
Abstract
Background
Tacrolimus (TAC) is an immunosuppressive macrolide that blocks T-cell activation by specifically inhibiting calcineurin. TAC was approved in Japan for the treatment of rheumatoid arthritis (RA) in 2005. However, the safety and effectiveness of TAC adding on to biological disease-modifying anti-rheumatic drugs (DMARDs) in the real clinical setting may not be clear enough.
Objectives
We report here the interim results of post marketing surveillance (PMS) of TAC adding on to biological DMARDs in RA patients who failed to show an adequate response to biological DMARDs.
Methods
Patients who had an inadequate response to biological DMARDs were enrolled. An inadequate response to biological DMARDs was defined as that all of the following conditions were met: a Simplified Disease Activity Index (SDAI) score of >3.3 when TAC was started; both the tender joint count and swollen joint count were the same or increased compared with those at 4–8 weeks prior to TAC; and biological DMARDs were used for at least 8 weeks prior to TAC. This study was conducted in compliance with the ministerial ordinance on “Good Post-Marketing Study Practice” (GPSP).
Results
The safety data collection and evaluation for 172 patients and effectiveness data collection and evaluation for 165 patients were reported. The mean age was 61.9 years. Adverse drug reactions occurred in 18 patients. The mean SDAI decreased from 20.1 at baseline to 11.7 at week 24.
Conclusions
TAC is well tolerated and effective when added on to biological DMARDs in RA patients who failed to achieve an adequate response to biological DMARDs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
It has been reported that biological disease-modifying anti-rheumatic drugs (DMARDs) fail to achieve adequate suppression of disease activity and that the effectiveness of biological DMARDs is reduced in some patients. |
Tacrolimus (TAC) adding on to biological DMARDs in patients who failed to achieve an adequate response to biological DMARDs was well tolerated and improved the clinical outcome. |
This study suggests that TAC in combination with biological DMARDs can be one of the options in patients with rheumatoid arthritis who failed to show an adequate response to biological DMARDs in the real clinical setting. |
1 Introduction
Rheumatoid arthritis (RA) is characterized by chronic polyarthritis and primary inflammation of joint synovium, and causes joint dysfunction because of destruction of bones and joints [1]. Inflammatory cytokines, such as Tumor Necrosis Factor-α (TNF-α), Interleukin-1 (IL-1), and IL-6, are produced in synoviocytes of RA, and these cytokines play important roles in the onset and progression of RA [2–5]. Tacrolimus (TAC) is an immunosuppressive macrolide discovered as a metabolite of Streptomyces tsukubaensis, an actinomycete, at the laboratory of Fujisawa Pharmaceutical Co., Ltd. (currently, Astellas Pharma Inc.) in 1984. TAC specifically blocks T-cell activation through inhibition of calcineurin, a dephosphorylation enzyme, and shows immunosuppressant effects including suppression of production of inflammatory cytokines and production of antibodies from B cells. Studies reported that TAC improved arthritis symptoms [6–9], and inhibited maturation and differentiation of osteoclasts [10] with these properties. TAC was approved for the indication of “RA (only in a case of inadequate response to conventional therapies)” in Apr 11, 2005 [10–14]. The subsequently conducted drug use-results survey [15], long-term specific drug use-results survey, and post-marketing clinical study [16] confirmed the safety and efficacy of TAC. Studies reported the recent use of add-on TAC to patients with RA with inadequate responses to methotrexate (MTX) and patients with RA who could not use a sufficient dose of MTX because of adverse drug reactions (ADRs) and complications [17, 18]. Although reduction of clinical symptoms used to be the therapeutic goal in the past, the emergence of biological disease-modifying anti-rheumatic drugs (DMARDs) has made remission an achievable goal in recent RA treatment. On the other hand, biological DMARDs fail to achieve adequate suppression of disease activity or the efficacy of biological DMARDs is reduced in some patients. Dose increase of biological DMARDs, switch to other biological DMARDs, and add-on use or dose increase of conventional anti-rheumatic drugs are therapeutic options for such patients, and there are studies reporting the usage of add-on TAC [19–23]. Infliximab and etanercept were the only marketed biological DMARDs at the time of the drug use-results survey and long-term specific drug use-results survey of TAC, and few patients were using TAC adding on to these two drugs [15]. Therefore, the safety and efficacy of TAC adding on to biological DMARDs have not been sufficiently confirmed in the real clinical setting. A specific drug use-results survey of TAC (clinicaltrials.gov identifier: NCT01870908) was conducted to evaluate the safety and effectiveness of add-on TAC in patients with RA who failed to show an adequate response to biological DMARDs. We report here the interim results of 172 patients analyzed for safety and 165 patients analyzed for effectiveness among 175 patients who had completed the observation period and had their data collected before October 2013.
