
Abstract
Background
A novel ibuprofen (IBU) formulation, Advil® Film-Coated Tablets (IBUNa), was developed.
Objective
Pharmacokinetic comparison of IBUNa versus other IBU formulations.
Study Design
Two randomized, single-dose, open-label, five-way crossover pharmacokinetic studies.
Setting
Inpatient research clinic.
Subjects
Seventy-one healthy adult volunteers.
Intervention
Study 1: In three periods, fasted subjects received 400-mg IBU dose equivalents as IBUNa 2 × 256 mg, Advil® Liqui-Gels® (IBULG) 2 × 200 mg, and Motrin® IB (IBUMot) 2 × 200 mg tablets. In two periods following a high-fat breakfast, subjects received 400-mg IBU dose equivalents as IBUNa 2 × 256 mg and IBULG 2 × 200 mg. Study 2: In five study periods, fasted subjects received 400-mg IBU dose equivalents as IBUNa 2 × 256 mg, Advil® FastGel® (IBUFG) 2 × 200 mg, Nurofen® (IBUNur) 2 × 200 mg, Advil® (IBUAdv) 2 × 200 mg, and Nurofen® Express containing IBU lysinate (IBULys) 2 × 342 mg.
Main Outcome Measure
Log-transformed area under the plasma concentration versus time curve to last observable concentration (AUCL) and maximum plasma concentration (C max) were the primary pharmacokinetic parameters; time to maximum measured plasma concentration (T max) was analyzed post hoc.
Results
IBUNa was bioequivalent to IBULG (fasted and fed) and IBUFG and IBULys (fasted) for rate (C max) and extent (AUCL) of IBU absorption. After fasting, AUCL was bioequivalent for IBUNa and IBUMot, IBUAdv, and IBUNur, but C max occurred significantly earlier with IBUNa. After fasting, median IBUNa T max was comparable to that for IBULG, IBUFG, and IBULys, but was much shorter than that for IBUMot, IBUNur, and IBUAdv. Food slowed absorption of IBUNa and IBULG similarly. All treatments were tolerated similarly.
Conclusion
IBUNa is absorbed faster but to a similar extent as standard IBU formulations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A novel formulation of ibuprofen sodium (IBUNa) is absorbed faster than (but to a similar extent to) standard ibuprofen (IBU) in healthy subjects; a clinical study found it to provide faster pain relief than standard IBU formulations in subjects with dental pain. |
IBUNa has a pharmacokinetic profile similar to that of other faster-absorbed formulations of IBU. |
All IBU formulations were well tolerated, most adverse events were mild in nature, and no significant safety findings were noted. |
1 Introduction
When treating acute pain, rapid onset of relief is desirable. With ibuprofen (IBU), one of the most widely used nonprescription analgesics available, pain relief is directly related to IBU plasma levels [1]. Although IBU is almost completely absorbed, allowing for nearly 100 % bioavailability, the rate of absorption depends on dissolution of the given formulation [2]. IBU, which is a carboxylic acid, shows low solubility in aqueous acidic media such as that which is found in the stomach [3]. As a result, meaningful pain relief typically takes approximately 45 min after ingestion of an over-the-counter (OTC) dose (400 mg) of standard IBU [4, 5]. Patients experiencing acute painful conditions such as headache, musculoskeletal pain, menstrual cramps, or dental pain would benefit from a faster onset of pain relief.
In an effort to provide more rapid pain relief, newer IBU formulations have been designed to dissolve more readily in the acidic environment of the stomach. Such formulations include IBU salt conjugates [e.g., IBU lysinate (IBULys), arginine (IBUArg), or sodium (IBUNa)] and gelatin capsules containing solubilized IBU that achieve maximum plasma concentrations (C max) that are higher and are reached earlier (time to maximum measured plasma concentration; T max) than those found with standard IBU tablets [2, 4, 6]. Clinically, IBUArg has been found to provide a faster onset of pain relief versus standard IBU [4, 5, 7], while both IBULys [8] and solubilized IBU [9–11] have demonstrated more rapid pain relief compared with acetaminophen. Recently, a novel tablet formulation of IBU sodium dihydrate has been developed that has a thin-film coating and is manufactured using a patent-pending process. This report details two studies evaluating the pharmacokinetic profile of this new formulation in comparison with both standard IBU tablets and rapidly absorbed IBU formulations.
