
Abstract
Introduction
Approximately one-third of patients with type 2 diabetes mellitus (T2DM) have concurrent renal impairment. There are limited therapeutic options for these patients. Fasiglifam is a G protein-coupled receptor 40 agonist that was under investigation for the treatment of T2DM. The objective of this study was to evaluate the potential effect of renal impairment on the pharmacokinetics and safety of a single dose of fasiglifam and its metabolite M-1.
Methods
This was a phase I, open-label, parallel-group study. Subjects with varying degrees of renal function received a single oral dose of fasiglifam 50 mg. Blood and urine samples were collected through 168 h postdose. Study endpoints were pharmacokinetic and safety variables.
Results
Fifty-three subjects were enrolled. Mean fasiglifam plasma concentrations were higher in subjects with mild renal impairment compared with other groups, but within each renal function cohort, plasma concentrations tended to decrease with decreasing renal function. Regression analyses indicated that fasiglifam exposure decreased and M-1 exposure increased with decreasing renal function. Predicted exposure values at about the midpoint of creatinine clearance for each renal impairment group differed by up to 21 % (fasiglifam) and 87 % (M-1) from that of the normal renal function group. Hemodialysis had no effect on fasiglifam or M-1 exposure. Fasiglifam renal clearance (CLR) was not affected, but M-1 CLR decreased with increasing impairment. No incidences of hypoglycemia were reported during the study.
Conclusion
Varying renal function status did not have a significant impact on the clearance of fasiglifam in this study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Fasiglifam is a potent and highly selective agonist of the G protein-coupled receptor 40 |
Varying degrees of renal function in subjects with type 2 diabetes mellitus did not have a significant impact on fasiglifam exposure |
1 Introduction
Diabetes mellitus is a global health concern, with a predicted prevalence rate of 552 million by the year 2030 [1]. Approximately 90 % of patients with diabetes have type 2 diabetes mellitus (T2DM) [2]. Up to 35 % of patients with T2DM have some form of renal impairment [3–5]. This prevalence may be even higher, as many patients have undiagnosed renal impairment [3, 4].
Renal impairment is a common occurrence among patients with T2DM [3] and may often impede and complicate adequate glycemic control by pharmacotherapy [6]. Insulin secretagogues such as sulfonylureas and glinides are known to cause hyperinsulinemia and are associated with the risk of hypoglycemia [7]. Dose adjustments are often recommended when these secretagogues are used to treat T2DM patients with renal impairment [6]. Other antidiabetic medications such as metformin or some dipeptidyl peptidase 4 inhibitors may be contraindicated or need a dose adjustment in T2DM patients with renal impairment because they are mainly eliminated by the kidneys [8, 9]. Thus, there remains an unmet medical need for effective and safe antidiabetic medications in T2DM patients with renal impairment, as many currently available drugs are contraindicated, not suitable for use in such a population, or have to be dose adjusted [10]. There is a need for an agent that can stimulate insulin secretion and can be used in patients with various levels of renal impairment without the concern of dose adjustment or the risk of hypoglycemia.
Fasiglifam (TAK-875), a potent and highly selective agonist of the G protein-coupled receptor 40 (GPR40), was under investigation as an adjunct to diet and exercise to improve glycemic control in patients with T2DM [11, 12]. GPR40 is highly expressed in pancreatic β cells and is activated by the binding of medium- or long-chain free fatty acids, which augments glucose-stimulated insulin secretion [13]. The novel mechanism of action of fasiglifam potentiates the activity of GPR40, thus stimulating insulin secretion only at elevated blood glucose concentrations [12, 14]. In contrast, insulin secretagogues, such as glinides and sulfonylureas, stimulate insulin secretion even at low blood glucose concentrations [7], which can lead to hypoglycemia. In phase II clinical trials, fasiglifam demonstrated clinically and statistically significant improvements in glycemic control in T2DM patients with inadequate control through diet and exercise with or without metformin [15–17]. Fasiglifam was well tolerated in healthy volunteers and subjects with T2DM, and the incidence of hypoglycemia was low and not different from placebo [15–19]. However, because of liver safety concerns, the clinical development of fasiglifam was terminated in December 2013. This decision was a result of liver test data in subjects indicating drug-induced liver injury.