2 Methods
2.1 Survey Patients
The survey was conducted in patients with RA who failed to show an adequate response to biological DMARDs. An inadequate response to biological DMARDs was defined if all of the following conditions were met: a Simplified Disease Activity Index (SDAI) [24] score of >3.3 when TAC was started; both the tender joint count (TJC) and swollen joint count (SJC) were the same or increased compared with those at 4–8 weeks prior to the start of TAC; and biological DMARDs were used for at least 8 weeks prior to the start of TAC.
2.2 Survey Methods
The survey employs a central registration method, and patients who are administered TAC after the investigator has signed off the survey agreement will be registered within 14 days after the first dose of TAC. The observation period was 24 weeks from the first day of administration of TAC, the investigation period was 2 years and 8 months, from August 2012 to March 2015, and the enrollment period was 2 years and 2 months, from August 2012 to September 2014. This survey was conducted in compliance with the ordinance on “Good Post-Marketing Study Practice” (GPSP), which was authorized by the Ministry of Health, Labour and Welfare in Japan (Ordinance No. 171, dated 20 December 2004). Institutional review board approval was obtained according to the rules of each institution as required for a post marketing surveillance (PMS) study, and a contract with all medical facilities participating in this study was constructed. Written informed consent was not obtained, as the PMS study in Japan is allowed to be conducted without informed consents.
2.3 Investigational Items and Timing
2.3.1 Patient Backgrounds
The investigational items are sex, age at the start of administration, body weight, RA duration, presence/absence of complication and name of complication, presence/absence of medical history and name of disease, Steinbrocker stage classification [25], Steinbrocker functional classification [25], rheumatoid factor [26], and anti-cyclic citrullinated peptide (anti-CCP) antibody [27].
2.3.2 Status of RA Therapy
For administration of TAC, the investigational items are daily dose, duration of treatment, and reason for discontinuation. For the status of RA therapy with other drugs, the investigational items are the dose of and duration of treatment with MTX, steroids, and biological DMARDs.
2.3.3 Safety Evaluation
The safety endpoints are presence/absence of adverse events (AEs) (including ADRs, laboratory values, and other abnormal laboratory values), AE term, date of onset, seriousness, action taken to TAC, symptomatic therapy, event outcome, date of outcome, causal relationship to TAC, and factors other than TAC. ADRs were defined as AEs in which the causal relationship to TAC was not ruled out by the investigator, and were tabulated by the System Organ Class (SOC) and Preferred Term (PT) using the Medical Dictionary for Regulatory Activities/Japanese (MedDRA/J) Version 16.1. SOCs are tabulated in the number of patients with events, and PTs are tabulated in the number of events. If events of the same PT occurred more than once in the same patient, they were counted as one event for tabulation. The following seven items were analyzed as key investigational items: “infection,” “renal impairment,” “glucose tolerance impaired,” “cardiac dysfunction,” “pancreatic dysfunction,” “psychoneurologic disorder,” and “lymphoma.”
