smooth mean and covariance functions, and \(\delta_{ij}\) is white noise with mean zero and variance \(\sigma_{\delta }^{2}\). Therefore, we used the notation \(X_{i} \left( {t_{ij} } \right)\) for the error-free covariate, and \(D_{ij}\) for the noisy covariate (i.e. observed dose profile).
Consistent with the methodology proposed by Kim et al. [35], the TTC at a particular time was modeled using an unknown bivariate function that depended on the value of the drug dosing at that time, as well as the time point itself. For illustration, we posit a model of the type:
where F \(( \cdot , \cdot )\) is a smooth and unknown bivariate function defined on ℝ \(\times T\) (ℝ: a set of real numbers), and \(\epsilon_{i} ( \cdot )\) is an error process independent of the covariate \(X_{i} (t)\). It is assumed that error process \(\epsilon_{i} ( \cdot )\) has mean zero and unknown autocovariance function \(G( \cdot , \cdot )\). These types of models are often called nonlinear functional concurrent models (FCM) [32, 35] and are considered part of function-on-function regression models [29,30,31, 36]. The form F \(( \cdot , \cdot )\) quantifies the unknown dependence between the TTC and the dose at any time \(t\) without limiting the level of complexity in their relationship. In principle, this model allows us to extend the effect of the covariate beyond standard linearity assumptions.
The model shown in Eq. (1) has two unknown components: the bivariate function,F\(( \cdot , \cdot )\), and the autocovariance function, \(G( \cdot , \cdot )\). In a practical situation where only the noisy covariate, \(D_{ij}\), is observed, one also needs to recover the true error-free covariate, \(X_{i} (t_{ij} )\), before estimating the unknown model components, and then follow the estimation procedure using the recovered covariate [35]. We describe the main steps and estimation of \(G( \cdot , \cdot )\) in the Electronic Supplementary Material (Online Resource 1).
2.2 Model Comparisons
To formally assess the effect of dose heterogeneity on TTC, both a simulation study and a phase III data analysis compared the predictive accuracy of our approach with two alternatives: (1) LME as the comparator; and (2) a functional principal component analysis (FPCA)-based model as a positive control (Table 1).
The FPCA procedure examines the main directions of variability within the curves as a method for understanding the major sources of variability in the data, and facilitates reconstruction of full trajectories based on a few observation points [34, 37, 38]. Due to theoretical properties, the FPCA-based model can predict the outcome variables with minimal loss of information, and the estimated/predicted results were therefore expected to be excellent. However, this approach does not incorporate the tacrolimus dose as a covariate in the analysis, and therefore functioned only as a positive control.
2.3 Simulation Study
We first investigated the finite sample performance of our model through Monte Carlo simulation. The model performance was assessed by generating 1000 samples each, under a variety of conditions. The steps followed in our Monte Carlo simulation experiments are illustrated in Table 2. For each sample, we generated training sets of size \(N_{\text{train}} \epsilon \{ 100, 200, 300\}\) and a test set of size \(N_{\text{test}} = 100\). The training sets included two different sampling designs for \(t\)(dense and sparse). The dense design assumed that each patient had 131 TTC assessments equally spaced in time over 171 days, i.e. \(\left\{ {t_{j} :j = 1, \ldots , 131} \right\} \in [1, 171]\), while the sparse design assumed that each patient had \(10 - 35\) randomly selected TTC assessments over 171 days, i.e. \(\{ t_{ij} :j = 1, \ldots , m_{i} \} \epsilon [0, 171]\) with \(m_{i} = 10 - 35\) for each i.
Using these six combinations of scenarios, we investigated the amount of information lost when the sample size is small, as well as when there is a large amount of ‘missingness’ in the covariate and response. Fitting Eq. (1) requires preprocessing steps because of the skewness in the response and the proposed estimation procedure [35]. As such, we first (1) log-transformed the outcome variable, \(y \mapsto { \log }(y + 1)\); (2) applied a pointwise centering and scaling transformation (i.e. standardization) to the covariate function; and then (3) smoothed out the dose profile using FPCA [35].
The primary measure of model performance is predictive accuracy, and this was assessed by in-sample and out-of-sample root mean squared prediction error (RMSPE); both are commonly used to assess goodness-of-fit and predictive accuracy, respectively. The in-sample RMSPE is given by:
where \(Y_{{i,{\text{train}}}}^{(r)} ( \cdot )\) and its estimate \(\hat{Y}_{{i,{\text{train}}}}^{(r)} ( \cdot )\) are from the rth Monte Carlo generation, the measure depending on the distance between the actual and predicted responses in the training set.
