
Abstract
Abrocitinib (Cibinqo®) is an oral small-molecule inhibitor of Janus kinase 1 (JAK1) being developed by Pfizer for the treatment of moderate-to-severe atopic dermatitis (AD). In September 2021, abrocitinib was approved in the UK and Japan for the treatment of moderate-to-severe AD in adults and adolescents 12 years and older who are candidates for systemic therapy. Abrocitinib has also received a positive CHMP opinion in the EU for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy. Regulatory applications for the drug have also been submitted for review to several other countries, including the USA and Australia. This article summarizes the milestones in the development of abrocitinib leading to this first approval for the treatment of moderate-to-severe AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Oral, small-molecule inhibitor of JAK1 being developed by Pfizer for the treatment of moderate-to-severe AD. |
Received its first approval on 9 September 2021 in the UK. |
Approved for the treatment of moderate-to-severe AD in adults and adolescents 12 years and older who are candidates for systemic therapy. |
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.16900546. |
1 Introduction
Atopic dermatitis (AD) is a common heterogeneous inflammatory condition of the skin typified by skin barrier dysfunction and pruritus, which can have a detrimental impact on quality of life [1, 2]. Its pathogenesis is complex and involves multiple inflammatory and signalling immune pathways, one of which is the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway [2]. The JAK-STAT signalling pathway mediates a number of immune pathways underlying AD [2] and is also thought to impact various downstream inflammatory cytokines, such as interleukins and interferons, as well as multiple growth factors [1]. There are four mammalian JAKs, including JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2) [1]. JAKs act in pairs, with specific pairing combinations activated by different cytokines binding to their receptor. The activated JAK pairs then activate distinct STAT proteins, which subsequently regulate genes leading to changes in cellular functions, and upregulation of proinflammatory cytokine and growth factor production [1, 2].
Abrocitinib (Cibinqo®) is an oral small-molecule inhibitor of JAK1 being developed by Pfizer for the treatment of moderate-to-severe AD. On 9 September 2021, abrocitinib was approved in the UK for the treatment of moderate-to-severe AD in adults and adolescents 12 years and older who are candidates for systemic therapy [3,4,5,6]. The recommended dosage is 100 or 200 mg once daily [3,4,5]. Abrocitinib was also approved in Japan for the same indication on 27 September 2021 [7]. Abrocitinib received a positive CHMP opinion in the EU on 14 October 2021 for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy [8]. Regulatory applications for abrocitinib have also been submitted for review to several other countries, including the USA, Australia [6]. Abrocitinib continues to be assessed in phase 2a and 3 studies for moderate-to-severe AD, and an expanded access programme is now available for this indication.

1.1 Patent Information
In July 2015, Pfizer filed a world patent for abrocitinib and other pyrrolo[2,3-d]pyrimidine derivatives useful for inhibiting JAK [9].
2 Scientific Summary
2.1 Pharmacodynamics
Abrocitinib is selective for JAK1 over JAK2 (28-fold), JAK3 (> 340-fold) and TYK2 (43-fold) in biochemical assays [3,4,5, 10]. Within cells, abrocitinib preferentially inhibits cytokine-induced phosphorylation of STAT by JAK pairs that include JAK1; signalling by JAK2/JAK2 and JAK2/TYK2 pairs is spared by the drug, although the clinical relevance of its selective inhibition of JAK enzymes is not yet known [3,4,5].
Treatment with abrocitinib reduced platelet counts in a dose-related fashion in patients with moderate-to-severe AD in placebo-controlled trials of up to 16 weeks’ duration [3,4,5]. These reductions reached nadir in the first 4 weeks, with platelet counts returning towards baseline thereafter with continued treatment.

In addition, abrocitinib was associated with a dose-related increase versus placebo in levels of low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and total cholesterol at week 4 in these trials, with these lipids remaining elevated at the treatment period’s final visit [3,4,5].