In healthy volunteers, single doses of fasiglifam (25–800 mg) were rapidly absorbed, reaching peak plasma concentrations in 3–4 h, with a terminal elimination half-life (t 1/2) of 28–30 h [19]. Increases in exposure were approximately dose dependent. Fasiglifam is metabolized to a minor inactive metabolite, fasiglifam M-1 [19]. Renal clearance (CLR) plays a minimal role in total fasiglifam clearance. Multiple dosing of fasiglifam (25–400 mg) in subjects with T2DM led to increased exposure at day 14 that was roughly three times that of the corresponding single dose, primarily because of lower clearance rates [17]. Although CLR of fasiglifam was found to be minimal in healthy volunteers, [19] renal impairment can inhibit some pathways of hepatic and intestinal metabolism [20].
The primary objective of this study was to evaluate the effect of renal impairment on the pharmacokinetics (PK) of a single oral 50-mg dose of fasiglifam and its metabolite M-1 in subjects with varying degrees of renal function.
2 Materials and Methods
2.1 Subjects
Male or female volunteers 18–80 years of age who were considered by the investigator to be healthy except for controlled hypertension or diabetes and problems associated with the primary diagnosis of renal impairment, with a body weight of ≥50 kg and a body mass index between 19.0 and 36.0 kg/m2 were eligible to enroll. Additional inclusion criteria included a creatinine clearance (CLCR) ≥90 mL/min for subjects with normal renal function, ≥60 to <90 mL/min for subjects with mild renal impairment, ≥30 to <60 mL/min for subjects with moderate renal impairment, and <30 mL/min for subjects with severe renal impairment [21]. Subjects with end-stage renal disease (ESRD) were on intermittent hemodialysis.
Exclusion criteria included, but were not limited to, a predisposition to easy bruising or bleeding disorders; a medical history of gastric or duodenal ulceration; any trauma within 1 week of screening; extensive ecchymosis; hemoptysis; gingival bleeding; hematemesis; repeated or significant nose bleeds; peri-orbital hematoma; retinal detachment; menorrhagia; hematuria; melanoma; an acute clinically significant illness within 30 days prior to day −1; a history of abdominal, thoracic, or nonperipheral vascular surgery within 6 months prior to day −1; a history of cancer other than basal cell or stage 1 squamous cell carcinoma of the skin that had not been in remission for ≥5 years prior to day −1; any protocol-specified prohibited medications or supplements; a serum creatinine kinase level >3 times the upper limit of normal; sustained systolic blood pressure ≥155 mmHg or ≤90 mmHg or a diastolic blood pressure ≥95 mmHg or ≤50 mmHg; resting pulse rate of <51 or >100 beats per minute; abnormal electrocardiogram (ECG) at screening or check-in day; alanine aminotransferase or aspartate aminotransferase level >1.5 times the upper limit of normal; active liver or gall bladder disease; or a history of drug/alcohol abuse.
2.2 Study Design
The study was conducted in compliance with the institutional review board regulations, Good Clinical Practice regulations and guidelines, and all applicable local regulations. This was a phase I, open-label, parallel-group study. The use of a single dose of fasiglifam in this study was chosen because the multiple-dose PK parameters of fasiglifam and M-1 are linear and time independent [17] and, therefore, can be extrapolated from single-dose PK studies. All subjects received a single oral dose of fasiglifam 50 mg (Takeda Pharmaceutical Co. Ltd, Osaka, Japan) in the morning after an overnight fast of ≥10 h; subjects with ESRD requiring dialysis fasted for ≥2 h. Subjects were admitted to the clinic on day −1 and were confined to the clinic until the collection of the 168-h postdose PK samples on day 8 and the completion of all scheduled procedures. Standardized meals containing 30 % fat were provided to all subjects during the confinement period.
Up to 64 subjects were planned for enrollment in the study: up to 32 subjects with normal renal function and eight within each category of renal impairment. Subjects with normal renal function were matched by sex, weight, age, and smoking status. Subjects with normal renal function could be matched to more than one subject with renal impairment, but to no more than one subject within each impairment group.