The out-of-sample RMSPE denoted by \(\sqrt {{\text{MSPE}}_{\text{OUT}} }\) is defined similarly. For the LME, we report similar measures using \(Y_{{ij,{\text{train}}}}^{(r)}\) and \(\hat{Y}_{{ij,{\text{train}}}}^{(r)}\) in place of \(Y_{{i,{\text{train}}}}^{(r)} (t_{ij} )\) and \(\hat{Y}_{{ij,{\text{train}}}}^{(r)} (t_{ij} )\), respectively.
Estimation accuracy was further assessed using the bias and variance of the estimated TTC. We used the integrated squared bias (IsBias), defined by \({\text{IsBias}} = \mathop \smallint \limits_{X} \mathop \smallint \limits_{T} \left\{ {\frac{1}{1000}\mathop \sum \nolimits_{r = 1}^{1000} \hat{F}^{(r)} (x,t) - F(x,t) } \right\}^{2} {\text{d}}x{\text{d}}t\), as a summary measure of bias, where \(\hat{F}^{(r)} (x,t)\) is the estimated bivariate function obtained from the rth simulation. The domain of the covariate is fixed as \(X = \left\{ {x | x \in [0,40]} \right\}\) throughout the simulations, to be consistent with the range of patients’ dose profiles in the actual phase III clinical data.
The performance of the pointwise prediction intervals (PIs) was evaluated in terms of average coverage probability (ACP), a measure that can inform how reliable the predicted TTC variance is across the simulations. To compute the ACP, the \(100(1 - \alpha )\%\) pointwise PIs \(C_{1 - \alpha , i}^{(r)} (t)\) for the TTC from the test set, \(Y_{{i,{\text{test}}}}^{(r)} (t)\), obtained at the rth Monte Carlo generation, was first calculated. Specifically, we defined the \(100(1 - \alpha )\%\)PI for the ith patient by \(C_{1 - \alpha , i}^{(r)} (t) = \hat{Y}_{{i,{\text{test}}}}^{(r)} (t) \pm z_{\alpha /2} \left[ {\mathop {\text{var}}\limits^{ \wedge } \{ Y_{{i,{\text{test}}}}^{(r)} (t) - \hat{Y}_{{i,{\text{test}}}}^{(r)} (t)\} } \right]^{{\frac{1}{2}}}\), where \(z_{\alpha /2}\) is the \(\alpha /2\) upper quantile of the standard normal distribution and \(\hat{Y}_{{i,{\text{test}}}}^{(r)} (t)\) is the predicted TTC from the rth simulation [39]. The ACP at the \((1 - \alpha\)) level can then be approximated by \({\text{ACP}}(1 - \alpha ) = \frac{1}{{1000 \cdot N_{\text{test}} }}\mathop \sum \limits_{r = 1}^{1000} \mathop \sum \nolimits_{i = 1}^{{N_{\text{test}} }} \mathop \smallint \limits_{T} I\left\{ {Y_{{i,{\text{test}}}}^{(r)} (t) \in C_{1 - \alpha , i}^{(r)} (t)} \right\}{\text{d}}t\), where \(I( \cdot )\) is the indicator function (i.e. Kronecker delta). Again, for the LME, we report a similar measure using \(Y_{{ij,{\text{test}}}}^{(r)}\) and \(\hat{Y}_{{ij,{\text{test}}}}^{(r)}\) in place of \(Y_{{i,{\text{test}}}}^{(r)} (t_{ij} )\) and \(\hat{Y}_{{i,{\text{test}}}}^{(r)} (t_{ij} )\), respectively. Note that best model performance is indicated with the nominal coverage probability equal to the estimated coverage probability (e.g. perfect ACP for a 95% PI would be 0.95).
2.4 Phase III Data Analysis
We used tacrolimus whole blood concentrations and dosing collected during the first 6 months following transplantation in the phase III registration studies for extended-release tacrolimus (Astagraf XL®; Astellas Pharma Inc., Tokyo, Japan; also known as Advagraf®, Graceptor®, and Prograf® XL). Studies 02-0-158 (NCT00064701) [40], FG-506E-12-03 (NCT00189839) [41], and PMR-EC-1210 (NCT00717470) [42] included data from a total of 960 patients who received at least one oral dose of extended-release or immediate-release tacrolimus (Prograf®; Astellas Pharma Inc., Tokyo, Japan) within 48 h after kidney transplantation.
All studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and the International Council for Harmonisation guidelines. Each study was approved by the Independent Ethics Committee or Institutional Review Board at each study site, and all patients provided written informed consent before enrollment.
