
Abstract
Despite recent advances in the treatment of metastatic prostate cancer, progression to a castration-resistant state remains inevitable for most and prognosis is limited. Genetic testing for homologous recombination repair pathway alterations is recommended for all patients with advanced prostate cancer given that a mutation is present in up to 25% of cases. Poly(ADP-ribose) polymerase (PARPis) are now approved for use in patients with metastatic castration-resistant prostate cancer who have progressed on an androgen receptor pathway inhibitor (ARPI) and harbour a germline or somatic homologous recombination repair mutation. Preclinical data support a synergistic effect with an ARPI and PARPi, and various ARPI-PARPi combinations have therefore been explored in phase III clinical trials. Despite heterogeneous findings, a clear hierarchy of benefit is evident, with patients harbouring a BRCA mutation deriving the greatest magnitude of benefit, followed by any homologous recombination repair mutation. The benefit in homologous recombination repair-proficient cohort is less clear, and questions remain about whether ARPI-PARPi combination therapy should be offered to patients without a homologous recombination repair mutation. With ARPIs now considered standard-of-care for metastatic hormone-sensitive prostate cancer, ARPI-PARPi combination therapy is currently being explored earlier in the treatment paradigm. The purpose of this review is to discuss the rationale behind ARPI-PARPi combination therapy, summarise the results of key clinical trials, and discuss clinical considerations and future perspectives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Androgen receptor pathway inhibitors (ARPI) and poly(ADP-ribose) polymerase inhibitors (PARPi) are already established treatment options for patients with metastatic prostate cancer. Given pre-clinical data suggesting synergy between the two agents, several studies have evaluated ARPI-PARPi combinations in metastatic castration-resistant prostate cancer. |
The greatest survival benefit with an ARPI-PARPi combination compared to an ARPI alone is seen in patients with metastatic castration-resistant prostate cancer who have a BRCA mutation. Improvements in survival are seen in the overall subgroup of patients with any homologous recombination repair mutation, though differential results are seen depending on the gene involved. |
Questions remain about the benefit of an ARPI-PARPi combination in patients without a homologous recombination repair mutation, and the optimal sequencing in the evolving prostate cancer treatment landscape. |
1 Introduction
Prostate cancer is the second most common cancer in men, with over 1.4 million new cases diagnosed globally in 2020 [1] and an incidence predicted to significantly increase to 2.9 million by 2040 [2]. The prevalence of metastatic prostate cancer, in particular, is rising [3], and remains incurable with high morbidity.
Since the discovery of testosterone dependency in prostate cancer in 1941 [4], androgen deprivation therapy (ADT) has formed the backbone of treatment for metastatic disease. The treatment landscape has significantly evolved over the last two decades, with multiple novel therapies now integrated into the therapeutic paradigm. Standard systemic treatments for metastatic hormone-sensitive prostate cancer (mHSPC) include taxane-based chemotherapy (docetaxel) and androgen receptor pathway inhibitors (ARPI) in combination with ADT. Despite these early interventions, however, progression to metastatic castration-resistant prostate cancer (mCRPC) remains inevitable for most. Several additional therapies are available to treat mCRPC, such as further taxane chemotherapy with cabazitaxel, an alternate ARPI, poly(ADP-ribose) polymerase inhibitors (PARPi) and targeted radioligand therapy such as [177Lu]Lu-PSMA and radium-223 [5]. Prognosis remains poor[6], however, and further research into mechanisms of treatment resistance and strategies to overcome these are crucial.
The transition to mCRPC is largely driven by alterations in the androgen receptor (AR). These alterations include AR ligand-binding domain mutations, AR overexpression, and AR splice-variants (AR-V), which can restore AR signalling despite ongoing androgen suppression from ADT [7]. As such, the AR signalling pathway forms a key therapeutic target and various ARPIs, such as enzalutamide and abiraterone acetate, have been evaluated and approved for use in mCRPC [8,9,10]. Some AR alterations, however, lead to constitutive activation of the AR pathway resulting in a state of androgen independence, with inherent resistance to ARPIs [11]. Other resistance mechanisms leading to castration resistance include aberrations in DNA repair genes (in particular, the homologous recombination repair [HRR] pathway) and loss of tumour suppressor genes [12, 13]. In patients with mCRPC, the prevalence of somatic and germline pathogenic HRR mutations is increased compared with in localised or hormone-sensitive disease, reaching 20–25% and 12%, respectively [14]. The BRCA1 and BRCA2 genes play critical roles in the HRR pathway, with germline and somatic BRCA aberrations being present in 5% and 11% of mCRPC, respectively [15]. Alterations in the HRR-related genes, especially BRCA, confer a poor prognosis [16,17,18,19,20]. Currently, the recommendation for genomic testing to assess BRCA and HRR mutation status is incorporated into most guidelines and consensus statements, though optimal timing and methods of HRR gene testing vary [21].
In patients with HRR-mutant (HRRm) prostate cancer, double-stranded DNA breaks are unable to be repaired via the HRR pathway. Consequently, alternative non-conservative pathways of DNA repair are preferentially utilised, such as non-homologous end joining or traditional single-strand DNA repair pathways (e.g. the poly(ADP-ribose) polymerase [PARP]-mediated nucleotide excision repair [NER] or base-excision repair [BER] pathways) [22,23,24]. Given such pathways do not use a homologous DNA sequence to guide repair, they may lead to the accumulation of other DNA alterations such as deletions, conferring an increased risk of cancer developing. If the non-conservative pathway is also inhibited or non-functional, DNA repair cannot occur and cell death ensues.
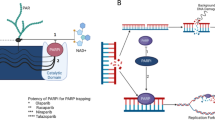
Poly(ADP-ribose) polymerase inhibitors have therefore emerged as a potential therapeutic option for HRRm patients, exploiting synthetic lethality through disruption of the NER/BER pathways (see Fig. 1). The lack of a functional NER/BER pathway results in increased degradation of single-strand DNA breaks into double-strand breaks, which are then unable to be repaired with high fidelity in HRRm cells. PARP1 and PARP2 are the two predominant enzymes involved with sensing DNA damage and facilitating repair. The catalytic function of each enzyme becomes activated at DNA damage sites, which leads to DNA repair through the NER/BER pathway. Once repaired, PARP releases itself from the DNA through a process called autoPARylation. Most PARPis target primarily PARP1 and PARP2, and to varying degrees also prevent release of PARP molecules from sites of DNA damage—a process known as PARP trapping [25]. The net result of these effects is prevention of DNA repair, leading to cell death.

Mechanism of action of poly(ADP-ribose) polymerase inhibitors (PARPi) in a homologous recombination repair pathway-deficient (HRRd) cell resulting in synthetic lethality. BER base excision repair pathway, BRCA1 BReast CAncer gene 1, BRCA2 BReast CAncer gene 2, NER nucleotide excision repair pathway. Created with BioRender.com
2 PARP Inhibitors as Monotherapy in Metastatic Castration-Resistant Prostate Cancer
Use of PARPis as monotherapy is now considered standard of care for patients with mCRPC harbouring either a somatic or germline pathogenic BRCA mutation. Several single-arm phase II trials evaluated various PARPis in HRRm patients with mCRPC and demonstrated a consistent benefit in the objective response rate (ORR). The TOPARP-B (olaparib) [26], TRITON-2 (rucaparib) [27], and TALAPRO-1 (talazoparib) [28] trials included patients with mono- and biallelic HRR alterations who had progressed after a prior taxane and ARPI (prior ARPI optional in TOPARP-B). The ORR in the BRCA1/2 cohorts in each study were 83.3%, 43.5% and 46%, respectively, with lower response rates seen for other HRR gene subgroups. Of note, the TOPARP-B trial adopted a non-standard definition of ORR, which included a composite of at least a 50% reduction in prostate-specific antigen level, conversion of circulating tumour cells from ≥ 5 per 7.5 mL of blood to < 5/7.5 mL, in addition to RECIST defined objective response. The GALAHAD trial (niraparib) only included patients with bi-allelic or germline HRR alterations, and the ORR for BRCA1/2 patients was 34.2% [29]. Consequently, two randomised trials were then performed comparing the use of a PARPi to standard-of-care treatment in mCRPC.
