
Abstract
Background
Standard treatment with sunitinib for patients with metastatic renal cancer provides an ‘on-off’ schedule (daily administration of a 50-mg capsule for 4 weeks, followed by a 2-week break; consecutive 6-week cycles). We developed an alternative intermittent schedule to reduce the toxicity and symptoms of tumor regrowth during the rest period and to allow prolonged continuation of therapy, maintaining dose intensity.
Objective
The objective of this study was to provide a retrospective evaluation of the feasibility, safety, and efficacy of an alternative schedule of sunitinib in patients who did not tolerate classical treatment.
Methods
Patients treated with the classical schedule with at least grade 2 toxicity or recurrence of symptoms during the rest period were switched to an alternative schedule (the same daily dose 5 consecutive days per week for 5 weeks and then the same daily dose on days 1, 3, and 5 in the sixth week; consecutive 6-week cycles).
Results
Twenty-five patients were enrolled. The median time from sunitinib initiation to schedule switch was 2.9 months. After the switch, the median therapy duration was 9.2 months. Rate of delay, corrected by cycle number, was 10% for both schedules. After the switch, 48.7% of patients obtained a toxicity reduction (hypertension −82%, stomatitis −71%, cutaneous toxicity −69%). A reduction in ‘on-off symptoms’ (−86%) was achieved. Overall response rate was 40% and the disease control rate was 80%. Median progression-free survival was 16.4 months and median overall survival was 41.3 months.
Conclusions
Despite the small sample size and retrospective nature, we demonstrated the feasibility, safety, and efficacy of the alternative schedule, allowing prolonged treatment and better quality of life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Standard treatment with the classical schedule of sunitinib for patients with advanced renal cancer is burdened by adverse events and the phenomenon of ‘on-off’ symptoms. |
We formulated a new alternative intermittent schedule of administration, retrospectively demonstrating its feasibility, safety, and efficacy in patients who did not tolerate the classical treatment. |
1 Introduction
Sunitinib malate is an oral, multi-targeted, tyrosine kinase inhibitor of vascular endothelial growth factor (VEGF) receptors (VEGFR-1, VEGFR-2, and VEGFR-3) among other receptor tyrosine kinases [1]. Sunitinib competitively inhibits the binding of adenosine triphosphate to the tyrosine kinase domain on targeted proteins at a concentration of 5–100 nM. It is metabolized by cytochrome P450 3A4 to an active metabolite, SU12662, as well as to further inactive products. The pharmacokinetics is not affected by food intake [2].
Sunitinib has been approved for metastatic renal cell carcinoma (mRCC) in all treatment settings and for gastrointestinal stromal tumor therapy after disease progression (or intolerability) to imatinib mesylate therapy. Moreover, clinical studies confirm the activity of sunitinib in other several solid tumor types [3].
The landmark trial with sunitinib in mRCC was a double-blinded, randomized, phase III study enrolling 750 treatment-naïve patients to receive sunitinib (experimental arm) or interferon-α (IFN-α, control arm) as first-line therapy. The primary endpoint was progression-free survival (PFS) and the trial was unblinded after a second interim analysis, demonstrating a significant benefit of sunitinib over IFN-α: patients treated with sunitinib showed improved median PFS (11 vs. 5 months, p < 0.001) and overall survival (OS, 26.4 vs. 21.8 months, p = 0.051). Furthermore, differential OS was likely to be underestimated, owing to the significant rate of crossover from IFN-α to sunitinib treatment after unblinding [1, 3].
The results of this study established sunitinib as a standard of care for the first-line treatment of mRCC; furthermore, the subsequent COMPARZ study confirmed the efficacy and toxicity profile of this drug in the same setting [4]. The standard mRCC treatment with sunitinib is characterized by an ‘on-off’ schedule, with daily oral administration of a 50-mg capsule for 4 weeks, followed by a 2-week break (4/2 schedule), with consecutive 6-week cycles [1, 3]. The dose of sunitinib can be modified according to toxicity; nevertheless, a daily dose <25 mg is frequently reported as ineffective [2]. Although initial preclinical studies were planned to provide continuous administration, the 4/2 schedule was then selected for human experimentation at the request of the regulatory entities, to allow recovery from potential adverse events (AEs) observed in animal models. Indeed, toxicological evaluation of such models revealed bone marrow depletion and toxic effects in rats and monkeys, as well as adrenal micro-hemorrhages in rats [3].