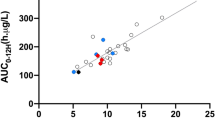
Between October 2003 and May 2004, a total of 6499 TTC (ng/mL) and drug dose (mg/day) measurements were collected at each follow-up time after transplantation (\(m_{i} = 1 - 10\)measurements per patient) (Fig. 1). To investigate the performance of the model, we first randomly split the dataset into a training set with \(N_{\text{train}} = 860\) patients, and a test set with the remaining \(N_{\text{test}} = 100\) patients.

Observed tacrolimus dose (left) and trough concentrations (right) obtained from phase III patient data. The time domain on the horizontal axis represents the evaluation time of each data point, defined as (blood drawn date) to (first dosing date) + 1. Three subjects have been highlighted to emphasize the complexity of observed patterns with the same color. TTC tacrolimus trough concentration
We then fitted each of the nonlinear FCM and two alternative models for two different durations of follow-up (short- vs. long-term). The long-term follow-up period considered the full data of 171 days, \(t_{ij} \in [0, 171]\), while the short-term case used the first 50 days post-transplantation, \(t_{ij} \in \left[ {0, 50} \right]\). The primary reason for examining these two follow-up periods was to explore patterns of TTC fluctuations that were more complex during the early days following transplantation rather than the later days. This was illustrated by examining the out-of-sample predictive accuracy from the short-term and long-term cases. To fit the nonlinear FCM, TTC assessments were log-transformed using previously proposed methods [35]. However, to aid interpretation, we compared the estimated/predicted TTC with the TTC observations in the original scale. For simplicity, the predictive accuracy evaluated from the log-transformed data and the original observed data is indicated by ‘log scale’ and ‘original scale’, respectively, throughout this paper.
Statistical analysis was implemented in the computing environment R (R Core Team, 2016) using the functions of the Mixed GAM Computation Vehicle (mgcv) package (version 1.8–2.3) [43] and the programming code provided by Kim et al. [35].
3 Results
3.1 Simulation Study
Table 3 shows the RMSPE, IsBias, and ACP for the nominal levels of 85%, 90%, and 95% when the number of patients in the training and test sets was \(N_{\text{train}} = 100, 200, 300\) and \(N_{\text{test}} = 100\), respectively. The results indicate that the nonlinear FCM Eq. (1) outperforms the standard LME in terms of predicting TTC and estimating its variance. This is true irrespective of whether there is a large amount of ‘missingness’ in the covariate and response. The relative percentage gain in prediction with respect to LME was:
where \(\sqrt {{\text{MSPE}}_{\text{OUT}}^{\text{NFCM}} }\)and \(\sqrt {{\text{MSPE}}_{\text{OUT}}^{\text{LME}} }\) were the out-of-sample prediction errors obtained by fitting Eq. (1) and LME, respectively. Values close to zero imply that both methods have similar performance, whereas larger values are indicative of greater improvement. Based on the results from Table 3, our model showed an average 16.5% improvement in prediction with respect to LME (range 11.9–21.8%).Compared with the bias obtained from the nonlinear FCM, the LME tended to produce a larger bias irrespective of the sample sizes and sampling designs. An investigation of coverage for predicted TTC showed that the nonlinear FCM achieved nominal coverage as the information increased. This could be explained by the observation that even a small sample size (i.e. \(N_{\text{train}} = 100\)) resulted in coverage probabilities approaching nominal levels. The LME consistently underestimates the coverage probabilities over different sample sizes and sampling designs. This is a poor characteristic of any estimation method as it does not improve with increased information in the data (i.e. nonconsistent statistical estimation).
Figure 2 shows the 95% prediction band obtained from a selected case in the simulation study. Note that predicted TTC from the LME is ‘off-centered’ from the data, as seen in the absence of observed data in the upper bound of the PI and lack of smooth estimates over time relative to the nonlinear FCM. Further investigation (although results for all other cases are not shown in this article) indicated that, on average, the nonlinear FCM provided reasonable confidence bands, and the results were remarkably robust to the choice of sample size and sampling design in the setting considered here. This implies that the undercoverage of the prediction bands from the LME is in fact a result of inaccurate characterization of TTC fluctuation, possibly caused by the strict linearity assumption imposed in the model.

Individual-specific TTC prediction (on log scale) and pointwise PI fitted by the estimation procedure of nonlinear FCM (left panel) and the LME (right panel). Results were obtained for the case of \(N_{\text{train}} = 100\) and sparse sampling design from the simulation study. The black dots are the observed TTC (on log scale) from a simulated test set. The red dashed lines and blue solid lines are the predicted TTC (on log scale) and the 95% prediction band obtained by fitting the nonlinear FCM and the LME, respectively. FCM functional concurrent model, LME linear mixed-effects model, PI prediction intervals, TTC tacrolimus trough concentration
The standard LME can be extended to account for a possible nonlinear relationship by incorporating additional effects of dose and time. To provide some insight, we investigated the finite sample performance of mixed effects models by incorporating a quadratic time effect; simulation results are displayed in Sect. C of the Electronic Supplementary Material. We found that the predictive performance and the coverage at different nominal levels did not improve across different simulation scenarios. Nevertheless, it is worthwhile noting that this may not always be the case in other clinical studies, depending on the actual data used in the analysis.
