
Abstract
Background
Fluid retention is a common adverse event in patients who receive endothelin (ET) receptor antagonist therapy, including the highly selective ETA receptor antagonist, atrasentan.
Objective
We performed longitudinal assessments of thoracic bioimpedance in patients with type 2 diabetes mellitus and nephropathy to determine whether a decrease in bioimpedance accurately reflected fluid retention during treatment with atrasentan.
Study Design
We conducted a randomized, double-blind, placebo-controlled study in 48 patients with type 2 diabetes mellitus and nephropathy who were receiving stable doses of renin angiotensin system inhibitors and diuretics.
Methods
Patients were randomized 1:1:1 to placebo, atrasentan 0.5 mg, or atrasentan 1.25 mg once daily for 8 weeks. Thoracic bioimpedance, vital signs, clinical exams, and serologies were taken at weeks 1, 2, 4, 6, and 8, with the exception of serum hemoglobin, which was not taken at week 1, and serum brain natriuretic peptide, which was only taken at baseline, week 4, and week 8.
Results
Alterations in bioimpedance were more often present in those who received atrasentan than in those who received placebo, though overall differences were not statistically significant. Transient declines in thoracic bioimpedance during the first 2 weeks of atrasentan exposure occurred before or during peak increases in body weight and hemodilution (decreased serum hemoglobin).
Conclusions
We conclude that thoracic bioimpedance did not reflect changes in weight gain or edema with atrasentan treatment in this study. However, the sample size was small, and it may be of interest to explore the use of thoracic bioimpedance in a larger population to understand its potential clinical use in monitoring fluid retention in patients with chronic kidney disease who receive ET receptor antagonists.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Thoracic bioimpedance did not reflect changes in weight or the development of edema in patients with type 2 diabetes mellitus and nephropathy treated with atrasentan. |
Thoracic bioimpedance should be tested in a larger population to understand its potential clinical use in assessing fluid retention in patients with chronic kidney disease who receive endothelin receptor antagonists. |
1 Introduction
Endothelin (ET) receptor antagonists are being used or investigated in disease states that result from pathological increases in activity of the ET system [1,2,3,4,5]. Atrasentan is a potent ETA receptor antagonist [6] that reduces proteinuria in patients with type 2 diabetes mellitus and nephropathy [1, 4]. Currently, atrasentan is in late clinical development as a treatment for diabetic nephropathy [7].
In clinical studies, the most frequent adverse events associated with ET receptor antagonists are weight gain, edema, and congestive heart failure (CHF) [8,9,10]. Identifying the extent of peripheral edema is the most commonly used method to assess fluid retention, but it poorly quantifies the amount of fluid retained and therefore has low specificity for predicting CHF. Thoracic bioimpedance is an approved noninvasive method to quantify fluid accumulation in a wide variety of clinical conditions, including CHF [11,12,13,14,15,16]. In settings of early or ongoing CHF, reduced thoracic bioimpedance is a sensitive indicator of pulmonary fluid accumulation even before the development of CHF symptoms [17].
This study evaluated the longitudinal effect of two low doses of atrasentan on fluid retention as measured by different parameters in patients with type 2 diabetes mellitus with nephropathy who were receiving maximum tolerated labeled doses of renin angiotensin system (RAS) inhibitors. The aims were to assess whether atrasentan increased thoracic fluid accumulation as measured by bioimpedance and to determine whether changes in bioimpedance correlated with changes in weight, peripheral edema, or diuretic usage.
2 Methods
2.1 Study Design
This was a phase IIb, randomized, double-blind, placebo-controlled, parallel-group, multi-center study to examine the safety of two doses of atrasentan in patients with type 2 diabetes mellitus with nephropathy who were currently receiving the maximum tolerated labeled daily dose of a RAS inhibitor.