Sunitinib was associated with a higher incidence of treatment-related AEs compared with IFN-α, with the most pronounced differences in the overall incidence of diarrhea (61 vs. 15%) and dysgeusia (46 vs. 15%). The most common AE of sunitinib, namely the fatigue, affects 50–70% of patients and may be disabling. Hypothyroidism occurs in 40–60% of patients. Bone marrow suppression with severe neutropenia is reported in 10% of patients. The most common grade 3 or 4 AE is represented by hypertension (12%), followed by fatigue (11%), diarrhea (9%), and hand–foot syndrome (9%) [1, 2]. In the sunitinib open access program, 8% of patients discontinued therapy because of serious AEs and a further 30% required dose reductions for toxicity [1].
In clinical practice, it is often noted that AEs can increase throughout each cycle and tend to worsen in the final 2 weeks of the treatment cycle [5]. The long-term impact of sunitinib-associated toxicities is greater the longer patients live [1].
Sunitinib and its active equipotent metabolite SU12662 have half-lives of 40–60 and 80–110 h, respectively; steady-state concentrations are achieved within 10–14 days and the maximum tolerated dose is 50 mg/day [3]. Sunitinib exhibited dose- and time-dependent anti-tumor activity in mice. Data from animals and from studies in patients with acute myeloid leukemia showed that the target plasma concentration of a drug capable of inhibiting platelet-derived growth factor-β and VEGFR-2 phosphorylation is established in the range of 50–100 ng/mL, corresponding to daily doses of 50 mg [3, 6]. Oral doses of sunitinib able to produce these plasma concentrations for at least 12 h of a 24-h dosing interval would lead to the inhibition of target receptors adequate to result in anti-angiogenic activity. The maintenance of continuous inhibition of the target receptors was not needed to achieve efficacy in murine models. The occurrence of dose-limiting toxicities was associated with sunitinib plasma concentrations superior to 100 ng/mL [6].
A phase II study with sunitinib 50 mg/day, administered with a 4/2 standard schedule to patients with mRCC, showed that average plasma concentration of the total drug reached therapeutic concentrations (>50 ng/mL) on day 14 of cycle 1, and that concentrations were sustained throughout treatment during the 4-week dosing period. The study also showed that the average plasma concentration of the total drug on days 14 and 28 of cycle 1 was comparable to those observed on day 28 of cycles 2 and 3, but plasma drug concentrations were not detectable on day 1 of cycle 2, suggesting a complete washout of the drug in the 2-week break [7]. The direct consequence of the latter observation may result in the phenomenon of tumor regrowth during the 2-week break, described both in clinical practice and in a preclinical setting [3, 8,9,10].
A meta-analysis published in 2010 by Houk et al. showed that increased plasma exposure to sunitinib is associated with improved clinical outcome: longer time to progression (TTP), longer OS, and a greater chance of disease response [11]. A further study, published in 2014 by Porta et al., assessed the link between dose intensity and OS: 291 patients were followed during a landmark period of 24 weeks after sunitinib treatment initiation. After adjusting for potential confounders, OS was significantly shorter among patients with a sunitinib dose intensity below 0.7 [hazard ratio (HR): 3.36, 95% confidence interval (CI) 1.49–7.55], but dose intensities below 0.8 and 0.9, respectively, were not associated with significantly shorter survival times following the 24-week landmark period. This evidence demonstrated that sunitinib dose reduction may result in an OS reduction. Moreover, OS was significantly shorter among patients who discontinued treatment because of AEs occurring within 24 weeks of the beginning of therapy (HR: 2.80, 95% CI 1.06–7.38) [12].
Alternative schedules of sunitinib administration have been explored in several clinical studies, with the goal of improving drug tolerability and dose intensity [5, 13,14,15,16,17,18,19,20,21,22,23]. Only two different regimens have been studied prospectively: the continuous daily dosing (CDD) schedule and the 2-week on, 1-week off schedule (2/1 schedule) [5, 20]. Prospective data about the CDD schedule of sunitinib were provided by the EFFECT trial, a randomized phase II study in which 292 patients with first-line mRCC were randomized to receive sunitinib at a daily dose of 50 mg with a 4/2 schedule or at a daily dose of 37.5 mg using the CDD regimen. Of note, the theoretical total dose in 6 weeks of the CDD schedule was 1575 mg (37.5 mg × 42 days), while for the 4/2 schedule it was 1400 mg (50 mg × 28 days) [3]. This study demonstrated no differences in terms of drug tolerability or patient-reported symptoms of disease, but showed a trend towards superiority of the 4/2 schedule over the CDD schedule in terms of TTP (9.9 months for 4/2 schedule vs. 7.1 months for CDD, HR 0.77, 95% CI 0.57–1.04, p = 0.09). No significant difference was observed in OS (23.1 months for the 4/2 schedule vs. 23.5 months for the CDD schedule, p = 0.615). Pharmacokinetic analysis and the outcome of this clinical trial suggested that treating patients at a lower dose intensity may result in reduced efficacy. Data also indicate that maintaining the daily dose intensity is more important than giving a minimal dose each day [5].