3.2 Phase III Data Analysis
The in-sample and out-of-sample RMSPE (Eq. 2) for the nonlinear FCM, LME, and FPCA-based model are summarized in Table 4. As expected, the FPCA-based model (positive control) always provided the smallest in-sample and out-of-sample prediction errors. The nonlinear FCM showed a dramatic improvement in out-of-sample prediction over the LME in all cases. For the long-term analysis, the nonlinear FCM had a relative percentage gain (Eq. 3) of 16.1% (log-scale analysis) over the LME. In the short term, the percentage gain was 20.4% (log-scale analysis). Therefore, the results corresponding to the early period \(t_{ij} \in \left[ {0, 50} \right]\) showed more improvement than those in the long term (i.e. \(t_{ij} \in [0, 171]\)), indicating that the TTC fluctuates a great deal during the early period.
The performance of nonlinear FCM was assessed through replication, such that we randomly split the data set into training and test sets 100 times and computed the out-of-sample RMSPE for each split. The boxplots in Fig. 3 (first row) show the sampling distribution of the RMSPE, indicating that fitting the nonlinear FCM preserves greater predictive accuracy than fitting the standard LME in the current data setting. This could be further illustrated by the estimated sample mean and sample standard deviation of the RMSPE presented in the figure. The bottom row of Fig. 3 shows the predicted TTC (left) given a dose profile (right) for a randomly selected single patient from the test data. This figure illustrates how the LME simply mirrors the dose profile without making allowances for the required, more complex, dose adjustment, thereby resulting in the loss of predictive accuracy.

Results of out-of-sample predictive performance obtained from phase III data analysis. The first row displays the out-of-sample root mean squared prediction errors obtained from the log-transformed data (left panel) and from the original observed data (right panel) for the case of \(t_{ij} \in [0, 171]\). The second row displays the individual-specific TTC prediction (left panel) and the corresponding dose profile (right panel) from a randomly selected patient. FCM functional concurrent model, FPCA functional principal component analysis, LME linear mixed-effects model, MSPE mean squared prediction error, SD standard deviation, TTC tacrolimus trough concentration
In summary, the results from Table 4 and Fig. 3 indicate that the LME could not capture the true underlying relationship between TTC and dose because of the nonlinear complexity of TTC in relation to tacrolimus dose.
Finally, Fig. 4 represents the estimated variance of log-scaled TTC, \(\{ {\text{Var}}\{ Y_{i} (t)|X_{i} \left( t \right)\}\) (top-left panel), and the estimated three-dimensional (3D) surface of \(F\left( { \cdot , \cdot } \right)\) from different viewpoints (top-right, bottom-left, and bottom-right panels). The estimated variance displayed in the top-left panel indicates that the variability of TTC is high during the first 50 days following transplantation. The estimated 3D surface typically aids in understanding the overall trend of TTC along the values of dose and time. The thick red line in the 3D surface highlights the estimated curve on particular days. When \(t = 10\)(i.e. on day 10), the amount of curvature was relatively large, indicating a strong nonlinearity between the dose and TTC (top-right and bottom-left panels). When \(t = 100\)(i.e. on day 100), the amount of TTC fluctuation was relatively low (bottom-right panel).

Estimated variance and bivariate surface obtained from phase III data analysis. The top-left panel displays the estimated variance of log-scaled TTC, \({\text{Var}}\{ Y(t)|X\left( t \right)\}\), and the top-right panel displays the estimated surface of \(F( \cdot , \cdot )\) along the values of dose and time; the red line represents the curve obtained by fixing the day as 10. The bottom-left panel shows the identical estimated surface from a different viewpoint, and the bottom-right panel represents the estimated surface of \(F( \cdot , \cdot )\); the red line indicates the curve on day 100. TTC tacrolimus trough concentration
4 Discussion
The current study used an analytical method based on functional data analysis [26,27,28] to describe the relationship between TTC and dose over time (not assuming a linear relationship). This model assumed that both TTC and tacrolimus dose are the functional response and functional covariate, respectively, with their relationship described using a flexible class of nonparametric functional regression models [26,27,28,29,30,31,32,33,34,35]. This framework does not include serial relationships of dosing changes based on preceding TTC values. While it is possible that this kind of relationship could be modeled with time-series methods (among others), doing so would be outside the current scope of effort evaluating the dose-adjusted IPV of TTC.