2.2 Patients
A total of 48 subjects were randomized 1:1:1 to placebo, atrasentan 0.5 mg, or atrasentan 1.25 mg once daily for 8 weeks. This study included doses of atrasentan within a previously defined range of doses that established the proof of concept for the ability of atrasentan to reduce urine albumin excretion [4]. A sample size of 48 was determined to have about 85% power to detect the overall treatment group differences in urine albumin excretion after 8 weeks of treatment with a one-sided significance level of α = 0.050. Inclusion criteria included the presence of type 2 diabetes mellitus, a urinary albumin:creatinine ratio (UACR) ≥300 and ≤3500 mg/g determined from the geometric mean of two first morning void urine specimens, and current treatment with a RAS inhibitor. The main exclusion factors were a history of moderate or severe peripheral edema, facial edema, pulmonary edema, pulmonary hypertension, or heart failure as defined by the American College of Cardiology/American Heart Association practice guidelines [18]. Subjects with a serum brain natriuretic peptide (BNP) concentration of ≥200 pg/ml at screening were also excluded.
After a screening period of up to 14 days, patients entered a run-in period of 4 weeks, during which they had to maintain an estimated glomerular filtration rate (eGFR) ≥30 and ≤75 ml/min/1.73 m2 and systolic blood pressure (SBP) ≥110 and ≤180 mmHg. For those in whom the dose of RAS inhibitor was not maximal, the run-in period was extended to 8–12 weeks to allow a stable period of 4 weeks at the maximum tolerated level of RAS inhibition. For entry into the treatment period, subjects had to receive an unadjusted dose of RAS inhibitor, UACR had to be ≥200 mg/g, and SBP had to be ≥110 and ≤180 mmHg. Diuretics were given unless medically contraindicated. Placebo or atrasentan treatment was then given for 8 weeks, after which it was withdrawn, and a follow-up study visit occurred 30 days later.
Vital signs, clinical exams, thoracic bioimpedance, serologies, and urine samples (for UACR) were taken at each study visit (week 1, 2, 4, 6, and 8 after randomization) with the exception of serum hemoglobin, which was not taken at week 1, and serum BNP, which was only taken at baseline, week 4, and week 8. Thoracic bioimpedance measurements were performed onsite using the ZOE® Fluid Status Monitor (Noninvasive Medical Technologies, Las Vegas, NV, USA) following supervised training from the manufacturer using a standardized method for all centers. The presence of thoracic fluid was estimated by the inverse relationship between fluid accumulation and reduced impedance (Ohms). The precision of thoracic bioimpedance measurements in healthy subjects is ±4% [17].
2.3 Statistical Methods
All analyses were conducted on the intention-to-treat (ITT) population, comprising subjects who were randomized and received at least one dose of study drug; data were analyzed on the randomly assigned treatment group. Differences between treatment groups for baseline characteristics and demographics were tested using one-way analysis of variance (ANOVA) or Fisher’s exact test. Changes between baseline and post-baseline visits on continuous measures were analyzed using a repeated measures analysis model. The association between a set of subjects’ baseline demographics and disease characteristics and the incidence of edema was evaluated using a logistic regression model.
3 Results
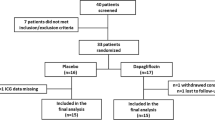
Of 199 patients screened, 48 met inclusion criteria and were randomized (Fig. 1). Each of the three groups contained 16 patients, and 15 (93.8%), 14 (87.5%), and 11 (68.8%) patients completed the study in the placebo, atrasentan 0.5 mg, and atrasentan 1.25 mg groups, respectively. The major reason for discontinuing from the study was adverse events. As shown in Table 1, there was a statistically significant difference between treatment groups for age, with a higher mean age in the atrasentan groups (mean age 69 ± 8 years in the atrasentan 0.5 mg group and 69 ± 7 in the atrasentan 1.25 mg group) than in the placebo group (mean age 62 ± 9 years; P = 0.022). There was also a statistically significant difference between groups for sex, with more males than females in the placebo group (87.5%) and the atrasentan 0.5 mg group (87.5%; P = 0.012).

Patient disposition
Other demographics and baseline characteristics were statistically similar among treatment groups (Table 1).