2 Methods
2.1 Study Design and Procedures
Two single-dose, randomized, open-label, inpatient, five-way crossover bioequivalence studies (Study 1, Study 2) were conducted to evaluate the pharmacokinetics of 400-mg dose equivalents of IBU administered in various different formulations. In both studies, subjects received all interventions according to a computer-generated random sequence provided by Pfizer Consumer Healthcare’s Biostatistics Department; treatment periods were separated by a washout period of at least 48 h. Subjects remained on site for the duration of each respective study.
In Study 1, conducted from July 29 to August 7, 2009, at PPD Development, LP (Austin, TX, USA), subjects received ibuprofen sodium dihydrate tablets [Advil® Film-Coated Tablets (IBUNa), Pfizer Consumer Healthcare, Madison, NJ, USA] 2 × 256 mg, solubilized IBU liquid capsules [Advil® Liqui-Gels® (IBULG), Pfizer Consumer Healthcare] 2 × 200 mg, and standard IBU tablets [Motrin® IB (IBUMot), McNeil Consumer Healthcare, Fort Washington, PA, USA] 2 × 200 mg in each of three study periods following an overnight fast. For the remaining two study periods, subjects received IBUNa tablets 2 × 256 mg or IBULG 2 × 200 mg within 20 min of a standardized high-fat breakfast. In Study 2, conducted from June 9 to 18, 2010, at Bio-Kinetic Clinical Applications, LLC (Springfield, MO, USA), subjects received IBUNa tablets 2 × 256 mg, solubilized IBU liquid capsules [Advil® FastGel® (IBUFG), Pfizer Consumer Healthcare] 2 × 200 mg, two formulations of standard IBU tablets [Nurofen® (IBUNur), Reckitt Benckiser, Slough, Berkshire, UK; and Advil® (IBUAdv), Pfizer Consumer Healthcare] 2 × 200 mg, and caplets of Nurofen® Express containing IBU lysinate (IBULys) (Reckitt Benckiser) 2 × 342 mg following an overnight fast.
Both protocols were approved by the appropriate institutional review board prior to study initiation, and both trials were conducted in compliance with International Conference on Harmonisation (ICH) standards for Good Clinical Practice and the Declaration of Helsinki and its amendments. All subjects provided written informed consent prior to the conduct of any study-related procedures.
2.2 Subjects
Subjects for Studies 1 and 2 were adult male and female (non-pregnant and non-lactating) volunteers in normal physical health, as determined by physical examination and laboratory evaluation, between 18 and 45 years of age and with a body mass index (BMI) of 18–29 kg/m2. Females of childbearing potential were required to be using reliable contraception. Excluded were individuals with a presence or history of any significant systemic medical disorder or condition felt to increase subject risk. Other exclusion criteria included hypersensitivity to aspirin, IBU, or other nonsteroidal anti-inflammatory drugs; alcohol or substance abuse within 2 years of enrollment; tobacco use within 6 weeks of enrollment; use of an investigational drug or participation in an investigational trial within 30 days of study initiation; and participation in another pharmacokinetic study or donation of blood/plasma within 4 weeks of the first treatment period, within 6 weeks of first treatment if >300 mL of blood was contributed, within 8 weeks of first treatment if >400 mL of blood was contributed, or within 10 weeks of first treatment if hemoglobin or hematocrit was noted to be abnormal.
Eligible subjects agreed not to take any medications (except oral contraceptives), nutritional supplements (except vitamin and mineral supplements), weight loss or energy products, herbal teas, or herbal supplements for 14 days prior to and during each study period. They also agreed not to ingest caffeine for 24 h or alcohol for 3 days prior to and during the study.