In a recent study, Ben Fredj et al. [44] developed a population pharmacokinetic model to describe the evolution of dose-adjusted TTC according to the time elapsed since transplant. They used a nonparametric adaptive grid approach to better detect outliers and unsuspected subpopulations. Although their model implicitly assumes that TTC and dose are linearly associated, the benefits of a nonparametric approach improved the prediction of dose-adjusted TTC over time. In contrast to our model, their model is suited for detecting changes in tacrolimus clearance and volume of distribution. Our framework is centered on calibrating the TTC fluctuation after making a proper dose adjustment.
The functional concurrent regression [32, 35] used in our model represents the relationship between TTC and dose using an unknown bivariate function \(F( \cdot , \cdot )\) to model the response based on the covariate and the time point. The bivariate function \(F( \cdot , \cdot )\) allowed for the description of a possibly complex nonlinear relationship between TTC and dose, and thus had the potential to improve predictive accuracy, especially when the data had a highly complex dependence structure. In this model, the linear dependence was a special case of their assumed relationship: \(F\left\{ {X_{i} \left( t \right), t} \right\} = \beta_{0} \left( t \right) + \beta_{1} \left( t \right)X_{i} (t),\) where \(\beta_{0} (t)\) and \(\beta_{1} (t)\) were the unknown time-varying coefficients.
Knowing this, the model in Eq. (1) could be viewed as a member of this wider class of functional regression models. Indeed, these constructs may be more appropriate to use in our data setting because of difficulties in subscribing strict linearity to the relationship between TTC and dose. In addition, our model framework was appealing in our data setting because the model in Eq. (1) did not impose a specific form of covariance structure, such as working independence or nonstationary error covariance structures, and could accommodate any type of structure. The proposed model estimation of the variance of \(Y_{i} (t)\) was therefore free of standard assumptions and could account for within-subject correlations in estimation and inference.
While the present study enlisted a previously described modeling procedure of nonlinear FCM [35] to analyze the complex nonlinear relationship between TTC and dose over time, it differed significantly from traditional TTC IPV models, which embrace an arbitrarily chosen stable dose as an important consideration. In contrast, the new, nonlinear FCM can accommodate a variety of realistic settings, such as nonlinear complexity between variables, continuously changing variability over time, sparse and irregular observations per patient, and data that are contaminated with measurement errors. However, it should be noted that preprocessing steps should be followed in order to deal with sparse and noisy functional data [35].
When testing our model against a simulated data set and phase III patient data from a large kidney transplant development program [40,41,42], our numerical investigation provided greater insight into the expected accuracy of nonlinear FCM. In cases where tacrolimus blood concentrations are predicted based on patients’ previous TTC and tacrolimus dose information, there was a 16.5% improvement in prediction using the nonlinear FCM versus the LME. This was largely due to the tendency of the LME model to improperly adjust estimates of TTC based on dosing due to the prespecified linearity assumption. In contrast, the flexibility of the new model accurately accounted for the variance of TTC during the periods of large fluctuations in dose. A 16.5% improvement in the prediction of TTC suggests that the nonlinear FCM has an enhanced ability to adjust the estimates of TTC to time and dose profiles. From a clinical perspective, since tacrolimus has a narrow therapeutic range, small changes in blood concentration can mean the difference between over-immunosuppression and its related adverse effects, such as susceptibility to infection, or under-immunosuppression, leading to graft rejection [45, 46]. The more accurate prediction of TTC and its variance with this robust analytical method could therefore be used to better inform treatment decisions.
The IPV function over time showed a noticeable increase around day 100 and a decrease in the following days (Fig. 4, top-left panel), possibly overlapping with recovery of a patient’s post-transplant hematocrit and albumin, and/or the minimization/discontinuation of corticosteroids, frequently observed around this time. Such an aspect is in line with several previous studies that discussed the effect of changes in hematocrit and albumin, or corticosteroid discontinuation on tacrolimus clearance in pharmacokinetic models [47,48,49]. Future research will not only focus on replicating the results of this model in an external, real-world data set but will also seek to confirm whether these observations persist.
Surprisingly, despite knowing that TTC variability experiences temporal fluctuation, there have been no prior attempts to quantify TTC variance as a function of time. From a methodological standpoint, building more generalizable and robust models would thus allow more accurate predictions of future TTC fluctuations. In view of that, before testing any association between tacrolimus IPV and transplant outcome, developing a robust estimator for describing TTC variance would allow for more effective detection of high-risk patients. Future research on this important topic is clearly warranted.