The PROfound trial was a phase III trial comparing olaparib 300 mg twice daily (BD) to physician’s choice of ARPI switch (enzalutamide or abiraterone acetate) in patients with mCRPC who had progressed on a prior ARPI. Patients were tested for 15 different HRR genes and were eligible if they had a pathogenic alteration detected. Patients harbouring a BRCA1, BRCA2 or ATM mutation formed Cohort A, with the remaining HRR mutations allocated to Cohort B. Approximately 65% of patients overall had received at least one prior taxane. The primary endpoint of radiographic progression-free survival (rPFS) in Cohort A was met (median rPFS 7.4 vs 3.6 months, hazard ratio [HR] 0.34 [95% confidence interval (CI) 0.25–0.47], p < 0.001), with a lower magnitude of benefit seen with Cohorts A and B combined (5.8 vs 3.5 months, HR 0.49 [95% CI 0.38–0.63], p < 0.001) [30]. The final overall survival (OS) favoured the experimental arm for Cohort A (19.1 vs 14.7 months, HR 0.69 [95% CI 0.50–0.97], p = 0.02), and in both cohorts to a lesser degree (17.3 vs 14.0 months, HR 0.79 [95% CI 0.61–1.03]) [31]. A post-hoc gene-by-gene analysis found that the ATM subgroup in Cohort A did not derive significant benefit from treatment with olaparib (median rPFS 5.4 months, HR 1.04 [95% CI 0.61–1.87], median OS 18.0 months, HR 0.93 [95% CI 0.53–1.75]) [32]. Further analysis of the BRCA subgroup found that the extent of OS benefit was greater in patients who were naïve to chemotherapy (median OS for previous taxane subgroup 17.4 vs 12.6 months, HR 0.64 [95% CI 0.39–1.08]; compared with no previous taxane subgroup, not reached [NR] versus 18.8 months, HR 0.30 [95% CI 0.10–0.78]) [33].
The TRITON-3 phase III trial compared rucaparib 600 mg BD to physician’s choice of either docetaxel or ARPI switch in patients with mCRPC harbouring a BRCA1, BRCA2 or ATM mutation [34]. The primary outcome of rPFS was met and favoured rucaparib, with the greatest benefit seen in the BRCA subgroup (median rPFS 11.2 vs 6.4 months, HR 0.50 [95% CI 0.36–0.69], p < 0.001) followed by the intention-to-treat group (10.2 vs 6.4 months, HR 0.61 [95% CI 0.47–0.80], p < 0.001). Importantly, the TRITON-3 trial is the only trial evaluating a PARPi in mCRPC that incorporated an upfront chemotherapy comparator. In this study, BRCA-mutant patients had longer rPFS if they received rucaparib after progression on a prior ARPI, compared with docetaxel (11.2 vs 8.3 months, HR 0.53 [95% CI 0.37–0.77]), which is consistent with the post-hoc analysis findings in the PROfound trial and suggests that a PARPi should be prioritised before chemotherapy for these patients. An exploratory analysis of the ATM subgroup did not show a significant rPFS benefit (8.1 vs 6.8 months, HR 0.95 [95% CI 0.59–1.52]).
As a result of these studies, in 2020 the US Food and Drug Administration (FDA) first approved olaparib for use as monotherapy in patients with mCRPC with a pathogenic germline or somatic HRR mutation, after progression on an ARPI [35]. This was followed by approval of rucaparib in patients with mCRPC harbouring a pathogenic germline or somatic BRCA mutation, who have received a prior ARPI and taxane [36]. The European Medicines Agency subsequently approved the use of olaparib as monotherapy for patients with mCRPC; however, the approval was restricted to include BRCA mutations only [37].
3 Rationale for Combining PARP Inhibitors with ARPIs
Preclinical in vitro models have demonstrated the potential for synergy between ARPIs and PARPis through a number of mechanisms (see Fig. 2), raising the question of whether their use in combination can be extended to a wider population of patients regardless of HRR mutation status. First, ARPIs inhibit downstream transcriptional activity of the AR, which includes several genes involved in the HRR pathway [38]. This induces a functional HRR-deficient state, or a ‘BRCAness’ phenotype, which then potentiates PARPi activity [38, 39]. Second, PARP enzymes are involved with recruiting the AR to its transcription site, thereby augmenting the AR signalling pathway and promoting an androgen-independent state [40, 41]. It follows then that PARP enzyme inhibition via a PARPi should reduce AR signalling activity. Poly(ADP-ribose) polymerase enzyme activity is also increased in advanced prostate cancer, particularly in mCRPC, which thereby may increase sensitivity to PARPis [38, 40, 42].

Mechanisms of action of poly(ADP-ribose) polymerase inhibitor (PARPis) and androgen receptor pathway inhibitors (ARPI) in advanced prostate cancer. AR-V AR splice variant, ARPI androgen receptor pathway inhibitor, AR androgen receptor, DHT dihydrotestosterone. ARPI suppresses AR activity (1), upregulates poly(ADP-ribose) polymerase (PARP) activity (2), and downregulates the homologous recombination repair (HRR) gene expression (3) thereby inducing a phenotype resembling HRR deficiency (BRCAness). PARPi disrupt single-strand DNA repair (4), leading to cytotoxic double-stranded DNA breaks, suppressing AR transcriptional activity (5), and may attenuate mechanisms of resistance to ARPIs mediated by co-deletion of RB1 and BRCA2 or AR-Vs (6). Created with BioRender.com
The addition of a PARPi may also attenuate mechanisms of resistance to ARPIs. One key mechanism that drives castration resistance is mutations in the AR, in particular, through the development of ligand-binding domain mutations and AR-Vs, which lack a functional ligand-binding domain [43]. AR splice-variants in particular are dependent on the catalytic function of PARP enzymes for transcriptional activation, with evidence demonstrating that PARP inhibition compromises the expression of AR-V-dependent genes and promotes sensitivity to ARPIs [44]. Another potential mechanism of resistance to an ARPI is through loss of the RB1 gene, which is commonly co-deleted with BRCA2 due to their proximity on chromosome 13q. RB1 and BRCA2 co-deletion has been reported in up to 50% of patients with metastatic prostate cancer and is associated with a more aggressive disease phenotype [45] with a less durable response to ARPIs [46]. The addition of a PARPi in cases of RB1-BRCA2 co-deletion may, therefore, potentially overcome the resultant ARPI resistance and improve efficacy.
4 Phase III Trials Evaluating an ARPI-PARPi Combination in Metastatic Castration-Resistant Prostate Cancer
Several early-phase trials have investigated the efficacy of PARPis and ARPIs in combination in patients with mCRPC. Notably, a randomised phase II trial first evaluating the combination of olaparib with abiraterone acetate enrolled 142 patients with mCRPC previously treated with docetaxel [47]. Patients were randomised regardless of HRR mutation status to receive either olaparib 300 mg BD or placebo in combination with abiraterone acetate 1000 mg daily and prednisolone 10 mg daily. The primary endpoint was investigator-assessed rPFS, which favoured the experimental arm in the intention-to-treat cohort (13.8 months vs 8.2 months, HR 0.65 [95% CI 0.44–0.97], p = 0.034). Analysis of the germline and circulating tumour DNA (ctDNA) plasma samples collected during this study found that the rPFS benefit of the ARPI-PARPi combination possibly extended to the non-HRRm cohort (HR 0.54 [95% CI 0.32–0.93]), suggesting that the synergistic effect of the PARPi-ARPI combination may exist irrespective of HRR mutational status [48]. Building on this pre-clinical and early clinical data, three pivotal phase III trials were performed evaluating different variations of PARPi-ARPI combinations in the first-line mCRPC setting — PROpel, MAGNITUDE and TALAPRO-2.