Abrocitinib was associated with a dose-dependent reduction from baseline in serum levels of inflammatory markers, including interferon-γ-induced protein-10 (IP-10) and high sensitivity C-reactive protein (CRP), in healthy volunteers in the 10-day multiple ascending-dose of a phase 1 study; subjects received either 30, 100, 200 or 400 mg once daily, or 100 or 200 mg twice daily) [11]. Similarly, in a phase 2 trial in which patients with moderate-to-severe AD received abrocitinib 10, 30, 100 or 200 mg or placebo once daily for 12 weeks, abrocitinib 200 mg/day was associated with significant (p < 0.05) reductions in levels of IP-10 (at week 2) and CRP (at most timepoints), as well as reductions in the disease biomarkers interleukin-31 and thymus- and activation-regulated chemokine (no further details reported) [12].
2.2 Pharmacokinetics
Abrocitinib is well absorbed, with oral absorption of over 91% and absolute oral availability of ≈ 60% [3,4,5]. The drug is rapidly absorbed after oral administration, reaching peak plasma concentrations within 1 h. Increases in exposure are proportional to dose up to 200 mg. Abrocitinib reaches steady-state concentrations in the plasma within 48 h when administered once daily. Taking abrocitinib with a high-fat meal does not impact exposure to the drug to any clinically relevant extent; abrocitinib can be administered without regard to food. Intravenous abrocitinib has a volume of distribution of ≈ 100 L. Distribution of abrocitinib and its two active metabolites M1 (3-hydroxypropyl) and M2 (2-hydroxypropyl) is equal between red blood cells and plasma, with ≈ 64% of circulating abrocitinib and ≈ 37% and ≈ 29% of circulating M1 and M2 being plasma protein bound [3,4,5]. The sum of the exposures of unbound M1, M2 and abrocitinib (adjusted for relative potencies) is known as the abrocitinib active moiety. In vitro, abrocitinib is metabolized mainly by CYP2C19 (≈ 53%) and CYP2C9 (≈ 30%) and to a lesser degree by CYP3A4 (≈ 11%) and CYP2B6 (≈ 6%). At steady state, M1 is a minor metabolite and M2 and the pharmacologically inactive M4 are major metabolites. Elimination of abrocitinib occurs mainly via metabolic clearance, with < 1% of a dose being excreted unchanged via the urine and elimination of M1, M2 and M4 occurring mainly via the urine. The drug has an elimination half-life of ≈ 5 h [3,4,5].
Patient characteristics, including bodyweight, sex, race, age [3,4,5] and CYP2C19/CYP2C9 genotype [3,4,5, 13], do not appear to impact abrocitinib exposure to any clinically meaningful extent. Indeed, a population pharmacokinetic analysis found that the mean steady-state abrocitinib exposure in adolescent patients (aged ≥ 12 to < 18 years) did not differ to any clinically relevant extent from that in adults at typical bodyweights [3,4,5]. Likewise, exposure to the abrocitinib active moiety is not altered to any clinically significant extent by mild or moderate hepatic impairment [3,4,5, 14] and is not expected to be altered to any clinically meaningful extent by mild renal impairment [3,4,5]. However, abrocitinib is contraindicated in patients with severe hepatic impairment (it has not been evaluated in this population) and requires dosage adjustment in moderate and severe renal impairment (as exposure to the abrocitinib active moiety may be increased). Abrocitinib has not been evaluated in patients with end-stage renal disease on renal replacement therapy [3,4,5].