It was estimated that 30 subjects were needed for each study to provide at least 80 % power to establish bioequivalence, assuming that the bioavailability of IBUNa was within 7.5–9.0 % of that for the reference, and the within-subject variability for Log C max was 0.178–0.187 or less based on previous studies; additional subjects were enrolled assuming an ~15 % dropout rate to ensure at least 30 subjects completed each study.
2.3 Bioanalysis
For IBU pharmacokinetic analyses, blood samples (3 mL each, collected into sodium heparin tubes) were drawn prior to dosing (hour 0) and at 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 75, and 90 min and at 2, 3, 4, 6, 8, 10, 12, and 16 h postdose. Once obtained, blood samples were mixed thoroughly, put on ice, and centrifuged within 30 min of collection. Following centrifugation, plasma was removed and stored at −20 °C until analyzed. Plasma was analyzed for racemic IBU using a validated method of high-performance liquid chromatography with tandem mass spectrometry detection. This method allowed for a lower limit of IBU quantitation of 0.2 μg/mL. Expressed as a percent coefficient of variation, the intra-assay precision was 0.542–3.36 %; the inter-assay precision was 1.11–2.79 %.
2.4 Pharmacokinetic Parameters and Analysis
Only those subjects providing evaluable data from at least two study treatment periods were included in the pharmacokinetic analyses of each study. Data were considered inevaluable if two consecutive plasma concentrations were missing for that period, predose plasma IBU concentration was >5 % of C max for that period, or if the subject experienced emesis at or before two times the median T max for that period. Concentrations below the limit of quantitation (0.2 μg/mL) were set to zero.
Untransformed pharmacokinetic parameters were derived using WinNonlin® version 5.1 (Pharsight, Inc., Mountain View, CA, USA). The following parameters were calculated based on actual sampling times: area under the plasma concentration versus time curve to last observable concentration (AUCL) and from time zero to infinity (AUCI), C max, T max, half-life, elimination rate constant, volume of distribution, and clearance.
Data for AUCL, AUCI, and C max (log transformed) were analyzed via analysis of variance (ANOVA) with effects for sequence, subject (sequence), period, and treatment terms in the model. For Study 1, fasting-state paired comparisons were conducted between IBUNa versus IBULG and IBUNa versus IBUMot; fed-state comparisons were made between IBUNa versus IBULG. For Study 2, IBUNa was compared with IBUFG, IBUNur, IBUAdv, and IBULys all in the fasting state. Additionally, IBUFG was also compared with IBUAdv. Bioequivalence was considered established if the two-sided 90 % confidence interval (CI) for the least squares means ratio of study drug to reference formulation was between 80 and 125 %. Post hoc analyses of T max were performed using the Wilcoxon rank-sum test; Hodges-Lehmann estimates were used to evaluate treatment differences. No other changes were made to the planned protocols following initiation of the studies. Statistical analyses were performed using SAS® version 9.2 (SAS Institute, Cary, NC, USA).
2.5 Safety Analysis
Safety was evaluated among all subjects who took at least one dose of study medication. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (Study 1, version 9.0; Study 2, version 13.0) and classified by severity and relationship to study medication. Prestudy and poststudy physical examinations and laboratory findings of clinical relevance were recorded.
3 Results
3.1 Subject Disposition and Baseline Demographics
Seventy-one healthy adults (N = 36 in Study 1 and N = 35 in Study 2) were randomized to receive study medication. Seven subjects discontinued prematurely, including four subjects in Study 1 (two voluntary withdrawals due to painful blood collections and two due to AEs) and three subjects in Study 2 (two due to AEs and one due to uncooperativeness). In Study 1, all four subjects who discontinued early (during treatment period 1) were excluded from all pharmacokinetic analyses; three additional subjects had data excluded for specific periods. In Study 2, one subject discontinued early (treatment period 1) and was excluded from all pharmacokinetic analyses; four additional subjects had data excluded from specific periods.
The demographic characteristics (Table 1) of subjects enrolled in Studies 1 and 2 were generally similar in terms of age, weight, height, and BMI; approximately equal proportions of male and female subjects participated. Most subjects in both studies were white.