2.3 Sample Collection
For the determination of plasma concentrations of fasiglifam and its metabolite M-1, blood samples (4 mL) were collected from all subjects at 15 min predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120, 144, and 168 h postdose. For subjects with ESRD, additional 4-mL arterial samples were collected at 1, 2, 3, and 4 h postdose (if subjects were still on dialysis). Three (10 mL) blood samples were collected 15 min predose from all subjects for protein-binding assessments. All voided urine was collected from subjects with normal renal function, and mild, moderate, and severe renal impairment at −12 to 0 h predose, and then at 24-h intervals up to 7 days postdose.
2.4 Plasma Protein Binding
The in vitro protein binding of fasiglifam was determined ex vivo using ultracentrifugation of predose plasma samples from the study subjects spiked with [14C] fasiglifam at a nominal concentration of 1 µg/mL.
2.5 Analytical Methods
Plasma and urine concentrations of fasiglifam and fasiglifam M-1 were measured using validated liquid chromatography with tandem mass spectrometry with a range of 5.00–10,000 ng/mL.
2.6 Pharmacokinetic Assessments
The PK parameters of fasiglifam and fasiglifam M-1 were derived using non-compartmental analysis methods with Phoenix WinNonlin Version 6.3 (Pharsight Corp, Cary, NC, USA). Measurements below the limit of quantification of 5.00 ng/mL were treated as 0 when deriving PK parameters. The PK parameters assessed for fasiglifam and fasiglifam M-1 (unless otherwise specified) included maximum observed plasma concentration (C max); time to C max (T max); area under the plasma concentration-time curve (AUC) from time 0 to the time of last quantifiable concentration (t), calculated using the linear trapezoidal rule (AUC t ); area under the plasma concentration-time curve from 0 to infinity calculated from AUC t + lqc/λz, where lqc is the last quantifiable concentration and λz is the terminal elimination rate constant, calculated as the negative of the slope of the log-linear regression of the natural logarithm concentration-time curve during the terminal phase (AUC∞); t 1/2 calculated as ln (2)/λz; apparent clearance after extravascular administration, calculated as dose/AUC∞ after a single dose (CL/F; calculated for fasiglifam only); apparent volume of distribution during the terminal phase after extravascular administration, calculated as (CL/F)/λz (Vz/F; for fasiglifam only); area under the unbound plasma concentration-time curve from time 0 to infinity, calculated as AUC∞ percent unbound (AUC∞,u ; calculated for fasiglifam only); maximum observed unbound plasma concentration (C max,u ; calculated for fasiglifam only); oral clearance of unbound fasiglifam, calculated as CL/F percent unbound (CL u /F; calculated for fasiglifam only).
The urinary parameters included total amount of drug excreted in urine from time 0 to time t (Ae t ); fraction of drug excreted in urine, calculated as (Ae t /dose) × 100 (f R ; calculated for fasiglifam only); and CL R calculated as Ae t /AUC t .
2.7 Safety Assessments
Physical examinations and 12-lead ECGs were performed on day −1, predose, and at study exit or early termination (ET). Vital signs were examined on day −1, predose on day 1, and 1, 2, 4, 8, 12, 24, and 48 h postdose, and at study exit or ET. Clinical laboratory evaluations comprising routine hematology, serum chemistry, and urinalysis were included in the safety assessments. Participants were also monitored for adverse events (AEs) and the incidence of hypoglycemia. Blood glucose was measured by fingerstick prior to breakfast (predose on day 1); at lunch and dinner during confinement, which lasted from day −1 to study exit on day 8; or if signs or symptoms of hypoglycemia occurred.
2.8 Statistical Analyses
The safety analysis set, which included all subjects who were enrolled in the study and received the study drug, was used for demographics and safety summaries. The PK set included all subjects with one or more measurable concentration(s) of fasiglifam. Descriptive statistics [N, mean, standard deviation (SD), percent coefficient of variation (%CV), median, minimum, and maximum] were used to summarize plasma concentrations of fasiglifam and fasiglifam M-1 by renal function group and collection time point. Similar descriptive statistics were used to summarize the plasma and urine PK parameters of fasiglifam and fasiglifam M-1, and the percent of unbound fasiglifam in plasma by renal function group. In addition, geometric means were calculated for C max and AUC. The relationship between renal function and fasiglifam T max, C max,u , AUC∞,u , λz, and CL u /F were evaluated with regression analyses. CLCR was a continuous variable in the regression model. Based on the results from the regression models for each PK parameter, individual parameter predictions and the 95 % prediction intervals were calculated for a hypothetical subject with CLCR at about the midpoint of each renal function category. Subjects with ESRD were not included in the regression analyses because CLCR was not determined in these subjects. Instead, an analysis of covariance with renal function as the factor and baseline weight as the covariate was used to compare the PK of ESRD subjects with that of normal subjects.