2.3.4 Effectiveness Evaluation
The effectiveness endpoints are SDAI, Disease Activity Score 28–C-reactive protein (DAS28-CRP) [28, 29], improvement as determined by the European League Against Rheumatism (EULAR) criteria [30], modified Health Assessment Questionnaire (mHAQ) scores [31], and serum matrix metalloproteinase-3 (MMP-3) [32] levels. Because data at 4–8 weeks prior to the start of TAC were collected as historical data in this survey, observation points were set to the following four time points: 4–8 weeks prior to the start of TAC, at the start of TAC, 12 weeks after the start of TAC (week 12), and 24 weeks after the start of TAC (week 24). Each effectiveness endpoint was analyzed in patients who were evaluated at the start of TAC and after the start of TAC (at week 12 or 24), and data evaluated during treatment with TAC and during treatment with biological DMARDs, which had been used at the start of TAC, were used.
2.4 Statistical Methods
Continuous variables were presented as mean ± standard deviation (SD), and nominal variables were presented as the number of patients (%). All analyses were performed using SAS® (version 9.2) in this survey. In the effectiveness evaluation, missing values after the start of TAC were imputed using the last observation carried forward (LOCF) method for distribution of SDAI disease activity, changes in SDAI scores over time, distribution of DAS28-CRP disease activity, changes in DAS28-CRP scores over time, improvement as determined by EULAR criteria, changes in TJC over time, changes in SJC over time, changes in Visual Analog Scale (VAS) by physicians over time, changes in VAS by patients over time, changes in C-reactive protein (CRP) over time, changes in mHAQ scores over time, and changes in serum MMP-3 levels over time. The comparisons of the data at each observation point versus the start of TAC were tested with Wilcoxon signed-rank test. All p values presented were two-sided, and a significance level of 5 % was used for each comparison.
3 Results
3.1 Disposition of Patients and Patient Backgrounds
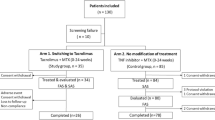
The disposition of patients is shown in Fig. 1. Of the 175 patients collected, 172 patients (excluding patients out of the scope of the survey and patients who did not come to the survey site after the first dose) were analyzed for safety, and 165 patients (excluding patients who were not evaluated for effectiveness) were analyzed for effectiveness.

Disposition of patients. One hundred seventy-five patients were collected, 172 patients were analyzed for safety, and 165 patients were analyzed for effectiveness. 1One patient who was out of the scope of the survey was also the patient who made no subsequent hospital visits after the initial administration. 2One patient had an SDAI of ≤3.3 when administration of TAC was started; one patient had (TJC at 4–8 weeks prior to the start of TAC) > (TJC at the start of administration of TAC); one patient either had a missing value for SDAI at the start of TAC or an unknown start date for administration of biological DMARDs. DMARDs disease-modifying anti-rheumatic drugs, SDAI Simplified Disease Activity Index, TAC tacrolimus, TJC tender joint count
The demographic characteristics of the 172 patients analyzed for safety are shown in Table 1. Of the patients, 151 (87.8 %) were female, and age (mean ± SD) was 61.9 ± 12.1 years, body weight (mean ± SD) 54.2 ± 10.0 kg, and RA duration (mean ± SD) 11.0 ± 8.3 years. A total of 132 patients (76.7 %) had complications; the most common complication was osteoporosis in 101 patients (58.7 %), followed by hypertension in 26 patients (15.1 %). A total of 41 patients (25.0 %) had a medical history; the most common medical history was appendicitis in four patients (2.4 %) and pneumonia in four patients (2.4 %), followed by gastric ulcer in three patients (1.8 %). The Steinbrocker stage classification was Stage III in 50 patients (30.9 %) and Stage IV in 50 patients (30.9 %). The Steinbrocker functional classification was Class 3 in 31 patients (19.4 %) and Class 4 in one patient (0.6 %). The rheumatoid factor was positive in 121 patients (80.1 %), and anti-CCP antibody was positive in 70 patients (86.4 %).