3.2 IBU Pharmacokinetic Results
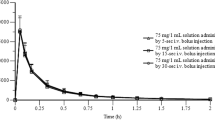
Mean IBU plasma concentration versus time curves and pharmacokinetic results for Study 1 are presented in Fig. 1 and Table 2, respectively. Using log-transformed data, IBUNa demonstrated an equivalent extent of absorption relative to IBUMot in the fasted state on the basis of AUCL; the 90 % CI for AUCI was also contained within the limits of bioequivalence. However, IBUNa reached a C max that was higher (90 % CI 125.2–145.5) than that for IBUMot. Additionally, the T max for IBUNa was 82.0 min faster (95 % CI 62.1–108.0, P < 0.001) (Table 3) than that for IBUMot.

Study 1: Mean ibuprofen (IBU) plasma concentrations from time 0 (predose) to hour 16 postdose in subjects administered 400-mg dose equivalents of IBU via a novel IBU sodium dihydrate tablet (Advil® Film-Coated Tablets; IBUNa) and Advil® Liqui-Gels® liquid capsules (IBULG) under fed and fasted conditions and Motrin® IB tablets (IBUMot) under fasted conditions
Comparisons of IBUNa and IBULG indicated bioequivalence under both fasted and fed conditions for both the rate and extent of IBU absorption. The 90 % CIs for the AUCI were in line with AUCL findings. As expected, the rate of IBU absorption with IBUNa and IBULG was slower in the fed state than in the fasted state (Fig. 1 and Table 2).
Mean IBU plasma concentration versus time curves and pharmacokinetic results for Study 2 are presented in Fig. 2 and Table 4. Compared with standard IBU formulations (IBUAdv and IBUNur), IBUNa had an equivalent extent of absorption (AUCL); the 90 % CIs for AUCI were also within the limits of bioequivalence. IBUNa was absorbed significantly faster compared with IBUAdv and IBUNur; C max was higher (90 % CI 117.5–136.4) for IBUNa versus IBUAdv and was also greater (90 % CI 121.3–140.5) for IBUNa versus IBUNur. As summarized in Table 3, the T max for IBUNa was faster than that for IBUAdv and IBUNur, (P < 0.001 for both comparisons).

Study 2: Mean ibuprofen (IBU) plasma concentrations from time 0 (predose) to hour 16 postdose in subjects administered 400-mg dose equivalents of IBU under fasted conditions via a novel IBU sodium dihydrate tablet (Advil® Film-Coated Tablets; IBUNa), Advil® FastGel® liquid capsules (IBUFG), Nurofen® Express caplets containing IBU lysinate (IBULys), standard Advil® tablets (IBUAdv), and Nurofen® tablets (IBUNur)
IBUNa was bioequivalent to both IBUFG and IBULys on the basis of C max and AUCL. There were no significant differences in T max between IBUNa and either IBUFG or IBULys.
3.3 Safety and Tolerability
Four subjects prematurely discontinued from these studies due to an AE (Study 1: one each for emesis and headache; Study 2: one each for hemorrhoids and contact dermatitis). Fifteen subjects from Study 1 reported 31 AEs; 14/31 (45.2 %) were considered treatment related, and all were rated as mild except for one report each of nausea, vomiting, headache, and blurred vision of moderate severity. The most common AE among all treatments in Study 1 was headache (six reports). In Study 2, 17 subjects reported 35 AEs; 9/35 (25.7 %) were considered treatment related, and all were rated as mild. The most common AE across all treatments in Study 2 was dizziness (12 reports). No severe or serious AEs occurred during either study. With the exception of the development of a mild ear infection that resolved spontaneously without any treatment in one subject in Study 2, there were no clinically significant laboratory, vital sign, or physical examination findings noted during either study. No new or unexpected safety concerns emerged in the IBUNa arms versus those of comparator IBU formulations; AE rates were similar across all treatments (data not shown).