The multicenter, RESTORE phase II trial enrolled 74 treatment-naïve patients with clear-cell mRCC, randomly assigned to receive 4/2 or 2/1 sunitinib schedules after stratification by the Memorial Sloan Kettering Cancer Center (MSKCC) risk group and the presence of measurable lesions. The failure-free survival rates at 6 months were 44% with the 4/2 schedule (n = 36) and 63% with the 2/1 schedule (n = 38). Neutropenia (all grades: 61 vs. 37%; grades 3–4: 28 vs. 11%) and fatigue (all grades: 83 vs. 58%) were more frequently observed with the 4/2 schedule. There was a strong tendency towards a lower incidence of stomatitis, hand–foot syndrome, and rash with the 2/1 schedule. Objective response rates (ORRs) were 47% for the 2/1 schedule and 36% for the 4/2 schedule. With a median follow-up of 30 months, the median TTP was 12.1 months for the 2/1 schedule and 10.1 months for the 4/2 schedule. This study, despite the limitations represented by a small sample size and the Asian-only ethnicity of patients, showed that sunitinib administered with a 2/1 schedule was associated with less toxicity and higher failure-free survival at 6 months than with a 4/2 schedule, without major differences in terms of ORR and TTP [20].
2 Materials and Methods
2.1 Purpose
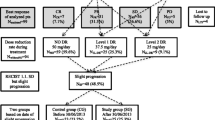
Considering the pharmacokinetic and clinical data reported above, at our centers, we opted for a possibly better tolerated alternative intermittent schedule of sunitinib administration (Fig. 1). While maintaining the same dose intensity of the classical schedule (28 capsules in 6 weeks), the new schedule could allow the patient both to be treated for a longer time, with fewer dose reductions, and to limit the symptomatic tumor regrowth [3]. We previously reported a small case series of patients treated with this modified schedule [3]. The aim of this retrospective updated analysis was the evaluation of feasibility, safety, and efficacy of this new sunitinib schedule in patients with mRCC who did not tolerate treatment with the classical 4/2 schedule.

Alternative intermittent sunitinib schedule showing the same dose intensity of the 4/2 schedule. W week
2.2 Patient Population
This retrospective analysis included consecutive patients with mRCC treated with sunitinib with the new alternative intermittent schedule at two Italian institutions (Medical Oncology Unit, University Hospital of Parma, Parma, Italy, and Medical Oncology Unit, Hospital of Cremona, Cremona, Italy), between June 2008 and May 2016. Patient informed consent was obtained.
Patients treated with the classical 4/2 schedule, without progressive disease (PD), who had at least a grade 2 toxicity (graded by the National Cancer Institute Common Terminology Criteria for AEs, Version 4.0), or who reported the recurrence of the disease’s symptoms during the 2-week treatment break, were switched to the new alternative intermittent schedule (see Fig. 1 for details) instead of a dose reduction as the first option.
The retrospective outcome analysis considered patients aged over 18 years, with histological confirmation of mRCC and evidence of measurable disease, based on Response Evaluation Criteria in Solid Tumors, Version 1.1 [24]. Patients were admitted to our hospitals, where they started treatment with sunitinib, initially administered with a classical 4/2 schedule. Additional eligibility criteria for the retrospective study included: availability of all data about toxicity and disease response (obtained from paper and electronic medical records); Eastern Cooperative Oncology Group Performance Status of 0–2; adequate hematologic, hepatic, and renal function; and informed written consent.
Patients were excluded from the final analysis if they had untreated brain metastases or any second malignancy within the previous 3 years (other than adequately treated basal cell carcinoma, squamous cell skin cancer, or in situ carcinomas). Additional exclusion criteria included a history of clinically significant cardiovascular disease, severe cardiac arrhythmia, prolongation of the corrected QT interval, uncontrolled hypertension, or other contraindications to receive sunitinib. No selection of patients was made according to treatment line or the number of prior therapies, to cover all settings of our clinical practice with sunitinib. For proper evaluation of toxicity modifications, we analyzed only patients who had completed at least two cycles of sunitinib (one with the traditional schedule and one with the alternative schedule).