3.2 Status of RA Therapy
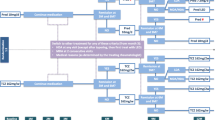
The status of administration of TAC is shown in Fig. 2. In the safety analysis set, the initial dose was 1.1 ± 0.5 mg/day, and the mean dose in the observation period was 1.3 ± 0.6 mg/day. The highest proportion was 1.0 mg/day throughout the observation period, and the dose was titrated up from the start of TAC (1.1 ± 0.5 mg/day) to week 24 (1.4 ± 0.7 mg/day). In the analysis by elderly (≥65 years) and non-elderly (<65 years) patients, the dose was titrated up from the start of TAC (elderly 1.1 ± 0.5 mg/day; non-elderly 1.2 ± 0.5 mg/day) to week 24 (elderly 1.3 ± 0.6 mg/day; non-elderly 1.5 ± 0.7 mg/day), and the mean daily dose in elderly patients was lower than that in non-elderly patients throughout the observation period (data not shown). Administration of TAC was continued up to week 24 in 87.2 % of patients, and the disposition of reason for discontinuation before week 24 was AEs in nine patients, unchanged/worsening of symptoms in nine patients, and request by patients in five patients. One patient out of “unchanged/worsening of symptoms” and one patient out of “patient’s request” are the same patient.

Status of administration of TAC from the start of TAC to week 24. The bar chart indicates distribution of dose, and the line chart indicates the mean dose. SD standard deviation, TAC tacrolimus
The status of RA therapy is shown in Table 2. Patients who had been treated with the first concomitant biological DMARD at the start of TAC showed the highest proportion, with 112 patients (65.1 %), followed by the second concomitant biological DMARD in 36 patients (20.9 %). Patients who had been using concomitant etanercept at the start of TAC showed the highest proportion, with 41 patients (23.8 %), followed by infliximab in 34 patients (19.8 %). The mean time to the first biological DMARD after onset of RA was 8.1 ± 7.6 years; “≥5 years” accounted for approximately 50 %, and “≥10 years” accounted for approximately 34.6 % (53 patients). The mean duration of treatment with biological DMARDs, which had been used at the start of TAC, before the start of TAC was 1.8 ± 1.8 years, and ≥50 % of patients had been using biological DMARDs for at least 1 year before TAC. MTX was coadministered in 116 patients (67.4 %), and steroids were coadministered in 95 patients (55.2 %). There was no significant change in the MTX dose from the start of TAC until week 24, and the mean MTX dose remained at 8.0–8.2 mg/week. Similarly, there was no significant change in the daily dose of oral steroids (prednisolone equivalent) from the start of TAC until week 24, and the mean daily dose remained at 4.1–4.4 mg/day.
3.3 Safety
3.3.1 Status of Development of ADRs
The status of development of ADRs in the 172 patients analyzed for safety is shown in Table 3. A total of 22 ADRs occurred in 18 patients (10.5 %). ADRs that occurred in at least two events were abdominal pain (two events), stomatitis (two events), and malaise (two events). Serious ADRs were herpes zoster (one event) and myocardial infarction (one event). Herpes zoster occurred at 19 days after the start of TAC, but administration of TAC was continued and the event was later resolved by an antiviral drug. The causal relationship to TAC was “unlikely related.” The patient who experienced myocardial infarction was 89 years old at the start of TAC, RA duration was 31 years, Steinbrocker stage classification was Stage IV, and Steinbrocker functional classification was Class 3. Myocardial infarction occurred at 103 days after the start of TAC, and the patient died on the same day. The causal relationship to TAC and factors other than TAC were “unknown.”
3.3.2 Key Investigational Items
“Infection,” “renal impairment,” “cardiac dysfunction,” and “glucose tolerance impaired” occurred in four patients (2.3 %), two patients (1.2 %), one patient (0.6 %), and one patient (0.6 %), respectively. “Pancreatic dysfunction,” “psychoneurologic disorder,” and “lymphoma” did not occur.