There were no changes in mean weight in the placebo group during the treatment period, whereas subjects in the two atrasentan groups experienced overall significant placebo-adjusted weight increases from baseline (Fig. 2a). Repeated measures analysis revealed placebo-adjusted weight gains of 1.7 and 1.6 kg in the atrasentan 0.5 and 1.25 mg groups, respectively. Most of the increases in weight occurred by the second week of treatment (placebo-adjusted weight gains of 1.5 and 2.3 kg in the 0.5- and 1.25-mg groups, respectively, at week 2; P ≤ 0.005). Weight returned to baseline levels in each of the atrasentan groups by the 30-day post-treatment visit.

Effect of atrasentan on weight, serum hemoglobin, incidence of peripheral edema and thoracic bioimpedance. 1 P<0.01, 2 P<0.02, 3 P<0.05 for change from baseline vs. placebo change from baseline. a Weight (kg); b serum hemoglobin (g/dl); c cumulative incidence of peripheral edema (%); d thoracic bioimpedance (Ohms). SE standard error
Patients in both atrasentan groups also had significant declines in serum hemoglobin compared with placebo (Fig. 2b), with values returning to baseline after drug discontinuation. Overall, there was a decrease of 0.47 g/dl in the 0.5-mg group (P = 0.017) and a decrease of 0.84 g/dl in the 1.25-mg group (P < 0.001) after placebo-adjusted repeated measures analysis. The nadir in hemoglobin occurred at 4 weeks in the 0.5-mg group (0.69 g/dl decrease, P < 0.001 vs. placebo) and at 6 weeks in the 1.25-mg group (0.90 g/dl decrease, P < 0.001 vs. placebo).
There was an increase in the cumulative incidence of peripheral edema in all groups, with the 1.25-mg group showing the largest increase (44%) by week 4 (Fig. 2c). The majority of cases of peripheral edema were regarded as mild, and only one patient discontinued the study because of edema (moderate). The mean duration of edema was similar in the placebo and 0.5-mg groups (21 and 25 days, respectively) and was longest in the 1.25-mg group (37 days). The development of edema was not associated with sex, age, race, baseline UACR, or chronic kidney disease (CKD) stage by logistics regression analysis. Diuretic use [81–97%, P = not significant (NS)] and type of diuretic (loop, 38–56%; distal, 35–44%) were equally distributed among the groups, and very few patients had a change in diuretic dose during the treatment period (one in placebo, none in the 0.5-mg group, and two in the 1.25-mg group).
Baseline thoracic bioimpedance measurements did not differ among the groups (Fig. 2d). Bioimpedance within the placebo group showed minor fluctuations from baseline (0.5–1.1 Ohms) throughout the treatment period. In contrast, the atrasentan groups showed mean reductions of 1.7 and 2.0 Ohms by week 1 (0.5 mg) and week 2 (1.25 mg), respectively, amounting to nadir mean declines of 7 and 11% from baseline, respectively. Following these declines, both atrasentan groups showed increases in bioimpedance of 16 and 21% at weeks 2 and 4 from their nadir, with values returning toward baseline by 6 and 8 weeks, respectively. Despite these fluctuations within the groups receiving atrasentan, the overall change in bioimpedance values was not significantly different from placebo when analyzed with a repeated measures model.
There were no significant differences in serum creatinine, eGFR, or BNP levels among the three groups during the treatment period. However, serum BNP levels did show a 25% increase from baseline at week 4 and a 46% increase at week 8 (P = 0.06) in the 1.25-mg group (Fig. 3). There were no differences in mean SBP among the groups, whereas the mean diastolic blood pressure was significantly reduced in the 1.25-mg group (−7.1 mmHg; P = 0.004 vs. placebo). Both doses of atrasentan reduced UACR from baseline by 28% (0.5 mg) and 35% (1.25 mg) within 2 weeks (P < 0.05 vs. placebo). These atrasentan-induced reductions were sustained over the 8 weeks of treatment, with values returning to baseline after 30 days off study drug, similar to findings in previous studies [1, 4].