2.3 Schedule
Our alternative intermittent schedule, shown in Fig. 1, allows the same dose intensity of a classical 4/2 schedule to be maintained: starting on Monday, one capsule is administered per day (50, 37.5, or 25 mg according to any previous dose adjustment) for 5 consecutive days per week (days 6 and 7 off) for 5 weeks and then one capsule is administered per day on days 1, 3, and 5 in the sixth week (days 2, 4, 6, and 7 off), for a total of 28 capsules in 6 weeks, every 42 days until disease progression (continuous repetition of 6-week cycles). Of note, this schedule does not allow a rest period for more than 48 h between a capsule intake and the next.
The decision to switch from the classical 4/2 schedule was at the discretion of the referring oncologist, also taking into account the desire of the informed patient to be still treated with sunitinib without (sometimes further) dose reduction or delay. Considering the possibility of errors or misunderstanding, patients were adequately instructed about timing and modalities of drug assumption, even with the help of a diary and of a clearly illustrated memo on a brochure. Further, alternative or subsequent dose reductions to 37.5 or 25 mg/day were considered on individual basis, depending on tolerability.
Patients who experienced sunitinib-related grade 3 or 4 toxicities stopped treatment until the return to grade 1 for non-hematologic or grade 2 for hematologic AEs, and then resumed treatment at the same or at a lower dose, at the clinician’s discretion. Patients requiring a further dose reduction below 25 mg/day, or a dose interruption longer than 6 weeks, owing to toxicity, were permanently discontinued from sunitinib therapy. Treatment was otherwise continued until disease progression or death.
2.4 Procedures, Baseline and During Treatment Evaluations
2.4.1 Endpoints
The primary co-endpoints were the retrospective evaluation of toxicity modifications after the switch from the 4/2 schedule to the alternative intermittent schedule and the feasibility of the latter (with monitoring of treatment compliance, dose reduction, and dose delay rates). Analysis of safety results was provided for all patients receiving at least one cycle of sunitinib with the alternative schedule. Secondary endpoints were OS and overall PFS measurements.
2.4.2 Baseline Assessment
The baseline assessment included: medical and personal history, Eastern Cooperative Oncology Group Performance Status evaluation, physical examination, arterial blood measurement, 12-lead electrocardiography, blood cell count and blood chemistry tests (including thyrotrophic-stimulating hormone, tetraiodothyronine, and phosphatemia), urinalysis, pregnancy test (if appropriate), total-body computed tomography or magnetic resonance imaging, bone scan, and any other examination suitable to measure target lesions.
2.4.3 Assessment During Treatment
Every 2 weeks at the first cycle of the 4/2 schedule and at the first cycle of the alternative schedule, and then every 6 weeks during treatment, a toxicity evaluation was provided (graded with the National Cancer Institute Common Terminology Criteria for AEs, Version 4) through collecting medical history, physical examination, blood and urine tests, and any other examination needed for toxicity assessment. Every 6–8 weeks during treatment (and ongoing long-term follow-up), we collected the medical history, physical examination, blood and urine analysis, electrocardiography, and disease evaluation with the adequate instrumental examinations compared with baseline, as assessed both by the radiologists and the clinicians using Response Evaluation Criteria in Solid Tumors, Version 1.1 [24].
Concerning the feasibility, we evaluated and compared these parameters with both schedules: duration of treatment; number of cycles; number of dose delays, and dose reductions (both total and corrected by the number of cycles). We also considered each dose delay and dose reduction to calculate the average effective daily dose intensity (mg) and the average relative dose intensity (%). Concerning the efficacy, we evaluated these parameters: ORR to treatment, disease control rate, median OS, and PFS.
2.5 Statistical Analysis
Data were collected from medical records. Patients’ characteristics were summarized using median and ranges for continuous variables, with frequency and percent for categorical variables. All statistical tests were two-sided, and p < 0.05 was considered as statistically significant. Duration of therapy was calculated from the date of sunitinib initiation (both from baseline and from the schedule switch) to discontinuation or death for any cause. Progression-free survival was calculated from the date of starting sunitinib to tumor progression assessed by Response Evaluation Criteria in Solid Tumors, Version 1.1 or to death for any cause. Overall survival was calculated from the date of starting sunitinib to death for any cause. Survivors with no disease progression at the last day of sunitinib administration were censored for progression on that day. The Kaplan–Meier method was used to estimate PFS and OS. We used the MSKCC and International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) scores to assess patients’ prognosis. SPSS Version 22.0 (IBM Corporation, Armonk, NY, USA) was used for all statistical calculations.