3.4 Effectiveness
The distribution of SDAI disease activity is shown in Fig. 3. The proportion of remission (SDAI ≤3.3) or low disease activity (3.3 < SDAI ≤ 11) was 46.3 % at 4–8 weeks prior to the start of TAC, 0.0 % for remission and 20.0 % for low disease activity at the start of TAC, 8.9 and 40.7 %, respectively, at week 12, and 13.3 and 45.2 %, respectively, at week 24, showing improvement. The mean (±SD) SDAI score was 13.6 ± 8.3 at 4–8 weeks prior to the start of TAC, 20.1 ± 10.3 at the start of TAC, 13.4 ± 9.4 at week 12, and 11.7 ± 9.1 at week 24; the mean SDAI score reduced over time after the start of TAC (Fig. 4). The distribution of DAS28-CRP disease activity is shown in Fig. 5. The proportion of remission (DAS28-CRP <2.3) or low disease activity (2.3 ≤ DAS28-CRP < 2.7) was 33.6 % at 4–8 weeks prior to the start of TAC, 3.7 % for remission and 6.7 % for low disease activity at the start of TAC, 24.4 and 14.8 %, respectively, at week 12, and 33.3 and 15.6 %, respectively, at week 24, showing improvement. The mean (±SD) DAS28-CRP score was 3.3 ± 1.1 at 4–8 weeks prior to the start of TAC, 4.0 ± 1.0 at the start of TAC, 3.2 ± 1.2 at week 12, and 2.9 ± 1.2 at week 24; the mean DAS28-CRP score reduced over time after the start of TAC (Fig. 6). Improvement as determined by EULAR criteria is shown in Fig. 7. Moderate response or good response was achieved in 60.7 % at week 12 and in 69.8 % at week 24. Changes over time in mean values (±SD) of TJC, SJC, VAS by physicians, VAS by patients, and CRP levels were also evaluated (Figs. 8, 9, 10, 11, 12). The TJC was 3.2 ± 3.8 at 4–8 weeks prior to the start of TAC, 5.5 ± 4.9 at the start of TAC, 3.1 ± 4.0 at week 12, and 2.7 ± 4.2 at week 24. The SJC was 2.8 ± 3.3, 4.7 ± 4.3, 2.7 ± 3.7, and 2.3 ± 3.0, respectively. The VAS by physicians was 3.2 ± 2.1, 4.0 ± 2.0, 2.9 ± 2.1, and 2.6 ± 2.0, respectively. The VAS by patients was 3.7 ± 2.4, 4.5 ± 2.5, 3.6 ± 2.6, and 3.3 ± 2.5, respectively. The CRP level was 1.1 ± 1.5, 1.4 ± 1.7, 0.8 ± 1.2, and 0.7 ± 1.0, respectively. All of them reduced over time after the start of TAC. The mean (±SD) mHAQ score, physical function evaluation by patients, was 1.0 ± 0.6, 1.2 ± 0.5, 1.0 ± 0.6, and 1.0 ± 0.6, respectively, and reduced over time after the start of TAC (Fig. 13). The mean (±SD) serum MMP-3 level was 160.2 ± 220.8, 193.8 ± 248.8, 153.0 ± 206.3, and 154.0 ± 221.0, respectively, and the values after the start of TAC were lower than those at the start of TAC (Fig. 14).