Effect of atrasentan on serum brain natriuretic peptide (BNP) levels. 1 P = 0.06 from change from baseline vs. placebo change from baseline
The incidence of AEs was similar across all treatment groups (Table 2). The most frequently reported AE was peripheral edema. More patients in the 1.25-mg group experienced serious AEs, with three patients in this group discontinuing because of an AE. One patient in the 1.25-mg group also experienced CHF after being hospitalized for headache. The CHF event occurred concurrently with pulmonary hypertension and coronary artery stenosis, 7 days after the last dose of atrasentan at treatment week 4 when mild peripheral edema was present. This patient’s bioimpedance values (Ohms) were as follows: 30.1 (baseline), 16.3 (week 1), 25.4 (week 2), and 26.2 (week 4).
4 Discussion
To our knowledge, this is the first longitudinal assessment of thoracic bioimpedance in patients with diabetes and nephropathy who were receiving an ETA receptor antagonist in combination with RAS inhibitors and diuretics. Overall, there were no significant differences in bioimpedance between the treatment groups and those receiving placebo, although early decreases in the atrasentan groups (7–11% decline) by week 2 were observed to coincide with the initial increases in body weight and declines in serum hemoglobin. Because these impedance changes were transient and not associated with pulmonary symptoms of fluid retention, the clinical relevance of these findings is unclear. However, recent studies in patients with acute myocardial infarction showed that a 12% reduction in thoracic bioimpedance was associated with pulmonary edema [19], suggesting that the early bioimpedance changes in our study, if sustained, could have become clinically apparent.
Because the majority of patients in this study were receiving diuretics before being randomized, it is possible they were partially protected against the potential fluid-retentive effects of atrasentan. Relevant studies in patients with CHF have demonstrated that furosemide treatment resulted in an 8% increase in thoracic bioimpedance, which correlated with increased urinary output (P = 0.004) [20]. It is also possible that endogenous BNP may have had a diuretic effect in this study. For example, patients receiving atrasentan 1.25 mg had a 25 and 46% increase in serum BNP at 4 and 8 weeks post-baseline, respectively, which may have resulted from an earlier increase in fluid retention induced by atrasentan. In this hypothetical scenario, the putative early increase in pulmonary fluid, identified by the 11% decrease in bioimpedance (1.25-mg group), could have stimulated cardiac production of BNP, which induced a diuresis that resolved further pulmonary fluid accumulation. The subsequent 21% rise in thoracic bioimpedance from nadir values would correlate with a decrease in pulmonary fluid. While the diuretic effect of exogenous BNP has been described in healthy individuals [21, 22], future studies are required to more fully assess a potential diuretic effect of endogenous BNP in response to ET receptor antagonist therapy in patients with diabetic nephropathy.
These results confirms those from earlier studies showing that atrasentan reduces serum hemoglobin and albuminuria while causing weight gain and peripheral edema in patients with diabetic nephropathy [1, 4]. The atrasentan doses for this study were chosen to minimize the risk for edema and, while the group receiving the higher atrasentan dose in this study had the most patients affected by edema (50%), a relevant number of patients in the placebo group (30%) also developed edema by the sixth week of treatment. This calls into question the clinical finding of mild peripheral edema as it applies to a CKD population. These results also confirm that most patients who receive atrasentan do not develop peripheral edema, at least in short-term treatment protocols. Importantly, the patients in this and other recent studies with atrasentan have been highly selected for not having a past history of CHF and for having serum BNP levels <200 pg/ml [1, 4]. Thus, the potential for atrasentan to cause fluid retention in an unselected population of patients with diabetes and nephropathy remains undefined.
Limitations of this study include the small sample size, the short duration of treatment, the absence of direct measures of pulmonary fluid retention, and the limited range of atrasentan doses tested. In addition, generalizability to a larger population of patients with diabetes and CKD is substantially diminished by the high screen fail rate for entry into the study. Despite these limitations, the frequent longitudinal measurements of thoracic bioimpedance enabled the detection of hypothetical physiological interactions induced by atrasentan that should be tested in a larger patient population, particularly with respect to the effects on patient well-being.
References
de Zeeuw D, Coll B, Andress D, Brennan JJ, Tang H, Houser M, et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol. 2014;25(5):1083–93.
Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–9.