3 Results
3.1 Patients Baseline Characteristics
From June 2008 to May 2016, we enrolled 25 eligible patients to receive the alternative intermittent schedule of sunitinib. The median follow-up was 25.2 months (range 2.1–73.0). Table 1 shows the patients’ baseline characteristics.
We enrolled 20 men and five women (ratio 4:1), with a median age of 65 years (range 33–82). Seventeen patients (68%) had clear cell carcinoma, four patients (16%) had papillary histology, two patients (8%) had a TFE3 translocated tumor, one patient (4%) had an undifferentiated carcinoma, and one patient (4%) had mixed histology (clear cell and chromophobe). According to the MSKCC score, six patients were good risk, 15 patients were intermediate risk, and four patients were poor risk. However, according to the IMDC score, six patients were good risk, 14 patients were intermediate risk, and five patients were poor risk.
At the time of the occurrence of at least grade 2 toxicity during therapy with the 4/2 schedule, three patients preferred to reduce the capsule dose and seven required a dose delay before switching to the intermittent schedule, while 15 patients were immediately switched to the new schedule without daily dose modifications. With regard to treatment lines, 15 patients were treatment naïve, three patients were pretreated with chemotherapy, four patients were pretreated with targeted therapy (among whom two were pretreated with chemo-immunotherapy plus bevacizumab as a part of a research protocol [25]), and three patients were pretreated with immunotherapy only (IFN-α and interleukin-2). Nineteen patients discontinued treatment with sunitinib because of PD. An elderly patient (82 years old) discontinued treatment after seven cycles (one with the 4/2 schedule and six with our modified schedule) for consent withdrawal, without clear PD or significant toxicity, after being subjected to cytoreductive nephrectomy. Similarly, another patient (75 years old) discontinued sunitinib treatment for consent withdrawal in the absence of clear PD or significant toxicity, after being subjected to local treatment (stereotactic radiotherapy of metastatic sites).
At the collection of data in May 2016, four patients were still receiving ongoing therapy with sunitinib (intermittent schedule) and ten patients were still alive. Fourteen patients received at least a subsequent line of therapy: seven with everolimus, three with axitinib, two with sorafenib, one with nivolumab, and one received chemotherapy. Seven of them received a further treatment line.
3.2 Treatment
The median time from the beginning of therapy to the schedule switch was 2.9 months (range 1.4–16.5), while the median duration of treatment after the switch was 9.2 months (range 0.5–32.4). Figure 2 shows the duration of therapy with each schedule. Overall, a median of nine sunitinib cycles (range 2–25) were administered. The median number of cycles with the 4/2 schedule was 2 (range 1–11), while the median number of cycles with the intermittent alternative schedule was 5 (range 1–20).

Overall and by schedule duration of sunitinib therapy for each patient (months). *Therapy ongoing at last follow-up
Table 2 shows delivered treatment, dose reductions, and delays. The number of patients who had a dose reduction was the same with both schedules. After the switch to the alternative schedule, there was an increase in treatment delays from 28 to 48% of patients. However, it should be considered that the overall number of administered cycles was higher with the modified schedule, resulting in an increased overall probability of toxicity. Correcting the number of delays according to the number of cycles, the rate of delays was 10% with the classical schedule and 11% with the alternative one. The average effective daily dose intensity with the 4/2 schedule was 46.1 mg and it was similar with the alternative schedule (44.3 mg). The average relative dose intensity with the 4/2 schedule was 92.3% and with the modified schedule was 88.7%.
3.3 Toxicities
Table 3 shows toxicity modifications. Toxicities more often observed, with both schedules, were asthenia (88%), diarrhea (76%), loss of appetite (76%), thrombocytopenia (76%), leukopenia (76%), neutropenia (68%), anemia (68%), arterial hypertension (68%), cutaneous toxicity (64%), pain (60%), stomatitis (56%), bleeding (56%), hypophosphatemia (40%), hypothyroidism (40%), and recurrence of disease’s symptoms during the rest period (28%). Other reported disorders were dysgeusia, nausea, vomiting, fever, peripheral edema, and increases in transaminases and bilirubin. We recorded three episodes of grade 4 toxicity (which may have life-threatening consequences): one episode of anemia (during treatment with the modified schedule) for which transfusions were needed (also owing to the disease itself); arterial hypertension (during treatment with the 4/2 schedule); and thrombocytopenia (during treatment with the 4/2 schedule). Hypertension and thrombocytopenia did not recur after the switch; a patient who previously experienced grade 4 hypertension reached a good pressure control with the modified schedule, with such a toxicity reduction to allow the discontinuation of anti-hypertensive therapy.