3 Results
3.1 Subject Disposition and Baseline Characteristics
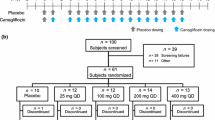
Of 168 subjects initially screened, 53 were enrolled and completed the study (Fig. 1). Subjects had a mean age of 57 years and the majority were white (66.0 %). Subject demographics and baseline characteristics were comparable among the various groups (Table 1). Of the 32 subjects with renal impairment, 14 (43.8 %) had concurrent T2DM (Table 1).

Disposition of subjects. CL CR creatinine clearance, ESRD end-stage renal disease. Normal renal function, CLCR ≥90 mL/min; mild renal impairment, CLCR = 60 to <90 mL/min; moderate renal impairment, CLCR = 30 to <60 mL/min; severe renal impairment, CLCR <30 mL/min. Note: Subjects with normal renal function could be matched to more than one subject with renal impairment, but to no more than one subject within each impairment group
3.2 Pharmacokinetic Analyses
Following oral administration of a single 50-mg dose of fasiglifam, mean plasma concentrations tended to be higher in subjects with mild renal impairment compared with subjects in other groups. Within the treatment groups with varying degrees of renal impairment, plasma concentrations tended to be lower with decreasing renal function throughout the time course (Fig. 2a). There was no consistent trend of increasing mean plasma concentrations of the inactive metabolite fasiglifam M-1 with increasing severity of renal impairment, although concentrations were generally higher in subjects with renal impairment compared with those with impaired renal function (Fig. 2b).

Mean concentration-time profile of a fasiglifam and b fasiglifam M-1 in a log-linear scale following administration of a single 50-mg dose of fasiglifam in subjects with varying degrees of renal function. aESRD requiring dialysis. CL CR creatinine clearance, ESRD end-stage renal disease. Normal renal function, CLCR ≥90 mL/min; mild renal impairment, CLCR = 60 to <90 mL/min; moderate renal impairment, CLCR = 30 to <60 mL/min; severe renal impairment, CLCR <30 mL/min
The mean percent unbound fasiglifam values in plasma were slightly higher in subjects with normal renal function (0.17 % unbound) than in subjects with mild, moderate, or severe renal impairment and subjects with ESRD requiring dialysis (0.11–0.15 % unbound).
Descriptive statistics for the noncompartmental PK parameter estimates (in plasma and urine) of fasiglifam and fasiglifam M-1 following the administration of a single 50-mg dose of fasiglifam to subjects with normal renal function, renal impairment, or with ESRD requiring dialysis are summarized in Tables 2 and 3, respectively. Fasiglifam plasma PK parameters were comparable between subjects with normal renal function and those with mild, moderate, or severe renal impairment. Among the urinary PK estimates of fasiglifam, the f R was estimated to be ≤0.35 % and CL R varied from 0.0015 to 0.0025 L/h across all subject groups. Mean plasma concentrations of fasiglifam M-1 were lower than those of the parent drug, with metabolite-to-parent AUC ratios from 0.11 to 0.55 (Table 3). Mean arterial and venous plasma concentrations of fasiglifam or fasiglifam M-1 were similar to each other during dialysis, but concentrations of fasiglifam M-1 were lower than those of the parent drug (Fig. 3).