Khanna D, Denton CP, Merkel PA, Krieg T, Le Brun FO, Marr A, et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 randomized clinical trials. JAMA. 2016;315(18):1975–88.
Kohan DE, Pritchett Y, Molitch M, Wen S, Garimella T, Audhya P, et al. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J Am Soc Nephrol. 2011;22(4):763–72.
Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903.
Opgenorth TJ, Adler AL, Calzadilla SV, Chiou WJ, Dayton BD, Dixon DB, et al. Pharmacological characterization of A-127722: an orally active and highly potent ETA-selective receptor antagonist. J Pharmacol Exp Ther. 1996;276(2):473–81.
Study of diabetic nephropathy with atrasentan (SONAR). https://clinicaltrials.gov/ct2/results?term=01858532&Search=Search. Accessed 18 July 2016.
Krum H, Viskoper RJ, Lacourciere Y, Budde M, Charlon V. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. Bosentan Hypertension Investigators. N Engl J Med. 1998;338(12):784–90.
Mann JF, Green D, Jamerson K, Ruilope LM, Kuranoff SJ, Littke T, et al. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21(3):527–35.
Weber MA, Black H, Bakris G, Krum H, Linas S, Weiss R, et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9699):1423–31.
Hung SC, Kuo KL, Peng CH, Wu CH, Lien YC, Wang YC, et al. Volume overload correlates with cardiovascular risk factors in patients with chronic kidney disease. Kidney Int. 2014;85(3):703–9.
Kotanko P, Levin NW, Zhu F. Current state of bioimpedance technologies in dialysis. Nephrol Dial Transplant. 2008;23(3):808–12.
Kushner RF. Bioelectrical impedance analysis: a review of principles and applications. J Am Coll Nutr. 1992;11(2):199–209.
Milzman D, Napoli A, Hogan C, Zlidenny A, Janchar T. Thoracic impedance vs. chest radiograph to diagnose acute pulmonary edema in the ED. Am J Emerg Med. 2009;27(7):770–5.
Packer M, Abraham WT, Mehra MR, Yancy CW, Lawless CE, Mitchell JE, et al. Utility of impedance cardiography for the identification of short-term risk of clinical decompensation in stable patients with chronic heart failure. J Am Coll Cardiol. 2006;47(11):2245–52.
Tsai YC, Tsai JC, Chen SC, Chiu YW, Hwang SJ, Hung CC, et al. Association of fluid overload with kidney disease progression in advanced CKD: a prospective cohort study. Am J Kidney Dis. 2014;63(1):68–75.
Sageman WS. Reliability and precision of a new thoracic electrical bioimpedance monitor in a lower body negative pressure model. Crit Care Med. 1999;27(9):1986–90.
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119(14):e391–479.
Shochat M, Shotan A, Blondheim DS, Kazatsker M, Dahan I, Asif A, et al. Usefulness of lung impedance-guided pre-emptive therapy to prevent pulmonary edema during ST-elevation myocardial infarction and to improve long-term outcomes. Am J Cardiol. 2012;110(2):190–6.
Freimark D, Arad M, Sokolover R, Zlochiver S, Abboud S. Monitoring lung fluid content in CHF patients under intravenous diuretics treatment using bio-impedance measurements. Physiol Meas. 2007;28(7):S269–77.
Holmes SJ, Espiner EA, Richards AM, Yandle TG, Frampton C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J Clin Endocrinol Metab. 1993;76(1):91–6.
van der Zander K, Houben AJ, Hofstra L, Kroon AA, de Leeuw PW. Hemodynamic and renal effects of low-dose brain natriuretic peptide infusion in humans: a randomized, placebo-controlled crossover study. Am J Physiol Heart Circ Physiol. 2003;285(3):H1206–12.
Author information
Authors and Affiliations
Contributions
AbbVie and DdZ designed the study. AbbVie executed the study with the help of the Steering Committee (DdZ, H-H P, HJLH, RC-R, DK, HM, VP, GR, SWT, RT). AbbVie and HJLH performed statistical analyses. PP contributed to recruiting patients. DJW, DdZ and HLH interpreted the trial results and wrote the first draft of the manuscript. All other authors (including AbbVie) assisted in trial execution, interpretation of trial results, and edited several versions of the manuscript.