In the phase III PROpel trial, 796 patients with mCRPC were randomised 1:1 to receive olaparib 300 mg BD or placebo, in combination with abiraterone acetate 1000 mg and prednisolone 10 mg daily [49, 50]. Patients were naïve to treatment in the mCRPC setting and were permitted to have received prior docetaxel if given in the localised or mHSPC settings, and an ARPI (other than abiraterone acetate) if ceased at least 12 months prior to randomisation. The primary endpoint was investigator-assessed rPFS, with OS as a key secondary endpoint. Patients were stratified according to location of metastatic sites, and prior exposure to docetaxel. HRR mutation status was not known at the time of enrolment and randomisation, and genomic testing was performed retrospectively using plasma ctDNA (using FoundationOne Liquid CDX) and/or tissue (using FoundationOne CDX) samples. Based on these results, patients were classified as either HRRm (see Table 1 for list of genes tested), non-HRRm or HRRm unknown. The primary analysis revealed that the median rPFS in the intention-to-treat cohort was significantly longer in the experimental arm compared with the control arm (24.8 vs 16.6 months, HR 0.66 [95% CI 0.54–0.81], p < 0.001). All pre-specified HRRm gene subgroups benefitted more from the experimental arm, with the greatest magnitude of benefit seen in patients with a BRCA mutation (HR 0.23 [95% CI 0.12–0.43]), followed by the overall HRRm subgroup (HR 0.50 [95% CI 0.34–0.73]) and then non-HRRm (HR 0.76 [95% CI 0.60–0.97]). The final OS analysis (median follow-up time of 36.6 months) also favoured the experimental arm in the intention-to-treat population (42.1 vs 34.7 months, HR 0.81 [95% CI 0.67–1.00], p = 0.054). The greatest OS benefit was again seen in the BRCA subgroup (HR 0.29 [95% CI 0.14–0.56]), with the benefit less clear in the non-HRRm subgroup (HR 0.89 [95% CI 0.70–1.14]) [49]. Though adverse events were in keeping with the known toxicity profile of either drug, the addition of olaparib to abiraterone acetate did increase toxicity overall. Most notably, anaemia was the most common adverse event occurring in 50% of patients compared with 18% in the control group (grade 3 or higher anaemia occurring in 16% vs 3%, respectively). Seventy-two (18%) patients required at least one blood transfusion in the experimental arm. A higher rate of venous thromboembolism (9% vs 4%), fatigue (39% vs 30%), nausea (31% vs 14%) and diarrhoea (21% vs 11%) were also noted. Following these results, olaparib in combination with abiraterone acetate and prednisolone was approved by the FDA for use in patients with mCRPC with a BRCA mutation only [51]. Conversely, the European Medicines Agency approved the combination in 2022 for use in all patients with mCRPC for whom chemotherapy is not clinically indicated [52].
The benefit of olaparib and abiraterone acetate compared to either agent alone in patients with inactivating germline or somatic BRCA1, BRCA2 or ATM mutant mCRPC was further evaluated in the ongoing phase II BRCAAway study (NCT03012321). In this trial, 165 patients were randomised to either abiraterone acetate, olaparib or the combination with cross-over permitted in the monotherapy arms. Importantly, the combination arm had a significantly longer median PFS of 39 months (compared with 14 months in olaparib arm, HR 0.32 [95% CI 0.14–0.75] and 8.4 months in the abiraterone acetate arm, HR 0.28 [95% CI 0.13–0.65]). These results possibly support combining abiraterone acetate and olaparib rather than using these drugs sequentially, though the study is limited by small patient numbers, and OS is yet to be reported [53].
The phase III MAGNITUDE trial similarly evaluated the combination of niraparib 200 mg daily versus placebo in combination with abiraterone acetate and prednisolone as first-line therapy for patients with mCRPC [54]. Patients could have received prior docetaxel in the mHSPC setting, and up to a 4-month lead-in of abiraterone acetate for mCRPC prior to enrolment. Patients underwent prospective testing for HRR status (see Table 1 for genes tested) using tissue and/or plasma samples. In contrast to the PROpel trial, these results then informed which cohort the patients would enrol into (HRRm vs non-HRRm) and were randomised 1:1 subsequently to either treatment arm. The primary endpoint was rPFS according to blinded independent central review. The first interim analysis and pre-planned futility analysis of the non-HRRm cohort found no benefit with adding niraparib to abiraterone acetate for the prespecified composite endpoint (first of prostate-specific antigen progression or rPFS) [HR 1.09; 95% CI 0.75–1.57] [55]. Consequently, futility was declared and the non-HRRm cohort was closed to enrolment following this analysis. The second interim analysis was performed after a median follow-up of 24.8 months for the BRCA1/2 subgroup and 26.8 months for the HRRm cohort (423 patients). The median rPFS in the BRCA1/2 subgroup was 19.5 versus 10.9 months (HR 0.55 [95% CI 0.39–0.78], p = 0.0007), with the benefit also extending to the HRRm cohort (HR 0.76 [95% CI 0.60–0.97], p = 0.028). A pre-specified analysis of patients harbouring a mutation in ATM, BRIP1, CDK12, CHEK2, FANCA, HDAC2 and PALB2 was performed, which demonstrated favourable rPFS outcomes for all patients except for those with ATM and CDK12 mutations. Overall survival at this timepoint was immature; however, favoured the experimental arm for the BRCA1/2 cohort but not the HRRm cohort (HR 0.88 [95% CI 0.58–1.34], p = 0.5505; vs HR 1.01 [95% CI 0.75–1.36], p = 0.948) [54]. A pre-specified inverse probability censoring weighting analysis of OS, which considered subsequent life-prolonging therapies including PARPi use, reported favourable survival outcomes in both cohorts for the combination (BRCA1/2 cohort: HR 0.54, [95% CI 0.33–0.90], p = 0.0181; HRRm cohort: HR 0.70 [95% CI 0.49–0.99], p = 0.0414) [56]. The final analysis with mature OS (unadjusted) after a median follow-up of 35.9 months focussed only on the BRCA1/2 cohort, and demonstrated a modest OS benefit favouring the experimental arm (30.4 vs 28.6 months, HR 0.79 [95% CI 0.55–1.12], p = 0.1828) [57]. These OS results are in contrast to the PROpel study, and potentially may be impacted by the subset (23.6% in experimental arm and 22.7% in the control arm) of patients in the experimental arm (HRRm population) who had received up to 4 months of abiraterone acetate before randomisation. Toxicity was higher in the experimental arm (frequency of grade ≥ 3 adverse events 72.2% vs 49.3%), with the most common ≥ grade 3 adverse events being anaemia (30.2% vs 8.5%) and hypertension (15.6% vs 12.3%). Following these results, the FDA approved the combination of niraparib and abiraterone acetate with prednisolone as first-line therapy for patients with BRCA-mutant mCRPC [58].
Third, the phase III TALAPRO-2 trial evaluated the combination of talazoparib with enzalutamide as a first-line treatment for patients with mCRPC [59, 60]. Patients were enrolled sequentially into two cohorts, the first being the HRR-unselected cohort (all-comers population), which included 169 patients with a HRR mutation and 636 without a known mutation. The second cohort selected for patients with an identified HRR mutation (n = 230), to form a combined cohort of 399 patients with HRRm mCRPC. Eligible patients could have had prior exposure to docetaxel and abiraterone acetate in the mHSPC setting. Patients underwent prospective testing for HRR mutation status (see Table 1 for genes tested) and were randomised 1:1 into the experimental or control arm. Stratification occurred according to HRR status (HRRm vs non-HRRm vs unknown) as well as prior exposure to docetaxel or abiraterone acetate in the mHSPC setting. The primary endpoint was rPFS according to a blinded independent central review (in both all-comers and in the HRRm cohort), with OS a key secondary endpoint. At the planned primary analysis of the all-comers cohort, the median rPFS was not yet reached in the experimental arm for the intention-to-treat cohort, with a clear trend favouring the combination (NR vs 21.9 months, HR 0.63 [95% CI 0.51–0.78], p = <0.0001), suggesting that the combination confers a clinically meaningful benefit regardless of HRR status [59]. The recent analysis of the combined HRRm cohort (n = 399) after a median follow-up of 17.5 and 16.8 months in the experimental and control arms, respectively, demonstrated that the combination arm had longer rPFS (NR vs 13.8 months, HR 0.45 [95% CI 0.33–0.61], p < 0.0001) [60]. The OS data remains immature, with a trend in favour of the combination (HRRm cohort: HR 0.69 [95% CI 0.46–1.03], p = 0.07; BRCA mutant cohort: HR 0.61 [95% CI 0.31–1.23], p = 0.16]).