Features and properties of abrocitinib
Alternative names | Cibinqo®; PF 04965842; PF-4965842 |
|---|---|
Class | Amines; anti-inflammatories; antipsoriatics; cyclobutanes; pyrimidines; pyrroles; skin disorder therapies; small molecules; sulfonamides |
Mechanism of action | JAK1 inhibitor |
Route of administration | Oral |
Pharmacodynamics | Selective for JAK1 over other JAKs; dose-dependently reduces markers of inflammation; reduces platelet counts and increases LDL-C, HDL-C and total cholesterol levels, in a dose-related fashion |
Pharmacokinetics | Rapidly absorbed after oral administration, reaching Cmax within 1 h; elimination half-life ≈ 5 h |
Most frequent adverse reactions | Nausea, headache, acne, herpes simplex, blood creatine phosphokinase increased > 5 × upper limit of normal, vomiting, dizziness, upper abdominal pain, herpes zoster |
ATC codes | |
WHO ATC code | D11A-H08 (abrocitinib) |
EphMRA ATC code | D11A (other dermatological preparations) |
Chemical name | N-(cis-3-(Methyl(7H-pyrrolo(2,3-d)pyrimidin-4-yl)amino)cyclobutyl)propane-1-sulfonamide |
2.2.1 Potential Drug Interactions
Given the role of CYP2C19 and CYP2C9 in abrocitinib metabolism, drugs that are strong inhibitors or inducers of these enzymes may increase or reduce exposure to the abrocitinib active moiety, respectively [3,4,5]. It is recommended that the abrocitinib starting dosage be reduced if used in combination with strong CYP2C19 inhibitors, whereas abrocitinib is not recommended for use in combination with moderate or strong CYP2C19 or CYP2C9 inducers [3,4,5].
In vitro, abrocitinib and its metabolites do not significantly induce or inhibit key CYP or UGT enzymes and, at clinically relevant concentrations, do not inhibit transporters, including BCRP, BSEP, MATE1/2K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 or OCT2 [3,4,5]. However, abrocitinib does inhibit P-gp; caution is therefore advised if using abrocitinib in combination with the P-gp substrate dabigatran (as exposure to dabigatran has been shown to increase) and drugs with narrow therapeutic indices that are P-gp substrates. Moreover, the M1 and M2 metabolites are OAT3 substrates; an increase in active moiety exposure was not clinically meaningful when abrocitinib was coadministered with probenecid, an inhibitor of OAT3. Abrocitinib does not interact with oral contraceptives to any clinically relevant extent [3,4,5].
2.3 Therapeutic Trials
2.3.1 Phase 3 Studies
2.3.1.1 JADE MONO-1
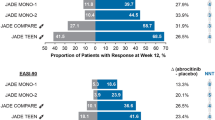
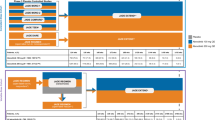
Abrocitinib monotherapy was effective in the treatment of moderate-to-severe AD in adults and adolescents aged ≥ 12 years in a randomized, double-blind, multicentre, phase 3 trial (NCT03349060; JADE MONO-1) [15]. Eligible patients had had an inadequate response to ≥ 4 weeks of topical corticosteroid or calcineurin inhibitor therapy, or were patients for whom topical therapies were medically inadvisable or whose AD required systemic therapy. Patients were randomized to receive abrocitinib 100 mg (n = 156) or 200 mg (n = 154) or placebo (n = 77) once daily for 12 weeks (use of oral antihistamines and topical non-medicated emollients was permitted); 21.7% of patients were aged < 18 years. At week 12, significantly (p < 0.005) more abrocitinib 100 or 200 mg/day recipients than placebo recipients had achieved each of the trial’s co-primary endpoints: an Investigator Global Assessment (IGA) response (score of 0 [clear] or 1 [almost clear] plus ≥ 2-grade improvement from baseline) [24% and 44% vs 8%] and a ≥ 75% improvement from baseline in Eczema Area and Severity Index (EASI) score (EASI-75 response) [40% and 63% vs 12%]. Findings for these outcomes were consistent when data were stratified by patient age (< 18 or ≥ 18 years) or baseline disease severity (moderate or severe) [15].