4 Discussion
Results of the two pharmacokinetic studies presented here demonstrate that this novel formulation of IBUNa was absorbed at a rate faster than standard IBU tablets and was comparable with rates of other fast-absorbed IBU formulations. IBUNa was bioequivalent to IBULG, IBUFG, and IBULys in terms of both the rate and extent of absorption in the fasted state. T max was approximately 5–10 min faster with IBUNa than IBULG in the fasted state. Feeding had a similar effect on the rate of absorption of both IBUNa and IBULG, as both C max and T max were similar between the two formulations in both the fasted and fed states. IBUNa was absorbed to the same extent as standard IBU when administered as IBUMot, IBUAdv, and IBUNur, but was absorbed more rapidly, with T max values of 30–35 min versus 120, 82.5, and 120 min, respectively. All IBU treatments were well tolerated.
IBU formulations that employ IBU dissolved in a gelatin capsule or conjugated to a salt allow healthy subjects to more rapidly absorb the product and in this way improve upon the relatively low solubility of standard tablets that are composed of IBU free acid. Since IBU is almost entirely absorbed, faster absorption does not increase the extent of absorption; hence, overall IBU exposure is similar to that of standard IBU, as shown in previous studies [2, 6]. Pharmacodynamic investigations have demonstrated that faster absorption of IBU arginate is associated with a faster onset of analgesia compared with standard IBU tablets [4, 5]. Furthermore, in a study modeling the pharmacokinetics and pharmacodynamics of an effervescent formulation of IBU, faster absorption of that formulation was also associated with faster onset of analgesia in patients with dental pain compared with standard IBU [12].
Similarly, IBUNa tablets have been designed with this same goal in mind. IBU is a carboxylic acid that does not rapidly dissolve in an acidic aqueous environment such as that of the stomach [3]. In vitro investigations have shown a significantly faster rate of dissolution for IBUNa compared with standard IBU tablets at acidic pH levels [13]. The current findings demonstrate that these novel IBUNa tablets, which have a thin-film coating and are manufactured using a patent-pending process, provide faster absorption with a more rapid attainment of peak IBU plasma concentrations compared with standard IBU tablets. Under fasted conditions, C max for IBUNa was approximately 30 % greater and T max occurred roughly 1–1.5 h sooner than for standard IBU tablets. Importantly, AUCL values for IBUNa and each of the standard IBU tablets tested were similar, indicating that conjugation of IBU with sodium salt does not alter the extent of IBU absorption and yields overall IBU exposure similar to conventional IBU tablets.
Our results are consistent with those of previous pharmacokinetic evaluations of older formulations of IBUNa. In a pair of open-label, randomized, single-dose, crossover studies conducted in healthy volunteers, Sorgel et al. [13] compared the pharmacokinetics of IBUNa with those of standard IBU tablets, IBULys, and IBULG (first study), as well as with IBUArg and IBULys (second study). These studies have shown that IBUNa had a significantly higher C max (47.6 vs. 36.8 µg/mL, P < 0.01) and shorter T max (0.6 vs. 1.4 h, P = 0.018) compared with standard IBU tablets and had no significant differences in absorption rate compared with IBULys, IBUArg, or IBULG [13]. Similarly, Dewland et al. [14] compared the single-dose pharmacokinetics (400-mg equivalents) of IBUNa with those of a novel IBU/poloxamer formulation and standard IBU tablets in healthy volunteers. While the overall extent of absorption was similar for all of the formulations, T max averaged 55 min shorter with IBUNa compared with standard IBU tablets (median of 35 vs. 90 min, respectively; P < 0.0002), and C max was approximately 30 % higher (41.47 vs. 31.88 µg/mL, respectively). It is worth noting that the T max values for IBUNa across both the current and previous pharmacokinetic studies are comparable—between 30 and 36 min.