Importantly, in the all-comers intention-to-treat cohort, 28% and 38% had an undetermined HRR or BRCA mutation status, respectively. A post hoc analysis of the BRCA and HRRm subgroups attempted to explore whether the high proportion of ‘undetermined’ patients may in fact be unidentified biomarker-positive patients and, therefore, potentially contribute to the overall benefit seen in the intention-to-treat cohort [61]. For the BRCA1/2 mutated cohort (10% of the intention-to-treat population), a clear benefit was seen in the experimental arm for both rPFS and OS (HR 0.24 [95% CI 0.12–0.49] and HR 0.53 [95% CI 0.28–1.03], respectively). Similar results were seen in the HRRm subgroup (HRs 0.51 [95% CI 0.36–0.72] and 0.68 [95% CI 0.47–0.99], respectively). Notably, an rPFS benefit was also seen in the non-BRCA1/2 mutated cohort (median 33.1 vs 22.1 months, HR 0.71 [95% CI 0.52–0.96]) and non-HRRm cohort (NR-22.1 months, HR 0.69 [95% CI 0.49–0.98]). An OS trend favouring the experimental arm was also seen in both the non-BRCA1/2 mutated and non-HRRm cohorts (HRs 0.76 [95% CI 0.56–1.03] and 0.88 [95% CI 0.63–1.23], respectively). For the BRCA1/2 and HRR undetermined cohort, the magnitude of benefit appeared smaller (higher HRs) than the BRCA1/2 mutated and HRRm cohorts, as well as the non-BRCA1/2 and non-HRRm cohorts (undetermined BRCA1/2 median rPFS NR-27.3 months, HR 0.75 [95% CI 0.53–1.07], undetermined HRRm median rPFS NR to 27.3 months, HR 0.73 [95% CI 0.47–1.13]). This analysis provides some reassurance that the inclusion of the ‘undetermined’ cohort in the non-BRCA1/2 mutated and non-HRRm population in the intention-to-treat analysis did not significantly impact results, and falsely augment the survival benefit seen in the all-comers cohort [61]. Adverse events occurred more frequently in the experimental arm (≥ grade 3 adverse events 75% vs 45%). Anaemia was again the most common adverse event and most common cause for a dose reduction, occurring in 66% of the experimental arm compared with 17% in the control arm. Following this study, the FDA approved the combination of talazoparib and enzalutamide as first-line treatment for HRRm mCRPC [62]. The combination has also recently been approved by the European Medicines Agency for use in all patients with mCRPC in whom chemotherapy is not indicated [63].
A pooled analysis of multiple trials evaluating PARPis as first-line therapy for patients with HRRm mCRPC was performed by the FDA [64]. This study analysed individual patient data from the PROfound, PROpel, MAGNITUDE, TALAPRO-1, TALAPRO-2 and TRITON-2 trials. Differential results were observed by specific gene, with treatment benefit appearing greatest with BRCA1, BRCA2, CDK12 and PALB2 mutations, and an apparent lack of benefit seen in CHEK2 and ATM mutations. In terms of ARPI-PARPi combinations, a meta-analysis of the PROpel, MAGNITUDE and TALAPRO-2 trials confirmed a clear hierarchy of benefit based on HRR mutation status [65]. Pooled results for the shared primary endpoint of rPFS found a 35% rPFS improvement in the all-comer population (HR 0.65, 95% CI 0.56–0.76, p < 0.001), with a differential magnitude of benefit depending on the subgroup by HRR status. The greatest rPFS benefit was seen in the BRCA1/2 mutated cohort (HR 0.32, 95% CI 0.17–0.61, p < 0.001), followed by the HRRm cohort (HR 0.55, 95% CI 0.39–0.77, p < 0.001), and non-HRRm (HR 0.74, 95% CI 0.61–0.90, p = 0.003). Toxicity was increased in the combination arm across the three studies, with a 45% increase in the relative risk of ≥ grade 3 treatment-related adverse events, in particular for ≥ grade 3 anaemia (31.9% vs 4.9%).
There is robust evidence supporting the use of an ARPI-PARPi combination in the BRCA1/2 mutated subgroup, and treatment intensification should be considered in these patients. A benefit is also seen in the HRRm patient population, though differential responses occur depending on the specific gene mutation present. This is reflected in some of the regulatory approvals restricting use of an ARPI-PARPi combination to BRCA-mutated patients only. The benefit in the all-comers and non-HRRm populations is less clear, and needs to be balanced with the increased toxicity seen with combination therapy.
Several trials are ongoing that will further evaluate an ARPI-PARPi combination in mCRPC and hopefully clarify which patients or subgroup benefit from treatment intensification (see Table 2). In particular, the CASPAR trial (NCT04455750) is a randomised phase III study in which patients receive rucaparib versus placebo in combination with enzalutamide as first-line treatment for mCRPC. Patients are enrolled regardless of HRR mutational status. This is the only study incorporating both rPFS and OS as co-primary endpoints [66].
5 Method of HRR Mutation Status Testing
Tissue testing remains the gold standard currently for assessing HRR alterations in mCRPC. Although obtaining a fresh metastatic tumour biopsy is ideal, from a pragmatic point of view sampling bone metastases or deep intra-abdominal/pelvic lymph node metastases can be challenging. As a result, archival tissue is typically used to identify HRR alterations and, in most pivotal trials of PARPi in mCRPC, was used for molecular testing. At the same time, the use of archival tissue can be limited by DNA degradation, given that it is often many years old and may not be representative of the metastatic clone [67, 68]. There is increasing awareness of the role that testing plasma ctDNA may play in identifying HRR aberrations. Although reliable detection of biallelic gene inactivation in the setting of low ctDNA fraction is a limitation of ctDNA [69], an analysis from TALAPRO-2 showed high agreement (95%) between HRR status of tumour tissue and ctDNA [70]. Last, testing for germline-only HRR mutations can be performed using blood or saliva samples
A key difference across the PROpel, MAGNITUDE and TALAPRO-2 studies was the variation in the method of HRR mutational testing. The PROpel trial retrospectively analysed the HRR status of each patient after enrolment and randomisation had occurred, with the intention-to-treat cohort being HRR-unselected. Almost all patients (98%) provided samples of tumour tissue (mostly archival) and/or blood samples for ctDNA analysis, and blood germline samples. The HRR mutation status was determined using tumour tissue for 68% of patients [71]. In contrast, the MAGNITUDE and TALAPRO-2 studies both prospectively analysed the HRR status prior to randomisation and used tissue samples in 69% and 100% of patients, respectively (in addition to ctDNA for some patients) [59, 70, 72]. The range of HRR-related genes tested in each study also varied (see Table 1). All studies utilised the FoundationOne®CDx panel for tumour tissue analysis, though the key HRR-related genes included as part of each trial varied. BRCA1, BRCA2, ATM, PALB2, CDK12 and CHEK2 mutations were included in the HRRm cohort of all three studies, while inclusion of genes in the RAD and FANC families was heterogeneous. This leads to lingering questions about whether patients harbouring these mutations benefit from combination therapy. Moving forwards, prospective studies should ideally include a consistent and wider range of HRR-related genes to provide further clarification on who derives benefit from combination ARPI-PARPi therapy.