Corresponding author
Ethics declarations
Ethical standards
The study was conducted in accordance with the International Conference on Harmonisation guidelines, applicable regulations, and the principles of the Declaration of Helsinki. The study protocol was approved by an independent ethics committee or institutional review board.
Informed consent
All patients provided written informed consent.
Funding
AbbVie funded the study and was responsible for the study design, research, analysis, and data collection. The authors were responsible for interpretation of data, for writing, reviewing, and approving the manuscript and for determining the final content. No payments were made to the authors for writing this paper. Editorial support was provided by Richard M. Edwards, PhD, of Complete Publication Solutions, LLC (North Wales, PA, USA), a CHC Group company, and was funded by AbbVie.
Conflict of interest
DJW is a consultant for AbbVie and has been a member of the independent data monitoring boards for the SONAR (Study Of diabetic Nephropathy with AtRasentan) and RADAR (Reducing residual Albuminuria in subjects with Diabetes and nephropathy With AtRasentan) clinical trials. RB serves on the speakers’ bureau of AbbVie, Merck, Bristol-Myers Squibb, Novo Nordisk, Daiichi Sankyo, Takeda, Eli Lilly, and Sanofi. PP is a consultant for AbbVie. DdZ is a consultant for and receives honoraria (to employer) from AbbVie, Astellas, AstraZeneca, Chemocentryx, J&J, Hemocue, Novartis, Reata, Takeda, and Vitae. RC-R has consultancy agreements with Roche, AbbVie, Amgen, AstraZeneca, and Boehringer Ingelheim; in the last 2 years he has served on the speakers’ bureau of Amgen, Roche, and Sanofi, and received grant support from Fibrogen and Janssen. DK is a consultant for AbbVie. HJLH is a consultant for and receives honoraria (to employer) from AbbVie, Astellas, AstraZeneca, Boehringer Ingelheim, Fresenius, and Janssen. HM is a consultant for AbbVie and Teijin; receives speaker honoraria from Astellas, Boehringer Ingelheim, Daiichi Sankyo, Dainippon Sumitomo, Kyowa Hakko Kirin, MSD, Pfizer, Takeda, and Tanabe Mitsubishi; and receives grant support from Astellas, Boehringer Ingelheim, Daiichi Sankyo, Dainippon Sumitomo, Kowa, Kyowa Hakko Kirin, MSD, Novartis, Novo Nordisk, Ono, Otsuka, Pfizer, Taishyo-Toyama, Takeda, Teijin, and Tanabe Mitsubishi. VP is a consultant for AbbVie, Astellas, AstraZeneca, Boehringer, Janssen, Roche, Servier, Merck, and Vitae. His employer receives funding/contracts for clinical trials from AbbVie, Baxter, Fresenius, Novartis, Pfizer, Resmed, Roche, Janssen, and Servier. YP is a former employee of Abbott and owns AbbVie stocks. GR is a consultant for Alexion, Reata, Bayer, Novartis, and AbbVie. All compensations are paid to his institution for research and educational activities. SWT is a consultant for AbbVie and received honoraria for academic talks from Servier. He is an investigator on both contract and investigator-initiated research projects with AstraZeneca, Bristol-Myers Squibb, Mitsubishi, and Pfizer. RT serves on advisory boards for Boehringer Ingelheim, Amgen, Relypsa, ZS Pharma, Bayer, Quintiles, and AstraZeneca. H-HP is a consultant for AbbVie. BC, DA, JJB, and MH are all AbbVie employees and own AbbVie stock.
Additional information
ClinicalTrials.gov identifier NCT01858532.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Webb, D.J., Coll, B., Heerspink, H.J.L. et al. Longitudinal Assessment of the Effect of Atrasentan on Thoracic Bioimpedance in Diabetic Nephropathy: A Randomized, Double-Blind, Placebo-Controlled Trial. Drugs R D 17, 441–448 (2017). https://doi.org/10.1007/s40268-017-0201-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0201-0