In the meantime, building more easily interpretable and robust models is essential, and the current study demonstrated such aspects through the phase III data analysis with the estimated 3D graph (Fig. 4). The estimated result could be interpreted in two different ways: (1) by fixing time; this graph helps understand how TTC is associated with the dose at a particular moment in a patient’s post-transplant course; and (2) by fixing dose; changes in TTC over time can be better understood. As this is a population-level estimation, the results provide information on how groups of patients may be affected by current therapy.
Although the proposed methodology is not new from an analytical standpoint, it is a novel application within the therapeutic area and can be applied to randomized phase III data. Additionally, the nonlinear relationship between TTC and dose is the first known example of applying nonlinear FCM in a real-world setting to recover complex relationships within a data set. In the present case, it has revealed that the true relationship underlying the response and covariate variables is in fact complex and nonlinear.
Predicting the appropriate tacrolimus dose to achieve a desired tacrolimus whole blood concentration is one future application for this model. Another application is a more accurate understanding of the effects of within-patient variability on transplant outcomes compared with standard LMEs. The influence of baseline covariates (e.g. age, sex, race) and time-dependent covariates (e.g. liver and renal function tests) can be better understood by additively incorporating relevant predictors in the nonlinear functional concurrent model (NFCM) framework, leading to more tailored immunosuppressive strategies. Given that nonparametric modeling allows for additive (multiple) covariates on a given response, this topic can be further addressed by extension of our tacrolimus model in a manner that has been discussed and investigated previously [35].
5 Conclusion
To overcome the limitations associated with linear models, we have applied a flexible functional regression model to quantify the complex nonlinear relationship between TTC and tacrolimus dose over time. This model has been validated using simulation analyses and assessed directly using extensive phase III clinical trial data from kidney transplant patients. Our model accurately accounted for TTC variability during periods of large dose fluctuations, such as is frequently encountered at an early stage following kidney transplantation.
This is the first known application of a functional regression model to assess complex relationships impacting TTCs in a clinical setting. This method has applicability in the context of future clinical trials, including real-world data sets, and will be featured as an outcome measure in the ongoing ASTOUND™ donor-specific antibody phase IV clinical trial. Future refinement of this approach may improve post-transplant treatment by predicting the dose required to achieve desired TTCs, thus helping to reduce within-patient variability and the associated risk of poor transplant outcomes.
References
Myers BD, Ross J, Newton L, Luetscher J, Perlroth M. Cyclosporine-associated chronic nephropathy. N Engl J Med. 1984;311:699–705.
Matas AJ, Gaston RS. Moving beyond minimization trials in kidney transplantation. J Am Soc Nephrol. 2015;26:2898–901.
Stegall MD, Gaston RS, Cosio FG, Matas A. Through a glass darkly: seeking clarity in preventing late kidney transplant failure. J Am Soc Nephrol. 2015;26:20–9.
Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R, Cosio FC, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation. 2010;90:68–74.
Lerut E, Kuypers DR, Verbeken E, Cleutjens J, Vlaminck H, Vanrenterghem Y, et al. Acute rejection in non-compliant renal allograft recipients: a distinct morphology. Clin Transplant. 2007;21:344–51.
Vanhove T, Vermeulen T, Annaert P, Lerut E, Kuypers DRJ. High intrapatient variability of tacrolimus concentrations predicts accelerated progression of chronic histologic lesions in renal recipients. Am J Transplant. 2016;16:2954–63.
Shishido S, Asanuma H, Nakai H, Mori Y, Satoh H, Kamimaki I, et al. The impact of repeated subclinical acute rejection on the progression of chronic allograft nephropathy. J Am Soc Nephrol. 2003;14:1046–52.
Cosio FG, Grande JP, Wadei H, Larson TS, Griffin MD, Stegall MD. Predicting subsequent decline in kidney allograft function from early surveillance biopsies. Am J Transplant. 2005;5:2464–72.
Moreso F, Ibernon M, Gomà M, Carrera M, Fulladosa X, Hueso M, et al. Subclinical rejection associated with chronic allograft nephropathy in protocol biopsies as a risk factor for late graft loss. Am J Transplant. 2006;6:747–52.
Heilman RL, Khamash HA, Smith ML, Chakkera HA, Moss AA, Reddy KS. Delayed allograft inflammation following alemtuzumab induction for kidney transplantation. Clin Transplant. 2013;27:772–80.
Naesens M, Kuypers DRJ, De Vusser K, Vanrenterghem Y, Evenepoel P, Claes K, et al. Chronic histological damage in early indication biopsies is an independent risk factor for late renal allograft failure. Am J Transplant. 2013;13:86–99.
Park WD, Griffin MD, Cornell LD, Cosio FG, Stegall MD. Fibrosis with inflammation at one year predicts transplant functional decline. J Am Soc Nephrol. 2010;21:1987–97.