Figure 3 shows the observed toxicities. The overall toxicity rate with the traditional schedule was 60%, whilst with the modified schedule it was 48%. There was a slight decrease of grade 1–2 toxicities (−8.7%) after the switch. Of note, grade 3–4 toxicities were significantly lower with the alternative intermittent schedule (−44.1%) and the rate of patients who obtained at least one grade toxicity reduction after the switch was 48.7%. The most significant toxicity reductions concerned arterial hypertension (−82%), stomatitis (−71%), and cutaneous toxicity (−69%). Further clinically meaningful data were represented by the clear reduction in the ‘on-off symptoms’ (−86%) of the disease.

Rate of patients with toxicities according to schedule
3.4 Clinical Activity
During treatment with the alternative schedule, the best response was represented by partial response for ten patients (40%), stable disease for ten patients (40%), PD for five patients (20%), with a total disease control rate of 80%. No patient had PD at the time of the switch proposal and therefore this population was positively selected.
Overall, the median PFS was 16.4 months (95% CI 11.3–21.5) (Fig. 4a). Of note, 75 and 27% of patients were alive without disease progression after 12 and 24 months, respectively. The median PFS according to the IMDC score (Fig. 4b) was 23.4 months for good-risk patients (95% CI 15.3–31), with 83 and 50% of patients alive without progression after 12 and 24 months, respectively; 16.1 months for intermediate-risk patients (95% CI 13.1–19.0), with 77 and 23% of patients alive and without progression after 12 and 24 months, respectively; and 5.6 months for poor-risk cases (95% CI 0–12.0), with 40% of patients alive and without progression after 12 months. Additionally, the MSKCC score stratified patients as expected, but without achieving a statistically significant difference. The median PFS according to the histological subtype was: 16.4 months (95% CI 10.7–22.0) for clear cell (and mixed) carcinoma; 22.4 months (95% CI 11.9–32.8) for papillary carcinoma; and 5.6 months (95% CI 0.7–10.4) for the others (carcinomas with TFE3 translocation and undifferentiated carcinomas).

Kaplan–Meier curves. Overall progression-free survival (PFS), from starting the sunitinib 4/2 schedule (a); PFS according to Heng score (blue line good risk; green line intermediate risk: orange line poor risk) (b); and overall survival (c)
The median OS was 41.3 months (95% CI 18.6–63.9) (Fig. 4c). Of note, 83, 61, and 28% of patients were alive at 12, 24, and 60 months, respectively. In this case as well, IMDC and MSKCC score stratification worked as expected, but without achieving a statistically significant difference.
4 Discussion
In this small retrospective study, we investigated the feasibility, safety, and activity of a new alternative intermittent administration schedule of sunitinib for patients with mRCC. At our institutions, patients with non-progressive mRCC who had at least a grade 2 toxicity, or reporting on-off symptoms, during treatment with the 4/2 schedule of sunitinib, could switch to the modified schedule to improve tolerability of treatment and quality of life, while maintaining the same dose intensity.
Concerning feasibility, several parameters (i.e., dose intensity measurement and number of cycles) led us to conclude that our modified schedule seems to be well tolerated and able to warrant the maintenance of a high adherence to therapy, supposedly resulting in the maintenance of anti-tumor activity. In fact, the median time from treatment start to the schedule’s switch was about 3 months and several patients received only a cycle with the traditional 4/2 schedule; the duration of treatment and the median number of cycles after the switch were significantly higher. The number of patients that had a dose reduction was the same with both schedules (12%). The dose could be reduced during therapy with the 4/2 schedule but, in our opinion, this possibility did not affect the results of the analysis because the schedule change was done without modifying the ongoing dose for each patient at the time of the switch. After the switch to the alternative intermittent schedule, there was an increase in the absolute number of treatment delays: however, it must be considered that the longer treatment duration with the modified schedule could have resulted in an increased probability of toxicity and treatment delay. We therefore corrected the number of delays for the absolute number of cycles, and we observed that the rate of delay was comparable before and after the switch.