Distribution of SDAI disease activity from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Disease activity was defined using SDAI scores as follows: remission, SDAI ≤3.3; low disease activity, 3.3 < SDAI ≤ 11; moderate disease activity, 11 < SDAI ≤ 26; high disease activity, 26 <SDAI. LOCF last observation carried forward, SDAI Simplified Disease Activity Index, TAC tacrolimus

Changes in SDAI scores (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Improvements were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, SD standard deviation, SDAI Simplified Disease Activity Index, TAC tacrolimus

Distribution of DAS28-CRP disease activity from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Disease activity was defined using DAS28-CRP scores as follows: remission, DAS28-CRP <2.3; low disease activity, 2.3 ≤ DAS28-CRP ≤ 2.7; moderate disease activity, 2.7 ≤ DAS28-CRP ≤ 4.1; high disease activity, 4.1 <DAS28-CRP. DAS28-CRP Disease Activity Score 28–C-reactive protein, LOCF last observation carried forward, TAC tacrolimus

Changes in DAS28-CRP scores (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Improvements were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). DAS28-CRP Disease Activity Score 28–C-reactive protein, LOCF last observation carried forward, SD standard deviation, TAC tacrolimus

Improvement rate at week 12 and at week 24 determined by EULAR criteria (DAS28-CRP), using LOCF method. DAS28-CRP Disease Activity Score 28–C-reactive protein, EULAR European League Against Rheumatism, LOCF last observation carried forward

Changes in TJC (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, SD standard deviation, TAC tacrolimus, TJC tender joint count

Changes in SJC (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, SD standard deviation, SJC swollen joint count, TAC tacrolimus

Changes in VAS (mean ± SD) by physicians from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, SD standard deviation, TAC tacrolimus, VAS Visual Analog Scale

Changes in VAS (mean ± SD) by patients from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, SD standard deviation, TAC tacrolimus, VAS Visual Analog Scale

Changes in CRP (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions in serum CRP levels were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). CRP C-reactive protein, LOCF last observation carried forward, SD standard deviation, TAC tacrolimus

Changes in mHAQ scores (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Improvements in mHAQ responses were seen at week 12 (p = 0.003) and at week 24 (p < 0.001). LOCF last observation carried forward, mHAQ modified Health Assessment Questionnaire, SD standard deviation, TAC tacrolimus

Changes in serum MMP-3 levels (mean ± SD) from 4 to 8 weeks prior to the start of TAC to week 24, using LOCF method. Significant reductions in serum MMP-3 levels were seen at week 12 (p < 0.001) and at week 24 (p < 0.001). LOCF last observation carried forward, MMP-3 matrix metalloproteinase-3, SD standard deviation, TAC tacrolimus
4 Discussion
This survey started in August 2012 to evaluate the safety and effectiveness of add-on TAC in patients with RA who failed to show an adequate response to biological DMARDs. This report is a summary of interim results of the demographic characteristics, status of RA therapy, and the safety and effectiveness in 172 patients analyzed for safety and 165 patients analyzed for effectiveness. In the patients analyzed for this survey, the mean age was 61.9 years and RA duration was 11.0 years, and patients were relatively at an old age with long-term RA. The mean daily dose of TAC was titrated up from the start of TAC (1.1 mg/day) to week 24 (1.4 mg/day), and the mean daily dose throughout the observation period was 1.3 mg. Other studies of the add-on use of TAC to patients with RA who failed to show an adequate response to biological DMARDs also reported the use of TAC at lower doses than the approved dose [19–21], and our interim results were similar to the results in these studies. Patients who had been treated with the first biological DMARD at the start of TAC showed the highest proportion at 65.1 %, and few patients repeatedly switched biological DMARDs. In the safety evaluation, the incidence of ADRs was 10.5 %. ADRs that occurred in at least two events were abdominal pain, stomatitis, and malaise (two events each). No ADR showed a significantly high incidence, and all of these ADRs could be expected from “Precautions for Use” in the package insert. Serious ADRs were herpes zoster and myocardial infarction (one event each). The outcome of myocardial infarction was death, and the causal relationship to TAC and factors other than TAC were “unknown.” The impact of predispositions of the patient was considered to have had a major impact because this was a patient with severe RA aged 89 years, who had a long-time RA duration of 31 years, Steinbrocker stage classification Stage IV, and Steinbrocker functional classification Class 3. The effectiveness was evaluated according to the remission criteria of the SDAI announced by the American College of Rheumatology and the EULAR in 2010. Remission or low disease activity was achieved in 49.6 % at week 12, and in 58.5 % at week 24. We will continue to evaluate the safety and effectiveness in large numbers of patients (e.g., tabulation by biological DMARD) to delineate the clinical significance of TAC adding on to biological DMARDs in patients who failed to achieve an adequate response to biological DMARDs, and ensure to provide information on appropriate use.
References
Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907–16.
Breedveld FC, Dayer JM. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2000;59:841–9.
Weyand CM. New insights into the pathogenesis of rheumatoid arthritis. Rheumatology. 2000;39(Suppl. 1):3–8.
Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995;38:151–60.
Kino T, Hatanaka H, Miyata S, Inamura N, Nishiyama M, Yajima T, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J Antibiot (Tokyo). 1987;40:1256–65.
Ochiai T, Nakajima K, Nagata M, Suzuki T, Asano T, Uematsu T, et al. Effect of a new immunosuppressive agent, FK 506, on heterotopic cardiac allotransplantation in the rat. Transplant Proc. 1987;19(1 Pt 2):1284–6.
Sakuma S, Kato Y, Nishigaki F, Sasakawa T, Magari K, Miyata S, et al. FK506 potently inhibits T cell activation induced TNF-α and IL-1b production in vitro by human peripheral blood mononuclear cells. Br J Pharmacol. 2000;130:1655–63.
Sakuma S, Kato Y, Nishigaki F, Magari K, Miyata S, Ohkubo Y, et al. Effects of FK506 and other immunosuppressive anti-rheumatic agents on T cell activation mediated IL-6 and IgM production in vitro. Int Immunopharmacol. 2001;1:749–57.
Magari K, Miyata S, Nishigaki F, Ohkubo Y, Mutoh S. Comparison of anti-arthritic properties of leflunomide with methotrexate and FK506: effect on T cell activation-induced inflammatory cytokine production in vitro and rat adjuvant induced arthritis. Inflamm Res. 2004;53:544–50.
Kim Y, Sato K, Asagiri M, Morita I, Soma K, Takayanagi H. Contribution of nuclear factor of activated T cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J Biol Chem. 2005;280(38):32905–13.
Kondo H, Abe T, Hashimoto H, Uchida S, Irimajiri S, Hara M, et al. Efficacy and safety of TAC (FK506) in treatment of rheumatoid arthritis: a randomized, double-blind, placebo-controlled dose-finding study. J Rheumatol. 2004;31:243–51.
Yocum DE, Furst DE, Kaine JL, Baldassare AR, Stevenson JT, Borton MA, et al. Efficacy and safety of tacrolimus in patients with rheumatoid arthritis: a double-blind trial. Arthritis Rheum. 2003;48:3328–37.
Furst DE, Saag K, Fleischmann MR, Sherrer Y, Block JA, Schnitzer T, et al. Efficacy of tacrolimus in rheumatoid arthritis patients who have been treated unsuccessfully with methotrexate : a six-month, double-blind, randomized, dose-ranging study. Arthritis Rheum. 2002;46:2020–8.
Kawai S, Hashimoto H, Kondo H, Murayama T, Kiuchi T, Abe T. Comparison of tacrolimus and mizoribine in a randomized, double-blind controlled study in patients with rheumatoid arthritis. J Rheumatol. 2006;33:2153–61.
Takeuchi T, Kawai S, Yamamoto K, Harigai M, Ishida K, Miyasaka N. Post-marketing surveillance of the safety and effectiveness of tacrolimus in 3,267 Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2014;24(1):8–16.
Kawai S, Takeuchi T, Yamamoto K, Tanaka Y, Miyasaka N. Efficacy and safety of additional use of tacrolimus in patients with early rheumatoid arthritis with inadequate response to DMARDs–a multicenter, double-blind, parallel-group trial. Mod Rheumatol. 2011;21(5):458–68.
Kitahama M, Nakajima A, Inoue E, Taniguchi A, Momohara S, Yamanaka H. Efficacy of adjunct tacrolimus treatment in patients with rheumatoid arthritis with inadequate responses to methotrexate. Mod Rheumatol. 2013;23(4):788–93.
Kanzaki T, Kawahata K, Kanda H, Fujio K, Kubo K, Akahira L, et al. Long-term therapeutic effects and safety of tacrolimus added to methotrexate in patients with rheumatoid arthritis. Rheumatol Int. 2013;33(4):871–7.
Naniwa T, Watanabe M, Banno S, Maeda T. Adding low dose tacrolimus in rheumatoid arthritis patients with an inadequate response to tumor necrosis factor inhibitor therapies. Rheumatol Int. 2009;29(11):1287–91.