O’Regan JA, Canney M, Connaughton DM, O’Kelly P, Williams Y, Collier G, et al. Tacrolimus trough-level variability predicts long-term allograft survival following kidney transplantation. J Nephrol. 2016;29:269–76.
Taber DJ, Su Z, Fleming JN, McGillicuddy JW, Posadas-Salas MA, Treiber FA, et al. Tacrolimus trough concentration variability and disparities in African American kidney transplantation. Transplantation. 2017;101:2931–8.
Damon C, Luck M, Toullec L, Etienne I, Buchler M, de Hurault Ligny B, et al. Predictive modeling of tacrolimus dose requirement based on high-throughput genetic screening. Am J Transplant. 2017;17:1008–19.
Rodrigo E, Segundo DS, Fernández-Fresnedo G, López-Hoyos M, Benito A, Ruiz JC, et al. Within-patient variability in tacrolimus blood levels predicts kidney graft loss and donor-specific antibody development. Transplantation. 2016;100:2479–85.
de Jonge H, Vanhove T, de Loor H, Verbeke K, Kuypers DRJ. Progressive decline in tacrolimus clearance after renal transplantation is partially explained by decreasing CYP3A4 activity and increasing haematocrit. Br J Clin Pharmacol. 2015;80:548–59.
Lieber SR, Volk ML. Non-adherence and graft failure in adult liver transplant recipients. Dig Dis Sci. 2013;58:824–34.
Borra LCP, Roodnat JI, Kal JA, Mathot RA, Weimar W, Van Gelder T. High within-patient variability in the clearance of tacrolimus is a risk factor for poor long-term outcome after kidney transplantation. Nephrol Dial Transplant. 2010;25:2757–63.
Sapir-Pichhadze R, Wang Y, Famure O, Li Y, Kim SJ. Time-dependent variability in tacrolimus trough blood levels is a risk factor for late kidney transplant failure. Kidney Int. 2014;85:1404–11.
Hsiau M, Fernandez HE, Gjertson D, Ettenger RB, Tsai EW. Monitoring nonadherence and acute rejection with variation in blood immunosuppressant levels in pediatric renal transplantation. Transplantation. 2011;92:918–22.
Gallagher HM, Sarwar G, Tse T, Sladden TM, Hii E, Yerkovich ST, et al. Erratic tacrolimus exposure, assessed using the standard deviation of trough blood levels, predicts chronic lung allograft dysfunction and survival. J Heart Lung Transplant. 2015;34:1442–8.
Supelana C, Annunziato RA, Schiano TD, Anand R, Vaidya S, Chuang K, et al. Medication level variability index predicts rejection, possibly due to nonadherence, in adult liver transplant recipients. Liver Transplant. 2014;20:1168–77.
Pollock-BarZiv SMS, Finkelstein Y, Manlhiot C, Dipchand AI, Hebert D, Ng VL, et al. Variability in tacrolimus blood levels increases the risk of late rejection and graft loss after solid organ transplantation in older children. Pediatr Tansplant. 2010;14:968–75.
Shuker N, Shuker L, van Rosmalen J, Roodnat JI, Borra LCP, Weimar W, et al. A high intrapatient variability in tacrolimus exposure is associated with poor long-term outcome of kidney transplantation. Transpl Int. 2016;29:1158–67.
Ramsay JO, Silverman BW. Functional data analysis with R and Matlab. 2nd ed. New York: Springer; 2005.
Ferraty F, Vieu P. Nonparametric functional data analysis: theory and practice. New York: Springer Science & Business Media; 2006.
Ramsay J, Hooker G, Graves S. Functional data analysis in R and Matlab. New York: Springer; 2009.
Wood SN. Generalized additive models: an introduction with R. Boca Raton: CRC; 2006.
Morris J. Functional regression. Annu Rev Stat Appl. 2015;2:321–59.
Scheipl F, Staicu A-M, Greven S. Functional additive mixed models. J Comput Graph Stat. 2015;24:477–501.
Maity A. Nonparametric functional concurrent regression models. Wiley Interdiscip Rev Comput Stat. 2017;9:e1394.
McLean MW, Hooker G, Staicu A-M, Scheipl F, Ruppert D. Functional generalized additive models. J Comput Graph Stat. 2014;23:249–69.
Yao F, Müller H-G, Wang J-L. Functional linear regression analysis for longitudinal data. Ann Stat. 2005;33:2873–903.
Kim J, Maity A, Staicu A-M. Additive nonlinear functional concurrent model. Stat Interface. 2018;11:669–85.
Kim J, Staicu A-M, Maity A, Carroll RJ, Ruppert D. Additive function-on-function regression. J Comput Graph Stat. 2018;27:234–44.