6 Choice of ARPI and PARPi: Does it Matter?
Several key differences exist between these three trials that may contribute to the heterogeneity in outcomes. The most obvious difference is the variation in drugs utilised. Both PROpel and MAGNITUDE used abiraterone acetate as the ARPI, whilst TALAPRO-2 used enzalutamide. Enzalutamide, as an AR antagonist (along with darolutamide and apalutamide), competitively inhibits androgen binding to the AR whilst also preventing translocation of the androgen-AR complex to the nucleus, and, therefore, inhibits transcription of downstream proliferative and cell survival pathways [73, 74]. In contrast, abiraterone acetate is an androgen biosynthesis inhibitor via irreversible inhibition of 17α-hydroxylase/C17,20-lyase (CYP17). Inhibition of CYP17 enzymes prevents testosterone synthesis, and thereby also reduces downstream transcription pathways of the AR due to androgen suppression [75]. A subset of DNA repair genes are under transcriptional control of the AR (including BRCA1, and genes from RAD and FANC families), so it follows that inhibition of the AR results in transcriptional downregulation of key HRR genes, inducing a consequent functional ‘HRR deficient’ phenotype [39, 76]. Whilst pre-clinical data are only available for enzalutamide to the best of our knowledge, it can be assumed that the same synergistic effect is seen with abiraterone acetate [38, 39]. The choice of ARPI therefore is not likely to impact the interaction with a PARPi, though differences in pharmacokinetics, drug interactions and toxicity must also be considered.
Conversely, the choice of PARPi is likely more impactful on outcomes given the variation in the degree of PARP1 and PARP2 inhibition, PARP trapping, drug interactions and toxicity (see Table 3). The inhibition constant (KI) for PARP1 is highest for talazoparib, followed by olaparib and then niraparib [77]. PARP2, in particular, is thought to have a crucial role in erythropoiesis [78], and therefore, potent enzyme inhibition may be a key driver of anaemia [79]. For PARP2, both olaparib and talazoparib exert a similar degree of inhibition (median 0.2 nM), with niraparib again the least potent PARPi. In vivo data suggest that talazoparib has the greatest potency in both BRCA− and BRCA+ cell lines [77]. All PARPi bind to the catalytic domain of PARP1 protein, resulting in interruption of the ADP-ribosylation process and trapping of PARP1 within chromatin. The strength of PARP1 trapping varies across different PARPi owing to differing molecular structures [80]. Talazoparib has the most potent PARP1 trapping capacity when compared to olaparib and niraparib [81]. In vitro data suggest the cytotoxicity resulting from PARP1 trapping leads to greater radiosensitisation, though there are no convincing data that this translates to increased efficacy when used in monotherapy and may lead to increased toxicity [81, 82]. Consequently, the highest incidence of ≥ grade 3 toxicity has been observed with talazoparib when combined with enzalutamide, and this study had the highest incidence of treatment discontinuation (see Table 1).
Importantly, the dose of PARPi when combined with an ARPI in some cases is reduced compared with the standard monotherapy dose. In the MAGNITUDE trial, niraparib 200 mg daily was administered in combination with abiraterone acetate compared to the standard monotherapy dose of 300 mg daily. This was because of an earlier phase Ib trial demonstrating increased toxicity when niraparib 300 mg daily was combined with either apalutamide 240 mg daily or abiraterone acetate 1000 mg with prednisolone 10 mg daily, and consequently, the recommended phase II dose when combined with abiraterone acetate was 200 mg daily[83]. The impact of this dose reduction may have compromised drug potency and therefore efficacy. In this study, niraparib exposures were lower in combination with apalutamide compared with when used alone. This was hypothesised to be due to apalutamide inducing the metabolism of niraparib via a drug–drug interaction. This effect was not seen when combined with abiraterone acetate, with niraparib exposure being dose proportional and in keeping with previous data for monotherapy exposure. Similarly, a lower dose of talazoparib (0.5 mg daily compared to 1 mg daily) was administered in TALAPRO-2 when combined with enzalutamide. This was based on a run-in study to TALAPRO-2 that identified a drug–drug interaction that resulted in a similar steady state of talazoparib exposure with 0.5 mg daily as seen with 1 mg daily when used as monotherapy [84].
7 Where Should We Sequence the ARPI-PARPi Combination in the Evolving Landscape of Metastatic Prostate Cancer?
Since the PROpel, MAGNITUDE and TALAPRO-2 trials commenced patient recruitment, there have been significant advances and changes made to the treatment landscape for prostate cancer. An ARPI in combination with ADT is now considered standard-of-care treatment for mHSPC. This complicates the interpretation of such first-line mCRPC studies, with the majority of patients in these studies being ARPI naïve prior to enrolment. Considering the efficacy of an ARPI switch is generally low [86], the impact of a prior ARPI on the efficacy of a subsequent ARPI-PARPi combination is an important question. The MAGNITUDE and TALAPRO-2 trials both allowed a prior alternate ARPI if given in the mHSPC setting. In the MAGNITUDE study, only a minority of patients had received an alternate ARPI for either mHSPC or nmCRPC (eight and five patients in each arm). In these subgroups, the median rPFS favoured the niraparib arm in both the HRRm and BRCA1/2 cohorts, though data are limited by very small numbers (HRRm cohort: not evaluable vs 4.3 months, HR 0.19 [95% CI 0.03–1.23]; BRCA1/2 cohort: not evaluable vs 4.3 months, HR 0.11 [95% CI 0.01–1.12]). In TALAPRO-2, 16 patients in the HRRm intention-to-treat cohort had received prior abiraterone acetate in both the experimental and the placebo arms. A subgroup analysis for rPFS reported outcomes for patients based on prior treatment/s and found that the benefit of talazoparib was maintained regardless of prior abiraterone acetate or docetaxel (HR 0.43 [95% CI 0.26–0.70]). Outcomes for patients who received prior abiraterone acetate were not independently reported. In the PROpel trial, only one patient received a prior ARPI for mHSPC. It is difficult to extrapolate from these data whether the ARPI-PARPi combination following progression on a prior ARPI remains an efficacious therapeutic option. The ongoing CASPAR and FUZUPRO trials may provide further insights given they both allow prior ARPI for mHSPC (see Table 2) [66].
Differential responses to treatment have previously been reported in prostate cancer depending on whether the treatment is sequenced before or after taxane chemotherapy. Between 20% and 23% of patients in the PROpel, MAGNITUDE and TALAPRO-2 trials had received prior docetaxel for mHSPC, and all trials stratified patients based on prior taxane exposure. In terms of rPFS, patients in the PROpel intention-to-treat population derived a similar benefit from the experimental arm regardless of prior docetaxel (HR 0.61 in the post-docetaxel cohort [95% CI 0.40–0.92], compared to HR 0.71 [95% CI 0.56–0.89] for docetaxel naïve patients). Similar outcomes were seen in the intention-to-treat population for OS (HR 0.76 in the post-docetaxel group [95% CI 0.52–1.11] vs HR 0.85 for the docetaxel-naïve group [95% CI 0.67–1.07]) [49]. In TALAPRO-2, a subgroup analysis for the post-docetaxel cohort specifically was not available in the all-comer intention-to-treat cohort. The HR for median rPFS for HRRm patients was similar in the overall (0.45 [95% CI 0.33–0.61]) and post-docetaxel (0.46 [0.31–0.69]) cohorts [60]. In the MAGNITUDE trial, however, the extent of benefit in terms of median rPFS was higher for HRRm patients who were taxane naïve (16.6 vs 13.8 months, HR 0.71 [95% CI 0.53–0.96]) compared with those who had received prior docetaxel (13.4 months vs 10.9 months in the placebo arm, HR 0.89 [95% CI 0.48–1.66]). Further, for the BRCA1/2 subgroup, patients who had received prior docetaxel did not appear to benefit from the experimental arm (median rPFS for post-docetaxel subgroup 13.4 months vs 13.7 months, HR 0.98 [95% CI 0.48–2.02]). For the taxane-naïve subgroup, median rPFS favoured the experimental arm (22.2 vs 10.9 months, HR 0.47 [95% CI 0.31–0.69]). These results suggest that HRRm patients may derive a greater benefit from the ARPI-PARPi combination if given prior to chemotherapy, particularly for patients harbouring a BRCA1/2 mutation. It is important to note that though these studies stratified outcomes based on prior docetaxel, they were not powered to analyse the pre-docetaxel and post-docetaxel cohorts as an endpoint. Further, in all three aforementioned studies, docetaxel was given for mHSPC, which typically is a limited course of up to six cycles, and often ceased before treatment resistance occurs. Importantly, TRITON-2 is the only trial to directly compare a PARPi to docetaxel given in the mCRPC setting. Data from both the PROfound and TRITON-2 studies support the use of PARPi monotherapy before docetaxel chemotherapy after progression on an ARPI for patients with a BRCA1/2 mutation [33, 34]. Further investigation with prospective studies is needed to answer the question of optimal treatment sequencing of an ARPI-PARPi combination with regard to docetaxel given in the mCRPC setting.