In this study, the average effective daily dose was 46.1 mg with the classical schedule and 44.3 mg with the alternative schedule, while the average relative dose intensity was 92.3 and 88.7%, respectively. As reported by Porta et al., in our population, OS was significantly shorter among patients with a sunitinib dose intensity below 0.7 (70%) (HR: 3.36, 95% CI 1.49–7.55), but dose intensities below 0.8 (80%) and 0.9 (90%) were not associated with significantly shorter survival times following the 24-week landmark period [12]. Thus, concerning delays, dose reductions, and dose intensity, we can assert that the performances of the two schedules are comparable. Concerning duration of treatment and number of cycles, our alternative schedule seems instead to be superior.
Interesting findings were observed about toxicity reduction. The overall rate of toxicities with the traditional schedule was greater (+12%) than with the modified schedule. Notably, after the switch to the modified schedule, there was a relevant decrease of grade 3–4 toxicities (−44.1%) and almost a half of patients obtained at least a one grade toxicity reduction. The most significant improvements concerned arterial hypertension, stomatitis, and cutaneous toxicity. Toxicity reduction was not corrected for the number of cycles; therefore, these interesting results could even be underestimated.
Of note, symptom control seems to be better with the alternative schedule, avoiding the alternate symptoms linked to the on-off schedule. Efficacy outcomes (ORR, disease control rate, PFS, and OS) of the alternative schedule are also interesting, but they should be considered with caution. In fact, they probably overestimate the activity of the modified schedule because of the retrospective nature of the study and the positive selection of patients, all without PD at the time of the switch (patients with primary refractory poor prognosis were consequently excluded). The enrolled patients had at least a grade 2 toxicity during the early cycles of therapy: this is a further selection bias because as reported in several studies, sunitinib-related AEs, in particular hypertension, are associated with improved clinical outcome in patients with mRCC, probably because of the pharmacokinetics and pharmacodynamics of the drug [26, 27].
However, we included some poor-risk patients and also two patients with a poor prognosis histology (TFE3 translocation). Moreover, ten patients (40%) were pretreated. Finally, our alternative schedule seems to provide the advantage of preventing tumor regrowth during the off-treatment period, instead observed in some cases with the standard schedule. With our modified schedule, each cycle of therapy is ‘spread’ over 6 weeks and the maximum time interval between a capsule intake and the subsequent is 72 h. Given the pharmacokinetics described above, theoretically this should not allow the drug to decrease at ineffective blood concentrations. The present study has the following limitations: data were retrospectively collected, the number of patients was limited, pharmacokinetics was not assessed, and quality of life was not measured with ad hoc tools (i.e., FACT questionnaire).
5 Conclusions
This study showed that the administration of sunitinib may be feasible, safe, and effective with our alternative intermittent schedule, maintaining the same dose intensity and activity of the standard 4/2 schedule. At the same time, the new solution seems to decrease treatment-related AEs and the regrowth phenomenon described during the rest period of the classical administration schedule. We believe that this alternative schedule deserves to be further investigated in future ad hoc prospective trials.
Change history
28 May 2018
In the Original Publication of the article, In Introduction part, 7th line, the value “5-100nM” has been published incorrectly. The correct value should read as “Plasma concentration of 50-100ng/ml”. In the Original Publication of the article, page 591, Table 2 has been published incorrectly. The corrected table is shown in the following page
References
Guida FM, Santoni M, Conti A, Burattini L, Savini A, Zeppola T, Cascinu S, Tonini G, Santini D. Alternative dosing schedules for sunitinib as a treatment of patients with metastatic renal cell carcinoma. Crit Rev Oncol Hematol. 2014;92(3):208–17.
Chabner BA, Brunton L, Knollmann BC, et al. Targeted therapies: tyrosine kinase inhibitors, monoclonal antibodies, and cytokines. In: Goodman & Gilman's The Pharmacological Basis of Therapeutics. 12th Ed. New York: Mc Graw Hill; 2011.
Buti S, Donini M, Lazzarelli S, Passalacqua R. A new modified schedule of sunitinib for metastatic renal cell carcinoma: a retrospective analysis. Acta Biomed. 2012;83:88–94.
Motzer RJ, Hutson TE, Cella D. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):722–31.
Motzer RJ, Hutson TE, Olsen MR, et al. Randomized phase II trial of sunitinib on an intermittent versus continuous dosing schedule as first-line therapy for advanced renal cell carcinoma. J Clin Oncol. 2012;30(12):1371–7.
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2000;9:327–37.