Goldsmith J, Greven S, Crainiceanu C. Corrected confidence bands for functional data using principal components. Biometrics. 2013;69:41–51.
Xiao L, Li Y, Ruppert D. Fast bivariate P-splines: the sandwich smother. J R Stat Soc Ser B Stat Methodol. 2013;75:577–99.
Rupper D, Wand M, Carroll R. Semiparametric regression. Cambridge: Cambridge University Press; 2003.
Silva HT, Yang HC, Abouljoud M, Kuo PC, Wisemandle K, Bhattacharya P, et al. Erratum: One-year results with extended-release tacrolimus/MMF, tacrolimus/MMF and cyclosporine/MMF in de novo kidney transplant recipients. Am J Transplant. 2007;7:595–608.
Krämer BK, Charpentier B, Bäckman L, Silva HT, Mondragon-Ramirez G, Cassuto-Viguier E, et al. Tacrolimus once daily (ADVAGRAF) versus twice daily (PROGRAF) in de novo renal transplantation: a randomized phase III study. Am J Transplant. 2010;10:2362–643.
Albano L, Banas B, Klempnauer JL, Glyda M, Viklicky O, Kamar N, et al. OSAKA trial: a randomized, controlled trial comparing tacrolimus QD and BD in kidney transplantation. Transplantation. 2013;96:897–903.
Wood S. Package ‘mgcv’: mixed GAM computation vehicle with automatic smoothness estimation (version 1.8–2.3). 2018.
Ben Fredj N, Woillard JB, Debord J, Chaabane A, Boughattas N, Marquet P, et al. Modeling of tacrolimus exposure in kidney transplant according to posttransplant time based on routine trough concentration data. Exp Clin Transplant. 2016;14:394–400.
Wallemacq P, Armstrong VW, Brunet M, Haufroid V, Holt DW, Johnston A, et al. Opportunities to optimize tacrolimus therapy in solid organ transplantation: report of the European consensus conference. Ther Drug Monit. 2009;31:139–52.
Kuypers DRJ, Claes K, Evenepoel P, Maes B, Vanrenterghem Y. Clinical efficacy and toxicity profile of tacrolimus and mycophenolic acid in relation to combined long-term pharmacokinetics in de novo renal allograft recipients. Clin Pharmacol Ther. 2004;75:434–47.
Woillard J-B, Saint-Marcoux F, Debord J, Åsberg A. Pharmacokinetic models to assist the prescriber in choosing the best tacrolimus dose. Pharmacol Res. 2018;130:316–21.
Saint-Marcoux F, Woillard J-B, Jurado C, Marquet P. Lessons from routine dose adjustment of tacrolimus in renal transplant patients based on global exposure. Ther Drug Monit. 2013;35:322–7.
Undre NA, Schäfer A. Factors affecting the pharmacokinetics of tacrolimus in the first year after renal transplantation. European Tacrolimus Multicentre Renal Study Group. Transplant Proc. 1998;30:1261–3.
Acknowledgements
This study was funded by Astellas Pharma Global Development, Inc. Cello Health MedErgy (Europe) assisted in editing the initial version of this manuscript under the direction of the authors, and provided editorial support throughout its development. Editorial support was funded by Astellas Pharma, Inc.
Author information
Authors and Affiliations
Contributions
All authors contributed sufficiently to this scientific work and therefore share collective responsibility and accountability for the results. Study design: JK, SW, NAU, RMK, JJS; Conduct and acquisition of data: JK, SW, NAU, RMK, JJS; Analysis and interpretation of data: JK, SW, NAU, FS, RMK, JJS; Writing of the manuscript: JK, JJS; Revising and final approval of the manuscript: JK, SW, NAU, FS, RMK, JJS.
Corresponding author
Ethics declarations
Funding
This study was funded by Astellas Pharma Global Development, Inc. Editorial support was funded by Astellas Pharma Inc.
Conflict of interest
Janet Kim, Sam Wilson, Nasrullah A. Undre, Fei Shi, Rita M. Kristy, and Jason J. Schwartz are employees of Astellas and have received a salary from Astellas.
Data Availability Statement
Access to anonymized individual participant-level data will not be provided for this trial as it meets one or more of the exceptions described on the http://www.clinicalstudydatarequest.com website under ‘Sponsor Specific Details for Astellas’.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kim, J., Wilson, S., Undre, N.A. et al. A Novel, Dose-Adjusted Tacrolimus Trough-Concentration Model for Predicting and Estimating Variance After Kidney Transplantation. Drugs R D 19, 201–212 (2019). https://doi.org/10.1007/s40268-019-0271-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-019-0271-2