8 Future Perspectives
Early treatment intensification remains a key focus in the management of advanced prostate cancer, with a shift towards implementing combination therapies at diagnosis of metastatic disease. As such, ARPI-PARPi combinations are now being studied in the mHSPC setting (see Table 4). In particular, the AMPLITUDE trial (NCT04497844) [87] is evaluating whether the combination of niraparib with abiraterone acetate improves rPFS in HRR-selected patients. TALAPRO-3 (NCT04821622) [88] follows on from TALAPRO-2 and explores the same combination in a HRR-selected mHSPC cohort. Importantly, two phase II trials are evaluating the combination of talazoparib with an ARPI in HRR-unselected mHSPC cohorts (NCT04734730, NCT04332744) [89, 90]. These studies will assist with identifying which patients or subgroups derive benefit from the combination treatment in an earlier setting.
Moreover, the development of selective PARP1 inhibitors may overcome the haematological toxicity that is thought to result predominantly from PARP2 inhibition, thereby potentially allowing dosing that results in greater PARP1 inhibition and improved efficacy while also improving drug tolerability. High-grade anaemia was the most common adverse effect in the PROpel, MAGNITUDE, and TALAPRO-2 trials, and resulted in PARPi dose reductions or discontinuation in some cases. Both PARP1 and PARP2 bind to sites of DNA damage, and olaparib, niraparib and talazoparib inhibit both enzymes to varying degrees. The mechanism resulting in synthetic lethality in HRR-deficient cells, however, is thought to be reliant solely on the loss of PARP1 activity [91, 92]. Next-generation PARPis that selectively inhibit and trap PARP1, therefore, have become an area of interest, with several currently in development. Saruparib (AZD5305) is a first-in-class selective inhibitor and trapper of PARP1, and demonstrated minimal haematological toxicity in pre-clinical models [93]. It has been evaluated in the phase I/IIa PETRA trial (NCT04644068) in multiple HRRm tumour types and demonstrated promising clinical activity with favourable tolerability [94]. Saruparib in combination with an ARPI in metastatic prostate cancer was shown to be well tolerated in the PETRANHA trial [95], and continues to be evaluated in the phase III EvoPAR-Prostate01 (NCT06120491) trial in both HRRm and non-HRRm cohorts.
9 Conclusions
Concurrent inhibition of AR and PARP with an ARPI-PARPi combination in patients with mCRPC leads to therapeutic lethal synergy due to several mechanisms. This translates to improved survival outcomes for some patients, as demonstrated by three pivotal, phase III randomised trials evaluating ARPI-PARPi combinations as first-line treatment for mCRPC. A hierarchy of benefit is evident, with patients harbouring a BRCA mutation gaining the most benefit from an ARPI-PARPi combination, followed by other selected pathogenic HRR mutations, and last, HRR-unselected patients or non-HRRm patients deriving the lowest magnitude of benefit. Given these differential responses, and the potential impact of prior docetaxel on efficacy, early screening for HRR mutations is crucial to guide treatment choices. Because of the rapidly changing treatment landscape for advanced prostate cancer, and the expectation that most patients will now receive an ARPI for mHSPC, the interpretation of studies such as PROpel, MAGNITUDE, and TALAPRO-2 is not straightforward. As more selective PARPis are developed and with several trials underway in both HRR-selected and HRR-unselected populations in mHSPC, we will hopefully gain further insights into the benefits of ARPI-PARPi combinations in prostate cancer. Identification of patients and subgroups who derive greatest benefit from treatment intensification will guide the treatment paradigm moving forwards.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
James ND, Tannock I, N’Dow J, Feng F, Gillessen S, Ali SA, et al. The Lancet Commission on prostate cancer: planning for the surge in cases. Lancet. 2024;403:1683–722.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972;22(4):232–40.
Posdzich P, Darr C, Hilser T, Wahl M, Herrmann K, Hadaschik B, et al. Metastatic prostate cancer: a review of current treatment options and promising new approaches. Cancers (Basel). 2023;15(2):461.
Khoshkar Y, Westerberg M, Adolfsson J, Bill-Axelson A, Olsson H, Eklund M, et al. Mortality in men with castration-resistant prostate cancer: a long-term follow-up of a population-based real-world cohort. BJUI Compass. 2022;3(2):173–83.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701–11.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97.
Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13(10):983–92.
Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152–60.
Cai M, Song X-L, Li X-A, Chen M, Guo J, Yang D-H, et al. Current therapy and drug resistance in metastatic castration-resistant prostate cancer. Drug Resist Updat. 2023;68: 100962.
Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4(3):365–80.
van Dessel LF, van Riet J, Smits M, Zhu Y, Hamberg P, van der Heijden MS, et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat Commun. 2019;10(1):5251.
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443–53.
Valsecchi AA, Dionisio R, Panepinto O, Paparo J, Palicelli A, Vignani F, et al. Frequency of germline and somatic BRCA1 and BRCA2 mutations in prostate cancer: an updated systematic review and meta-analysis. Cancers (Basel). 2023;15(9):2435.
Fettke H, Dai C, Kwan EM, Zheng T, Du P, Ng N, et al. BRCA-deficient metastatic prostate cancer has an adverse prognosis and distinct genomic phenotype. EBioMedicine. 2023;95: 104738.
Kohli M, Tan W, Zheng T, Wang A, Montesinos C, Wong C, et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine. 2020;54: 102728.
Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-B: a prospective cohort study of the impact of germline DNA repair mutations on the putcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37(6):490–503.
Olmos D, Lorente D, Alameda D, Cattrini C, Romero-Laorden N, Lozano R, et al. Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Ann Oncol. 2024;35:458–72.
Warner E, Herberts C, Fu S, Yip S, Wong A, Wang G, et al. BRCA2, ATM, and CDK12 defects differentially shape prostate tumor driver genomics and clinical aggression. Clin Cancer Res. 2021;27(6):1650–62.
Tuffaha H, Edmunds K, Fairbairn D, Roberts MJ, Chambers S, Smith DP, et al. Guidelines for genetic testing in prostate cancer: a scoping review. Prostate Cancer Prostatic Dis. 2023. https://doi.org/10.1038/s41391-023-00676-0.
Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20(17):4704–16.
Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–72.
Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61(12):4842–50.
Patel PS, Algouneh A, Hakem R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: where we stand. Oncogene. 2021;40(17):3001–14.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162–74.
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38(32):3763–72.
de Bono JS, Mehra N, Scagliotti GV, Castro E, Dorff T, Stirling A, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol. 2021;22(9):1250–64.
Smith MR, Scher HI, Sandhu S, Efstathiou E, Lara PN Jr, Yu EY, et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2022;23(3):362–73.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–102.
Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383(24):2345–57.
Bono JSD, Matsubara N, Penel N, Mehra N, Kolinsky MP, Bompas E, et al. Exploratory gene-by-gene analysis of olaparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): PROfound. J Clin Oncol. 2021;39(6_Suppl.):126.