Patients suffering from acute pain desire pain relief as quickly as possible. Previous studies have shown that the rate and extent of IBU absorption may be impaired during pain episodes when IBU is taken in its standard oral formulation, but that fast-dissolving IBU formulations fare much better in this regard [15, 16]. The current investigation did not characterize the pharmacokinetic/pharmacodynamic profile of analgesia following IBUNa administration. However, a clinical efficacy study using this same IBUNa formulation found it to provide a faster onset of analgesia compared with standard IBU in subjects with dental pain [17]. In that 8-h inpatient study examining the effect of the current IBUNa formulation on postsurgical dental pain using the third molar dental extraction model, IBUNa was associated with a significantly earlier time to meaningful pain relief (median 42.4 min) in comparison with placebo (>8 h, P < 0.001), pooled IBUAdv/IBUMot (median 55.3 min, P < 0.001), and IBUMot (median 60.7 min, P < 0.001) and was marginally faster than IBUAdv (median 52.0 min, P = 0.075) [17].
Two other randomized studies using previous IBUNa formulations in the third molar extraction model of dental pain showed similar findings [18, 19]. In the QUIKK trial, first perceptible pain relief occurred 6 min earlier (P = 0.004) with a previous IBUNa formulation than with standard IBU tablets according to stopwatch assessments, although the time to substantial pain relief was not significantly different between the formulations [18]. Patient diary assessments indicated that significantly more patients treated with IBUNa reported “some” to “complete” pain relief at 15 min (43 vs. 29 % for standard IBU tablets, P < 0.001) and at 30 min (82 vs. 63 %, P < 0.001) [18].
In a study by Schleier et al. [19], the first sign of perceptible pain relief occurred within 15 min for 52.5 % of patients treated with a previous IBUNa formulation vs. 35.9 % of those treated with standard IBU tablets (P < 0.001). Substantial pain relief was attained after a median of 42 versus 56 min with IBUNa versus standard IBU [19]. In addition, reduction in pain intensity occurred to a greater degree and was faster with IBUNa, such that pain intensity was reduced by 50 % after an average of 30 min with IBUNa versus 57 min with standard IBU tablets (P < 0.02) [19]. Taken together, these data indicate that the faster absorption achieved with various formulations of IBUNa translates into more rapid pain relief.
The current studies are limited in that they were performed in healthy volunteers, and therefore, the results obtained may not be generalizable to those with underlying comorbidities or to those with active pain. A previous study found that pain was associated with an inhibition of absorption of IBU and a decrease in the conversion of racemic IBU to the active S-(+) enantiomer [15]. Nonetheless, data from clinical studies utilizing the dental pain model have shown a more rapid onset of analgesia with this novel IBUNa formulation [17] as well as previous formulations [18, 19] of IBUNa versus standard IBU formulations, suggesting that although the absorption and conversion of racemic IBU to the active S-(+) enantiomer with this formulation may be delayed by pain, the faster-absorbed formulation still provides faster onset of analgesia than standard formulations in the presence of pain. Lastly, because subjects were not allowed to take concomitant medications while participating in these studies, the potential for drug–drug interactions could not be assessed. However, these would be expected to be the same as those known for standard IBU.
5 Conclusions
A newly developed, novel tablet formulation of IBUNa was absorbed more rapidly but achieved similar exposure in comparison with standard IBU tablets in healthy volunteers. In addition, IBUNa was absorbed at the same rate as other rapidly absorbed IBU formulations.
References
Laska EM, Sunshine A, Marrero I, Olson N, Siegel C, McCormick N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin Pharmacol Ther. 1986;40:1–7.
Klueglich M, Ring A, Scheuerer S, et al. Ibuprofen extrudate, a novel, rapidly dissolving ibuprofen formulation: relative bioavailability compared to ibuprofen lysinate and regular ibuprofen, and food effect on all formulations. J Clin Pharmacol. 2005;45:1055–61.
Levis KA, Lane ME, Corrigan OI. Effect of buffer media composition on the solubility and effective permeability coefficient of ibuprofen. Int J Pharm. 2003;253:49–59.
Desjardins P, Black P, Papageorge M, et al. Ibuprofen arginate provides effective relief from postoperative dental pain with a more rapid onset of action than ibuprofen. Eur J Clin Pharmacol. 2002;58:387–94.
Black P, Max MB, Desjardins P, Norwood T, Ardia A, Pallotta T. A randomized, double-blind, placebo-controlled comparison of the analgesic efficacy, onset of action, and tolerability of ibuprofen arginate and ibuprofen in postoperative dental pain. Clin Ther. 2002;24:1072–89.