Mean fasiglifam and fasiglifam M-1 arterial and venous plasma concentration-time curves in log-linear scale following the administration of a single 50-mg dose of fasiglifam to subjects with ESRD during dialysis. ESRD end-stage renal disease
Regression analyses were performed on the fasiglifam PK parameters T max, C max,u , AUC∞,u , CL u /F, and λz vs CLCR for subjects with normal renal function and with mild, moderate, or severe renal impairment (Table 4).The median times to reach C max were similar across subjects with normal renal function and mild, moderate, or severe renal impairment. Regression analyses showed a nonstatistically significant linear relationship between T max and CLCR. Regression analyses showed a nonstatistically significant trend between C max,u , AUC∞,u , or CL u /F, and CLCR. A 19 % decrease in the predicted C max,u values was estimated with a decline in renal function from normal to severe (p = 0.111). This was accompanied by a 21 % decrease in predicted AUC∞,u values (p = 0.114) and a 40 % increase in the predicted CL u /F values (p = 0.063) with declining renal function.
Linear regression analyses of fasiglifam M-1 C max, AUC∞, and λz vs CLCR showed statistically significant linear trends. The predicted C max of fasiglifam M-1 progressively increased from 75.2 to 110.1 ng/mL, as the renal function decreased from normal to severe, representing an approximate 47 % increase from the mean C max of the subjects with normal renal function. For fasiglifam M-1 λz, the predicted decrease with decreasing renal function was estimated to be approximately 27 % when compared with the normal renal function group. Linear regression analyses of T max on CLCR did not show a statistically significant linear relationship.
Scatter plots of fasiglifam C max,u and AUC∞,u are shown in Fig. 4a and 4b, respectively. The data are in accordance with the regression analysis in showing that C max,u and AUC∞,u did not significantly differ between subjects with normal renal function and those with mild, moderate, or severe renal impairment.

Scatter plots of fasiglifam a C max,u and b AUC∞,u following the administration of a single 50-mg dose of fasiglifam to subjects with varying degrees of renal function. Horizontal bar represents the mean. aESRD requiring dialysis. CL CR creatinine clearance. Normal normal renal function, CLCR ≥90 mL/min; Mild mild renal impairment, CLCR = 60 to <90 mL/min; Moderate moderate renal impairment, CLCR = 30 to <60 mL/min; Severe severe renal impairment, CLCR <30 mL/min; ESRD end-stage renal disease; AUC ∞,u area under the plasma concentration-time curve from time 0 to infinity for unbound drug; C max,u maximum observed plasma concentration for unbound drug
The comparison of the results from the analysis of covariance for the ESRD group with the normal group (results not shown) is similar to the comparison of arithmetic means provided in Table 2.
3.3 Safety Analyses
Thirteen of 53 subjects (24.5 %) had treatment-emergent AEs (TEAEs): four (19.0 %) in the normal renal function group, two (25.0 %), three (37.5 %), and three (37.5 %) in the mild, moderate, and severe impairment groups, respectively, and one (12.5 %) in the ESRD group. All TEAEs were experienced by single subjects except headache, nasopharyngitis, and nausea, which were experienced by two subjects each. The majority of TEAEs were mild in intensity. Two subjects had TEAEs that were considered moderate in intensity: abdominal pain (one subject with mild renal impairment) and increased blood pressure (one subject with moderate renal impairment). One serious AE, an acute myocardial infarction, occurred in a 74-year-old male subject with T2DM, hyperlipidemia, and mild renal impairment. This event occurred 18 days after administration of a single dose of fasiglifam 50 mg and resolved after 4 days, was not considered to be related to study drug, and did not lead to study discontinuation. No TEAEs or laboratory test results were reported in any subjects that led to study discontinuation. None of the subjects developed hypoglycemia in the study.
4 Discussion and Conclusion
In the present study, we evaluated the impact of varying degrees of renal impairment on the PK of fasiglifam regarding the use of this novel agent in patients with T2DM and chronic kidney disease. No major differences in the main PK parameters were found.
As the differences in protein binding between renal function groups were small, major differences in systemic clearance between healthy subjects and those with renal impairment based on the protein binding of the parent drug would not be expected. Generally, it would be expected that, following the administration of a drug to a subject with renal impairment, plasma exposure would be higher compared with that in a subject with normal renal function owing to the decrease in renal clearance of the drug. However, in this study, the renal clearance of fasiglifam was not substantially different between subjects with normal renal function and those with renal impairment. In addition, both mean C max,u and AUC∞,u values for fasiglifam trended lower as renal function decreased from mild to severe. Although regression analyses showed nonstatistically significant linear relationships, as renal function decreased from normal to severe, the predicted C max,u and AUC∞,u values decreased by as much as 21 % from those observed in subjects with normal renal function. Nonetheless, because the differences in mean C max,u and AUC∞,u values were relatively small between the renal function groups, the changes in exposure to fasiglifam in subjects with mild, moderate, or severe renal impairment may not be considered clinically meaningful, but would need to be confirmed after long-term clinical studies have been conducted.