Mori S. Additional use of tacrolimus after switching to tocilizumab therapy in patients with primary lack of efficacy of infliximab therapy for rheumatoid arthritis. Mod Rheumatol. 2012;22(6):947–50.
Miyata M, Asano T, Satoh S. Effect of additional administration of tacrolimus in patients with rheumatoid arthritis treated with biologics. Fukushima J Med Sci. 2011;57(2):54–9.
Fujibayashi T, Takahashi N, Kida D, Kaneko A, Hirano Y, Fukaya N, et al. Comparison of efficacy and safety of tacrolimus and methotrexate in combination with abatacept in patients with rheumatoid arthritis; a retrospective observational study in the TBC Registry. Mod Rheumatol. 2015. doi:10.3109/14397595.2015.1029238.
Takahashi N, Fujibayashi T, Kida D, Hirano Y, Kato T, Kato D, et al. Concomitant methotrexate and tacrolimus augment the clinical response to abatacept in patients with rheumatoid arthritis with a prior history of biological DMARD use. Rheumatol Int. 2015. doi:10.1007/s00296-015-3283-4.
Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011;63(3):573–86.
Steinbrocker O, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. J Am Med Assoc. 1949;140(8):659–62.
van Rossum MA, Zwinderman AH, Boers M, Dijkmans BA, van Soesbergen RM, Fiselier TJ, et al. Radiologic features in juvenile idiopathic arthritis: a first step in the development of a standardized assessment method. Arthritis Rheum. 2003;48(2):507–15.
Gilliam BE, Chauhan AK, Low JM, Moore TL. Measurement of biomarkers in juvenile idiopathic arthritis patients and their significant association with disease severity: a comparative study. Clin Exp Rheumatol. 2008;26(3):492–7.
Matsui T, Kuga Y, Kaneko A, Nishino J, Eto Y, Chiba N, et al. Disease Activity Score 28 (DAS28) using C-reactive protein underestimates disease activity and overestimates EULAR response criteria compared with DAS28 using erythrocyte sedimentation rate in a large observational cohort of rheumatoid arthritis patients in Japan. Ann Rheum Dis. 2007;66(9):1221–6.
Inoue E, Yamanaka H, Hara M, Tomatsu T, Kamatani N. Comparison of Disease Activity Score (DAS)28-erythrocyte sedimentation rate and DAS28- C-reactive protein threshold values. Ann Rheum Dis. 2007;66(3):407–9.
Wells G, Becker JC, Teng J, Dougados M, Schiff M, Smolen J, et al. 3.Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis. 2009;68(6):954–60.
Pincus T, Summey JA, Soraci SA Jr, Wallston KA, Hummon NP. Assessment of patient satisfaction in activities of daily living using a modified Stanford Health Assessment Questionnaire. Arthritis Rheum. 1983;26(11):1346–53.
Yamanaka H, Matsuda Y, Tanaka M, Sendo W, Nakajima H, Taniguchi A, et al. Serum matrix metalloproteinase 3 as a predictor of the degree of joint destruction during the six months after measurement, in patients with early rheumatoid arthritis. Arthritis Rheum. 2000;43(4):852–8.
Acknowledgments
These survey results were originally published in the Journal of New Remedies and Clinics (a Japanese journal), and have been reproduced here with the permission of the publisher, Iyaku-Joho-Kenkyujo, Inc. We would like to thank the many physicians who cooperated and provided us with valuable data for this survey.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was sponsored by Astellas Pharma Inc., Tokyo, Japan. KI, KS and TY are employees of Astellas Pharma Inc.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ishida, K., Shiraki, K. & Yoshiyasu, T. Evaluation of the Safety and Effectiveness of Add-On Tacrolimus in Patients with Rheumatoid Arthritis Who Failed to Show an Adequate Response to Biological DMARDs: The Interim Results of a Specific Drug Use-Results Survey of Tacrolimus. Drugs R D 15, 307–317 (2015). https://doi.org/10.1007/s40268-015-0106-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-015-0106-8