Uemura H, Shinohara N, Yuasa T, et al. A phase II study of sunitinib in Japanese patients with metastatic renal cell carcinoma: insights into the treatment, efficacy and safety. J Clin Oncol. 2010;40:194–202.
Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–45.
Abrams TJ, Murray LJ, Pesenti E, et al. Preclinical evaluation of the tyrosine kinase inhibitor SU11248 as a single agent and in combination with “standard of care” therapeutic agents for the treatment of breast cancer. Mol Cancer Ther. 2003;2:1011–21.
Wolter P, Beuselinck B, Pans S, et al. Flare-up: an often unreported phenomenon nevertheless familiar to oncologists prescribing tyrosine kinase inhibitors. Acta Oncol. 2009;48:621–4.
Houk BE, Bello CL, Kang D, Amantea MA. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res. 2009;15:2497–506.
Porta C, Levy A, Hawkins R, et al. Impact of adverse events, treatment modifications, and dose intensity on survival among patients with advanced renal cell carcinoma treated with first-line sunitinib: a medical chart review across ten centers in five European countries. Cancer Med. 2014;3(6):1517–26.
Escudier B, Roigas J, Gillessen S, et al. Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27(25):4068–75.
Barrios CH, Hernandez-Barajas D, Brown MP, et al. Phase II trial of continuous once-daily dosing of sunitinib as first-line treatment in patients with metastatic renal cell carcinoma. Cancer. 2012;118(5):1252–9.
Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer. 2014;50(6):1084–9.
Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes in patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol. 2014;191(3):611–8.
Bjarnason GA, Khalil B, Hudson JM, et al. Outcomes in patients with metastatic renal cell cancer treated with individualized sunitinib therapy: correlation with dynamic microbubble ultrasound data and review of the literature. Urol Oncol. 2014;32(4):480–7.
Neri B, Vannini A, Brugia M, et al. Biweekly sunitinib regimen reduces toxicity and retains efficacy in metastatic renal cell carcinoma: a single-center experience with 31 patients. Int J Urol. 2013;20(5):478–83.
Kondo T, Takagi T, Kobayashi H, et al. Superior tolerability of altered dosing schedule of sunitinib with 2-weeks-on and 1-week-off in patients with metastatic renal cell carcinoma: comparison to standard dosing schedule of 4-weeks-on and 2-weeks-off. J Clin Oncol. 2014;44(3):270–7.
Lee JL, Kim MK, Park I, et al. Randomized phase II trial of sunitinib four weeks on and two weeks off versus two weeks on and one week off in metastatic clear-cell type renal cell carcinoma: RESTORE trial. Ann Oncol. 2015;26(11):2300–5.
Pan X, Huang H, Huang Y, et al. Sunitinib dosing schedule 2/1 improves tolerability, efficacy, and health-related quality of life in Chinese patients with metastatic renal cell carcinoma. Urol Oncol. 2015;33(6):268.e9–15.
Bracarda S, Iacovelli R, Boni L, et al. Sunitinib administered on 2/1 schedule in patients with metastatic renal cell carcinoma: the RAINBOW analysis. Ann Oncol. 2015;26(10):2107–13.
Kaltra S, Rini BI, Jonasch E. Alternate sunitinib schedules in patients with metastatic renal cell carcinoma. Ann Oncol. 2015;26(7):1300–4.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Buti S, Lazzarelli S, Chiesa MD, et al. Dose-finding trial of a combined regimen with bevacizumab, immunotherapy, and chemotherapy in patients with metastatic renal cell cancer: an Italian Oncology Group for Clinical Research (GOIRC) study. J Immunother. 2010;33:735–41.
Rixe O, Billemont B, Izzedine H. Hypertension as a predictive factor of sunitinib activity. Ann Oncol. 2007;18(6):1117.
Rini B, Cohen DP, Chen I, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763–73.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the conduct of this study.
Conflict of interest
Sebastiano Buti, Maddalena Donini, Melissa Bersanelli, Alessia Gattara, Francesco Leonardi, and Rodolfo Passalacqua have no conflicts of interest directly relevant to the content of this study.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of our institutional research committees and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Because it was a retrospective study, a formal consent was not required. Despite this, each enrolled patient signed a standard informed consent before receiving sunitinib therapy.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Buti, S., Donini, M., Bersanelli, M. et al. Feasibility, Safety, and Efficacy of an Alternative Schedule of Sunitinib for the Treatment of Patients with Metastatic Renal Cell Carcinoma: A Retrospective Study. Drugs R D 17, 585–596 (2017). https://doi.org/10.1007/s40268-017-0209-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0209-5