Key clinical trials of abrocitinib
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor (collaborator) |
|---|---|---|---|---|---|---|
Abrocitinib; placebo | Moderate to severe AD (adolescents/adults) | 3 | Completed | Multinational | NCT03349060; Eudra2017-003651-29; JADE MONO-1 | Pfizer |
Abrocitinib; placebo | Moderate to severe AD (adolescents/adults) | 3 | Completed | Multinational | NCT03575871; Eudra2018-001136-21; JADE MONO-2 | Pfizer |
Abrocitinib; placebo | Moderate to severe AD (adolescents/adults) | 3 | Completed | Multinational | NCT03627767; EudraCT2018-000501-23; JADE REGIMEN | Pfizer |
Abrocitinib + BTT; dupilumab + BTT; placebo + BTT | Moderate to severe AD (adults) | 3 | Completed | Multinational | NCT03720470; EudraCT2018-002573-21; JADE COMPARE | Pfizer |
Abrocitinib + BTT; dupilumab + BTT | Moderate to severe AD (adults) | 3b | Completed | Multinational | NCT04345367; EudraCT2019-004013-13; JADE DARE | Pfizer |
Abrocitinib +/− BTT; placebo +/− BTT | Moderate to severe AD (adolescents/adults) | 3 | Enrolling | Multinational | NCT03422822; EudraCT2017-004851-22; JADE EXTEND | Pfizer |
Abrocitinib + BTT; placebo + BTT | Moderate to severe AD (adolescents) | 3 | Completed | Multinational | NCT03796676; EudraCT2018-003804-37; JADE TEEN | Pfizer |
Abrocitinib; placebo | Moderate to severe AD (adults) | 2a | Recruiting | USA, Canada | NCT03915496; JADE MOA | Pfizer |
Abrocitinib; placebo | Moderate to severe AD (adults) | 2b | Completed | Multinational | NCT02780167; EudraCT2015-005513-72 | Pfizer |
Abrocitinib | Moderate to severe AD (adolescents/adults) | EAP | Available | Multinational | NCT04564755; EudraCT2020-003610-12; JADE REAL | Pizer |
Significant (p < 0.03) benefit was also evident with abrocitinib 100 or 200 mg/day versus placebo for all key secondary endpoints, including the proportion of patients who achieved a ≥ 4-point improvement from baseline in Peak Pruritus Numerical Rating Scale (PP-NRS) score (i.e. a PP-NRS4 response) at week 2, 4 or 12 and the least-squares mean (LSM) change from baseline in Pruritus and Symptoms Assessment for Atopic Dermatitis (PSAAD) total score at week 12 [15]. In addition, other patient-reported outcomes of AD symptoms/severity (Patient-Oriented Eczema Measure [POEM] total score) and quality of life (Dermatology Life Quality Index [DLQI] total score in patients aged ≥ 18 years; Children’s DLQI [CDLQI] total score in patients aged 12–17 years) were generally improved with abrocitinib compared with placebo at 12 weeks (based on 95% CIs) [15].
2.3.1.2 JADE MONO-2
The efficacy of abrocitinib monotherapy in the treatment of moderate-to-severe AD in adults and adolescents aged ≥ 12 years was also demonstrated in JADE MONO-2 (NCT03575871), a phase 3 trial with a design identical to that of JADE MONO-1 [16]. Patients were randomized to receive abrocitinib 100 mg (n = 158) or 200 mg (n = 155) or placebo (n = 78) once daily for 12 weeks. Overall, 10.2% of patients were aged < 18 years. A significantly (p < 0.001) greater proportion of patients in the abrocitinib 100 and 200 mg/day groups than in the placebo group had achieved an IGA response (28.4% and 38.1% vs 9.1%) and an EASI-75 response (44.5% and 61.0% vs 10.4%) at week 12 (co-primary endpoints). When data for these outcomes were stratified by patient age (< 18 or ≥ 18 years), the findings were generally consistent with those of the primary analysis. Abrocitinib 100 or 200 mg/day was also associated with significant (p ≤ 0.0002) benefit versus placebo with regard to the proportion of patients who achieved a PP-NRS4 response at week 2, 4 or 12 and the LSM change from baseline in PSAAD total score at week 12 (key secondary endpoints). Significant improvements in other patient-reported measures of AD symptoms/severity (POEM; Patient Global Assessment [PtGA]) and quality of life (DLQI in patients aged ≥ 18 years; CDLQI in patients aged 12–17 years) were also generally seen at 12 weeks with abrocitinib versus placebo (based on 95% CIs) [16].