Schettler T, Paris S, Pellett M, Kidner S, Wilkinson D. Comparative pharmacokinetics of two fast-dissolving oral ibuprofen formulations and a regular-release ibuprofen tablet in healthy volunteers. Clin Drug Investig. 2001;21:73–8.
Mehlisch DR, Ardia A, Pallotta T. A controlled comparative study of ibuprofen arginate versus conventional ibuprofen in the treatment of postoperative dental pain. J Clin Pharmacol. 2002;42:904–11.
Mehlisch DR, Jasper RD, Brown P, Korn SH, McCarroll K, Murakami AA. Comparative study of ibuprofen lysine and acetaminophen in patients with postoperative dental pain. Clin Ther. 1995;17:852–60.
Hersh EV, Levin LM, Cooper SA, et al. Ibuprofen liquigel for oral surgery pain. Clin Ther. 2000;22:1306–18.
Olson NZ, Otero AM, Marrero I, et al. Onset of analgesia for liquigel ibuprofen 400 mg, acetaminophen 1000 mg, ketoprofen 25 mg, and placebo in the treatment of postoperative dental pain. J Clin Pharmacol. 2001;41:1238–47.
Packman B, Packman E, Doyle G, et al. Solubilized ibuprofen: evaluation of onset, relief, and safety of a novel formulation in the treatment of episodic tension-type headache. Headache. 2000;40:561–7.
Li H, Mandema J, Wada R, et al. Modeling the onset and offset of dental pain relief by ibuprofen. J Clin Pharmacol. 2012;52:89–101.
Sorgel F, Fuhr U, Minic M, et al. Pharmacokinetics of ibuprofen sodium dihydrate and gastrointestinal tolerability of short-term treatment with a novel, rapidly absorbed formulation. Int J Clin Pharmacol Ther. 2005;43:140–9.
Dewland PM, Reader S, Berry P. Bioavailability of ibuprofen following oral administration of standard ibuprofen, sodium ibuprofen or ibuprofen acid incorporating poloxamer in healthy volunteers. BMC Clin Pharmacol. 2009;9:19.
Jamali F, Kunz-Dober CM. Pain-mediated altered absorption and metabolism of ibuprofen: an explanation for decreased serum enantiomer concentration after dental surgery. Br J Clin Pharmacol. 1999;47:391–6.
Jamali F, Aghazadeh-Habashi A. Rapidly dissolving formulations for quick absorption during pain episodes: ibuprofen. Int J Clin Pharmacol Ther. 2008;46:55–63.
Brain P, Leyva R, Doyle G, Kellstein D. Onset of analgesia and efficacy of ibuprofen sodium in postsurgical dental pain: a randomized, placebo-controlled study versus standard ibuprofen. Clin J Pain. 2014. (in press).
Norholt SE, Hallmer F, Hartlev J, et al. Analgesic efficacy with rapidly absorbed ibuprofen sodium dihydrate in postsurgical dental pain: results from the randomized QUIKK trial. Int J Clin Pharmacol Ther. 2011;49:722–9.
Schleier P, Prochnau A, Schmidt-Westhausen AM, et al. Ibuprofen sodium dihydrate, an ibuprofen formulation with improved absorption characteristics, provides faster and greater pain relief than ibuprofen acid. Int J Clin Pharmacol Ther. 2007;45:89–97.
Acknowledgments
These studies were sponsored by Pfizer Consumer Healthcare. Medical writing support was provided by John H. Simmons, MD, and Lauren Cerruto of Peloton Advantage, LLC, and was funded by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial Registration
As these are pharmacokinetic studies, trial registration was not done.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Legg, T.J., Laurent, A.L., Leyva, R. et al. Ibuprofen Sodium Is Absorbed Faster than Standard Ibuprofen Tablets: Results of Two Open-Label, Randomized, Crossover Pharmacokinetic Studies. Drugs R D 14, 283–290 (2014). https://doi.org/10.1007/s40268-014-0070-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-014-0070-8