In contrast to the parent drug, mean plasma concentrations of fasiglifam M-1 increased in subjects with mild, moderate, and, most notably, severe renal impairment. Linear regression analyses showed a statistically significant linear trend between M-1 C max and renal function, as measured by CLCR. However, the observed increases in fasiglifam M-1 exposure in subjects with renal impairment may be of little clinical relevance, because in vitro studies have shown that fasiglifam M-1 does not bind to recombinant GPR40 (unpublished data). Additional data are needed to further understand if fasiglifam M-1 has any biological impact. The observed levels of fasiglifam M-1 exposure in subjects with mild, moderate, or severe renal impairment was lower than that seen when doses of 400 or 800 mg fasiglifam were administered to healthy volunteers [19]. Renal clearance of fasiglifam and fasiglifam M-1 in subjects with normal renal function or with mild, moderate, or severe renal impairment varied from 0.001 to 0.002 L/h and 0.009 to 0.029 L/h, respectively. Although renal impairment did not appear to affect the CL R of fasiglifam, it had a more substantial effect on the CL R of metabolite M-1. The decrease in CL R of M-1 is likely responsible for the observed increase in M-1 exposure with decreasing renal function.
The effect of renal insufficiency on CL R in individuals with normal renal function or some degree of renal impairment observed in this investigation is in line with a previous study that showed that the CL R of fasiglifam and fasiglifam M-1 in healthy subjects was negligible [19]. Taken together, these results suggest that fasiglifam and fasiglifam M-1 are primarily eliminated by nonrenal routes, including hepatic metabolism. The minimal impact of renal insufficiency on CL R may offer a therapeutic advantage to fasiglifam as this suggests that it may be administered to patients with renal impairment without further dose adjustments. Larger clinical trials in patients with T2DM and renal impairment who are receiving multiple doses of fasiglifam are needed to assess the overall efficacy and safety in this patient population before any clinical recommendations can be made.
Recent estimates suggest that diabetes is the leading cause of kidney failure and accounted for 44 % of new cases of ESRD in 2008 [22]. In this study, when subjects with ESRD requiring dialysis were administered a single 50-mg oral dose of fasiglifam, arterial and venous plasma concentrations of fasiglifam and its metabolite were nearly equivalent during dialysis. These results suggest that only a small fraction, if any, of fasiglifam or fasiglifam M-1 is lost from the blood during dialysis.
A single dose of fasiglifam was well tolerated in this study population of subjects with varying degrees of renal function, and AEs varied from mild to moderate in intensity. Previous studies have shown a low incidence of hypoglycemia in T2DM patients who were administered multiple doses of fasiglifam (6.25–400 mg) [15, 17]. This study adds to the increasing body of evidence suggesting a low occurrence of hypoglycemia associated with fasiglifam, as no incident of hypoglycemia was observed after a single dose of fasiglifam in any of the subjects with normal renal function, renal impairment, or ESRD.
In conclusion, renal function status did not have a significant impact on clearance of fasiglifam in this study, suggesting that dose adjustments would not be required for patients with renal impairment. Additionally, hemodialysis had no effect on plasma exposure of fasiglifam or its metabolite. Since the completion of this study, the fasiglifam clinical development program has been terminated as a result of liver safety concerns. This study, however, may provide useful data to aid in the development of other similar antidiabetic compounds.
References
IDF Diabetes Atlas. International Diabetes Federation. http://www.idf.org/diabetesatlas/5e/the-global-burden. Accessed 30 May 2014.
National Diabetes Statistics, 2011. National Institutes of Health. National Diabetes Information Clearinghouse (NDIC). NIH Publication No. 11-3892. http://diabetes.niddk.nih.gov/dm/pubs/statistics/index.htm. Accessed 5 June 2014.