Mateo J, de Bono JS, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for the treatment of patients with metastatic castration-resistant prostate cancer and alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J Clin Oncol. 2024;42(5):571–83.
Fizazi K, Piulats JM, Reaume MN, Ostler P, McDermott R, Gingerich JR, et al. Rucaparib or physician’s choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719–32.
FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer [press release]. US Food and Drug Administration; 2020.
Anscher MS, Chang E, Gao X, Gong Y, Weinstock C, Bloomquist E, et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA-mutated metastatic castrate-resistant prostate cancer. Oncologist. 2021;26(2):139–46.
Lynparza approved in the EU for the treatment of BRCA-mutated metastatic castration-resistant prostate cancer [press release]. AstraZeneca; 2020.
Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8(1):374.
Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10(480):eaam7479.
Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2(12):1134–49.
Makhov P, Fazliyeva R, Tufano A, Uzzo RG, Kolenko VM. Examining the effect of PARP-1 inhibitors on transcriptional activity of androgen receptor in prostate cancer cells. Methods Mol Biol. 2023;2609:329–35.
Gui B, Gui F, Takai T, Feng C, Bai X, Fazli L, et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc Natl Acad Sci USA. 2019;116(29):14573–82.
Yin Y, Li R, Xu K, Ding S, Li J, Baek G, et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res. 2017;77(18):4745–54.
Kounatidou E, Nakjang S, McCracken SRC, Dehm SM, Robson CN, Jones D, et al. A novel CRISPR-engineered prostate cancer cell line defines the AR-V transcriptome and identifies PARP inhibitor sensitivities. Nucleic Acids Res. 2019;47(11):5634–47.
Chakraborty G, Armenia J, Mazzu YZ, Nandakumar S, Stopsack KH, Atiq MO, et al. Significance of BRCA2 and RB1 co-loss in aggressive prostate cancer progression. Clin Cancer Res. 2020;26(8):2047–64.
Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;116(23):11428–36.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19(7):975–86.
Carr TH, Adelman C, Barnicle A, Kozarewa I, Luke S, Lai Z, et al. Homologous recombination repair gene mutation characterization by liquid biopsy: a phase II trial of olaparib and abiraterone in metastatic castrate-resistant prostate cancer. Cancers (Basel). 2021;13(22):5830.
Saad F, Clarke NW, Oya M, Shore N, Procopio G, Guedes JD, et al. Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(10):1094–108.
Clarke NW, Armstrong AJ, Thiery-Vuillemin A, Oya M, Shore N, Loredo E, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evid. 2022;1(9):EVIDoa2200043.
Fallah J, Xu J, Weinstock C, Brave MH, Bloomquist E, Fiero MH, et al. FDA approval summary: olaparib in combination with abiraterone for treatment of patients with BRCA-mutated metastatic castration-resistant prostate cancer. J Clin Oncol. 2024;42(5):605–13.
Lynparza in combination with abiraterone approved in the EU as 1st-line treatment for patients with metastatic castration-resistant prostate cancer [press release]. AstraZeneca, 2022.
Hussain MHA, Kocherginsky M, Agarwal N, Zhang J, Adra N, Paller CJ, et al. BRCAAWAY: a randomized phase 2 trial of abiraterone, olaparib, or abiraterone + olaparib in patients with metastatic castration-resistant prostate cancer (mCRPC) with DNA repair defects. J Clin Oncol. 2022;40(16_Suppl.):5018.
Chi KN, Rathkopf D, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Niraparib and abiraterone acetate for metastatic castration-resistant prostate cancer. J Clin Oncol. 2023;41(18):3339–51.
Chi K, Rathkopf D, Smith M, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol. 2022;40:12.
Chi KN, Sandhu S, Smith MR, Attard G, Saad M, Olmos D, et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: second interim analysis of the randomized phase III MAGNITUDE trial. Ann Oncol. 2023;34(9):772–82.
Chi KNN, Castro E, Attard G, Smith MR, Sandhu SK, Efstathiou E, et al. LBA85 Niraparib (NIRA) with abiraterone acetate plus prednisone (AAP) as first-line (1L) therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and homologous recombination repair (HRR) gene alterations: three-year update and final analysis (FA) of MAGNITUDE. Ann Oncol. 2023;34:S1326.
FDA approves new combined treatment for advanced prostate cancer. Cancer. 2023;129(24):3841–2.
Agarwal N, Azad AA, Carles J, Fay AP, Matsubara N, Heinrich D, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. Lancet. 2023;402(10398):291–303.
Fizazi K, Azad AA, Matsubara N, Carles J, Fay AP, De Giorgi U, et al. First-line talazoparib with enzalutamide in HRR-deficient metastatic castration-resistant prostate cancer: the phase 3 TALAPRO-2 trial. Nat Med. 2024;30(1):257–64.
Shore N, Agarwal N, Azad A, Carles J, Fay A, Matsubara N, et al. Post hoc analysis of rPFS and OS from the TALAPRO-2 (TP-2) study: genomic subgroups based on likelihood of BRCA or HRR gene alteration status. J Clin Oncol. 2024;42:136.
Heiss BL, Chang E, Gao X, Truong T, Brave MH, Bloomquist E, et al. US Food and Drug Administration approval summary: talazoparib in combination with enzalutamide for treatment of patients with homologous recombination repair gene-mutated metastatic castration-resistant prostate cancer. J Clin Oncol. 2024;42(15):1851–60.
European Commission approves Pfizer’s Talzenna® in combination with Xtandi® for adult patients with metastatic castration-resistant prostate cancer [press release]. BusinessWire2024.
Fallah J, Xu J, Weinstock C, Gao X, Heiss BL, Maguire WF, et al. Efficacy of poly(ADP-ribose) polymerase inhibitors byiIndividual genes in homologous recombination repair gene-mutated metastatic castration-resistant prostate cancer: a US Food and Drug Administration pooled analysis. J Clin Oncol. 2024;42:1687–98.
Sayyid RK, Klaassen Z, Berlin A, Roy S, Brandão LR, Bernardino R, et al. Poly(adenosine diphosphate-ribose) polymerase inhibitor combinations in first-line metastatic castrate-resistant prostate cancer setting: a systematic review and meta-analysis. BJU Int. 2023;132(6):619–30.
Rao A, Heller G, Ryan C, VanderWeele D, Lewis L, Tan A, et al. Alliance A031902 (CASPAR): a randomized, phase (ph) 3 trial of enzalutamide with rucaparib/placebo as novel therapy in first-line metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2022;40:TPS194-TPS.
Scott RJ, Mehta A, Macedo GS, Borisov PS, Kanesvaran R, El Metnawy W. Genetic testing for homologous recombination repair (HRR) in metastatic castration-resistant prostate cancer (mCRPC): challenges and solutions. Oncotarget. 2021;12(16):1600–14.
Antonarakis ES, Gomella LG, Petrylak DP. When and how to use PARP inhibitors in prostate cancer: a systematic review of the literature with an update on on-going trials. Eur Urol Oncol. 2020;3(5):594–611.
Kwan EM, Wyatt AW, Chi KN. Towards clinical implementation of circulating tumor DNA in metastatic prostate cancer: opportunities for integration and pitfalls to interpretation. Front Oncol. 2022;12:1054497.
Azad A, Fizazi K, Matsubara N, Fong P, Carles J, Shore N, et al. Use of circulating tumor DNA (ctDNA) to complement tumor tissue homologous recombination repair (HRR) gene alteration testing in TALAPRO-2, a phase 3 study of talazoparib (TALA) + enzalutamide (ENZA) versus placebo (PBO) + ENZA as first-line (1L) treatment in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2023;41:5056.
Armstrong A, Saad F, Thiery-Vuillemin A, Oya M, Shore N, Mehra N, et al. 1370P Detection of mutations in homologous recombination repair (HRR) genes in tumour tissue (TT) and circulating tumour DNA (ctDNA) from patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) in the phase III PROpel trial. Ann Oncol. 2022;33:S1168.