Meyers JL, Candrilli SD, Kovacs B. Type 2 diabetes mellitus and renal impairment in a large outpatient electronic medical records database: rates of diagnosis and antihyperglycemic medication dose adjustment. Postgrad Med. 2011;123:133–43.
Middleton RJ, Foley RN, Hegarty J, Cheung CM, McElduff P, Gibson JM, et al. The unrecognized prevalence of chronic kidney disease in diabetes. Nephrol Dial Transplant. 2006;21:88–92.
Rodriguez-Poncelas A, Garre-Olmo J, Franch-Nadal J, Diez-Espino J, Mundet-Tuduri X, Barrot-De la Puente J, et al. Prevalence of chronic kidney disease in patients with type 2 diabetes in Spain: PERCEDIME2 study. BMC Nephrol. 2013;14:46.
Avogaro A, Schernthaner G. Achieving glycemic control in patients with type 2 diabetes and renal impairment. Acta Diabetol. 2013;50(3):283–91.
Matthews DR, Cull CA, Stratton IM, Holman RR, Turner RC. UKPDS 26: sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet Med. 1998;15:297–303.
Glucophage® (metformin hydrochloride) tablets: full prescribing information. Princeton: Bristol-Myers Squibb Company; 2009.
Januvia® (sitagliptin) tablets: full prescribing information. Whitehouse Station: Merck & Co., Inc.; 2013.
National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 update. Am J Kidney Dis. 2012;60:850–86.
Negoro N, Sasaki S, Mikami S, Ito M, Suzuki M, Tsujihata Y, et al. Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist. ACS Med Chem Ltrs. 2010;1:290–4.
Tsujihata Y, Ito R, Suzuki M, Harada A, Negoro N, Yasuma T, et al. TAK-875, an orally available G protein-coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose-dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. J Pharmacol Exp Ther. 2011;339:228–37.
Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–7.
Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther. 2012;340:483–9.
Kaku K, Araki T, Yoshinaka R. Randomized, double-blind, dose-ranging study of TAK-875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care. 2013;36:245–50.
Araki T, Hirayama M, Hiroi S, Kaku K. GPR40-induced insulin secretion by the novel agonist TAK-875: first clinical findings in patients with type 2 diabetes. Diabetes Obes Metab. 2012;14:271–8.
Leifke E, Naik H, Wu J, Viswanathan P, Demanno D, Kipnes M, et al. A multiple-ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clin Pharmacol Ther. 2012;92:29–39.
Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–11.
Naik H, Vakilynejad M, Wu J, Viswanathan P, Dote N, Higuchi T, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamic properties of the GPR40 agonist TAK-875: results from a double-blind, placebo-controlled single oral dose rising study in healthy volunteers. J Clin Pharmacol. 2012;52:1007–16.
Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74.
Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137–47.
American Diabetes Association. Statistics about diabetes. Data from the 2011 National Diabetes Fact Sheet (released January 26, 2011). http://www.diabetes.org/diabetes-basics/diabetes-statistics/. Accessed 30 May 2014.
Acknowledgments
This study was wholly funded by Takeda Development Center Americas, Inc., Deerfield, IL, USA. Editorial assistance was provided by Deepa Mothey, PhD, and Meryl Gersh, PhD, of AlphaBioCom, LLC, King of Prussia, PA and funded by Takeda Pharmaceuticals International, Inc., Deerfield, IL, USA.
Conflicts of interest
All authors are employees of Takeda Development Center Americas, Inc., Deerfield, IL, USA.
Author contributions
Michael Mayer was involved in the study design, pharmacokinetic calculations, and data interpretation. Sai Nudurupati provided statistical analysis and contributed to the study design and data interpretation. Xuejun Peng provided medical oversight for the study. John Marcinak was responsible for the safety aspects of this study and contributed to the study design. All authors contributed to the writing of the manuscript, provided critical review of drafts, and provided final approval for manuscript submission.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Mayer, M., Nudurupati, S., Peng, X. et al. Evaluation of the Pharmacokinetics and Safety of a Single Oral Dose of Fasiglifam in Subjects with Normal or Varying Degrees of Impaired Renal Function. Drugs R D 14, 273–282 (2014). https://doi.org/10.1007/s40268-014-0066-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-014-0066-4