Attard G, Sandhu SK, Rathkopf D, Castro E, Saad M, Smith MR, et al. 1806P Efficacy of niraparib and abiraterone acetate plus prednisone (NIRA+AAP) in homologous recombination repair gene-altered (HRR+) metastatic castration-resistant prostate cancer (mCRPC) by tissue and/or plasma assays in the MAGNITUDE trial. Ann Oncol. 2023;34:S976–7.
Vander Ark A, Cao J, Li X. Mechanisms and approaches for overcoming enzalutamide resistance in prostate cancer. Front Oncol. 2018;8:180.
Schalken J, Fitzpatrick JM. Enzalutamide: targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int. 2016;117(2):215–25.
Jacob A, Raj R, Allison DB, Myint ZW. Androgen receptor signaling in prostate cancer and therapeutic strategies. Cancers (Basel). 2021;13(21):5417.
Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3(11):1245–53.
Rudolph J, Jung K, Luger K. Inhibitors of PARP: number crunching and structure gazing. Proc Natl Acad Sci USA. 2022;119(11):e2121979119.
Farrés J, Llacuna L, Martin-Caballero J, Martínez C, Lozano JJ, Ampurdanés C, et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015;22(7):1144–57.
Lin X, Gupta D, Vaitsiankova A, Bhandari SK, Leung KSK, Menolfi D, et al. Inactive Parp2 causes Tp53-dependent lethal anemia by blocking replication-associated nick ligation in erythroblasts. bioRxiv. 2024.
Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science. 2020;368(6486):eaax6367.
Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, et al. PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res. 2019;17(2):409–19.
Laird JH Jr, Lok BH, Ma J, Bell A, De Stanchina E, Poirier J, et al. PARP trapping by talazoparib is a potent mechanism of radiosensitization in small cell lung cancer cell lines and patient-derived xenografts. Int J Radiat Oncol Biol Phys. 2018;102(3):e184–5.
Saad F, Chi KN, Shore ND, Graff JN, Posadas EM, Lattouf JB, et al. Niraparib with androgen receptor-axis-targeted therapy in patients with metastatic castration-resistant prostate cancer: safety and pharmacokinetic results from a phase 1b study (BEDIVERE). Cancer Chemother Pharmacol. 2021;88(1):25–37.
Agarwal N, Azad A, Shore ND, Carles J, Fay AP, Dunshee C, et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Future Oncol. 2022;18(4):425–36.
Antolin AA, Ameratunga M, Banerji U, Clarke PA, Workman P, Al-Lazikani B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci Rep. 2020;10(1):2585.
Khalaf DJ, Annala M, Taavitsainen S, Finch DL, Oja C, Vergidis J, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019;20(12):1730–9.
Rathkopf D, Chi K, Olmos D, Cheng H, Agarwal N, Graff J, et al. AMPLITUDE: a study of niraparib in combination with abiraterone acetate plus prednisone (AAP) versus AAP for the treatment of patients with deleterious germline or somatic homologous recombination repair (HRR) gene-altered metastatic castration-sensitive prostate cancer (mCSPC). J Clin Oncol. 2021;39:TPS176-TPS.
Agarwal N, Saad F, Azad AA, Mateo J, Matsubara N, Shore ND, et al. TALAPRO-3 clinical trial protocol: phase III study of talazoparib plus enzalutamide in metastatic castration-sensitive prostate cancer. Future Oncol. 2024;20(9):493–505.
Kasparian S, Burnham L, Kittles R, Sun Z, Bedell F, Tran J, et al. Identifying androgen receptor (AR) and genomic characteristics that define populations of patients with mHSPC who benefit from early PARP inhibition therapy with talazoparib. J Clin Oncol. 2021;39(15_Suppl.):TPS5097-TPS.
Mateo J, Borque Á, Castellano D, Castro E, Duran M, Font A, et al. A randomized phase 2 trial to evaluate the antitumor activity of enzalutamide (EZ) and talazoparib (TALA) for the treatment of metastatic hormone-naïve prostate cancer (mHNPC): ZZFIRST. J Clin Oncol. 2022;40:TPS209-TPS.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, et al. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun. 2018;9(1):746.
Illuzzi G, Staniszewska AD, Gill SJ, Pike A, McWilliams L, Critchlow SE, et al. Preclinical characterization of AZD5305, a next-generation, highly selective PARP1 inhibitor and trapper. Clin Cancer Res. 2022;28(21):4724–36.
Yap TA, Im S-A, Schram AM, Sharp A, Balmana J, Baird RD, et al. Abstract CT007: PETRA: first in class, first in human trial of the next generation PARP1-selective inhibitor AZD5305 in patients (pts) with BRCA1/2, PALB2 or RAD51C/D mutations. Cancer Res. 2022;82(12_Suppl.):CT007-CT.
Azad A, Voskoboynik M, Joshua AM, Weickhardt AJ, Sankey P, Pacey S, et al. PETRANHA: phase 1/2 study of AZD5305 + novel hormonal agents in patients with metastatic prostate cancer: interim safety and pharmacokinetic results. J Clin Oncol. 2024;42(4_Suupl.):123.
Zhuang J, Zhang S, Qiu X, Guo H. 175TiP A prospective phase II study to investigate the efficacy and safety of olaparib plus abiraterone and prednisone combination therapy in mHSPC patients with HRR gene mutation. Ann Oncol. 2022;33:S1502.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Louise Kostos is supported by a University of Melbourne Australian Government Research Training Program Scholarship. No other external funding was used in the preparation of this article.
Conflicts of Interest
Louise Kostos has received honoraria for speaking duties from Bayer and Astra Zeneca. Ben Tran declares grants and personal fees from Amgen, Astra Zeneca, Astellas, Bristol Myers Squibb, Merck Sharpe Dome, Merck Serono, Pfizer, Janssen, Bayer, Ipsen, Roche and Sanofi. Arun A. Azad declares the following lifetime disclosures: consultant: Aculeus Therapeutics, Astellas, Janssen, Novartis. Honoraria: Aculeus Therapeutics, Amgen, Arvinas, Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Ipsen, Janssen, Merck Serono, Merck Sharpe Dohme, Novartis, Noxopharm, Pfizer, Sanofi, Telix, Tolmar. Scientific advisory board: Amgen, Arvinas, Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Ipsen, Janssen, Merck Serono, Merck Sharpe Dohme, Novartis, Noxopharm, Pfizer, Sanofi, Telix, Tolmar. Travel and accommodation: Amgen, Astellas, Bayer, Hinova, Janssen, Merck Serono, Novartis, Pfizer, Tolmar. Research funding: Aptevo Therapeutics (institutional), Astellas (investigator), Astellas (institutional), Astra Zeneca (institutional), Astra Zeneca (investigator), Bionomics (institutional), Bristol Myers Squibb (institutional), Eli Lilly (institutional), Exelixis (institutional), Gilead Sciences (institutional), Glaxo Smith Kline (institutional), Hinova (institutional), Ipsen (institutional), Janssen (institutional), MedImmune (institutional), Merck Serono (investigator), Merck Serono (institutional), Merck Sharpe Dohme (institutional), Novartis (institutional), Pfizer (institutional), Sanofi Aventis (institutional), SYNthorx (institutional). Steering Committee Member: Astra Zeneca, Arvinas, Astellas, Exelixis, Janssen and Pfizer.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
No datasets were generated or analysed during the current study.
Code Availability
Not applicable.
Authors’ Contributions
LK wrote the initial draft of the manuscript. AA and BT reviewed and edited the manuscript. AA provided manuscript oversight.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kostos, L., Tran, B. & Azad, A.A. Combination of PARP Inhibitors and Androgen Receptor Pathway Inhibitors in Metastatic Castration-Resistant Prostate Cancer. Drugs (2024). https://doi.org/10.1007/s40265-024-02071-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s40265-024-02071-y