2.3.1.3 JADE REGIMEN
Maintenance of abrocitinib monotherapy-induced responses was evaluated in adults and adolescents aged ≥ 12 years with moderate-to-severe AD in a responder-enriched, phase 3, withdrawal study (NCT03627767; JADE REGIMEN) [17]. Eligibility criteria included an inadequate response to ≥ 4 consecutive weeks of medicated topical therapy or required systemic treatments for AD control. This multicentre trial had three phases: (1) a 12-week open-label induction phase in which patients received abrocitinib 200 mg once daily; (2) a 40-week maintenance withdrawal phase in which patients who had achieved an IGA and EASI-75 response in phase 1 were randomized to receive abrocitinib 100 or 200 mg or placebo once daily in a double-blind fashion; and (3) a 12-week open-label rescue phase in which patients with loss of response that required rescue (protocol-defined flare: defined as ≥ 50% loss of initial EASI response at week 12 with a new IGA score ≥ 2 during the maintenance phase) received abrocitinib 200 mg/day plus medicated topical therapy (corticosteroids, calcineurin inhibitors or topical PDE-4 inhibitors). Oral antihistamines and topical non-medicated emollients were allowed throughout the study. In total, 798 of 1233 patients achieved a protocol-defined response after receiving 12 weeks of abrocitinib 200 mg/day in phase 1 and entered into the maintenance phase [17].
During the maintenance phase, the proportion of patients who had a protocol-defined flare was numerically lower with abrocitinib 200 mg/day (44 of 266; 16.5%) and 100 mg/day (105 of 265; 39.6%) than with placebo (207 of 267; 77.5%) [17]. A significant (p < 0.0001) reduction in flare risk was evident with abrocitinib 200 mg/day (hazard ratio [HR] 0.10; 95% CI 0.07–0.14) and 100 mg/day (HR 0.27; 95% CI 0.21–0.34) versus placebo, and with abrocitinib 200 versus 100 mg/day (HR 0.36; 95% CI 0.26–0.52). A total of 351 patients entered the rescue phase after receiving abrocitinib 200 mg/day (n = 43), abrocitinib 100 mg/day (n = 104) or placebo (n = 204) in the maintenance phase. After 12 weeks of rescue treatment with abrocitinib 200 mg/day plus topical corticosteroids in these respective groups, 55.0%, 74.5% and 91.8% of patients recaptured an EASI-75 response, 36.6%, 58.8% and 81.6% recaptured an IGA response and 30.0%, 35.3% and 73.2% recaptured a PP-NRS4 response [17]. Findings in adolescent patients (aged < 18 years) were consistent with those in the overall trial population [3,4,5].
2.3.1.4 JADE TEEN
Abrocitinib was an effective addition to topical therapy in adolescents (aged 12 to < 18 years) with moderate-to-severe AD in a randomized, double-blind, placebo-controlled, phase 3 study (NCT03796676; JADE TEEN) [18]. Patients eligible for this multicentre trial had inadequately responded to ≥ 4 consecutive weeks of medicated topical therapy in the last 6 months or had received systemic therapy in the last 6 months or were candidates for systemic therapy. Patients received abrocitinib 100 mg (n = 95) or 200 mg (n = 94) or placebo (n = 96) once daily for 12 weeks in conjunction with topical therapy (specifically, standardized regimens of medicated topical therapy and non-medicated topical emollients). Both coprimary endpoints of the trial were met, with significantly (p < 0.02) more abrocitinib 100 or 200 mg/day than placebo recipients achieving an IGA response (41.6% and 46.2% vs 24.5%) or an EASI-75 response (68.5% and 72.0% vs 41.5%) at 12 weeks [18].
Among key secondary endpoints, both abrocitinib dosages provided significant (p < 0.02) benefit over placebo in terms of the proportion of patients who achieved a PP-NRS4 response at week 2, with the benefit remaining significant (p < 0.002 vs placebo) at weeks 4 and 12 with abrocitinib 200 mg/day [18]. Abrocitinib 100 mg/day did not provide significant benefit versus placebo for this measure at week 4; consequently, due to sequential hypothesis testing, the difference between abrocitinib 100 mg/day and placebo for PP-NRS4 response at week 12 and the difference between each abrocitinib dosage and placebo in the LSM change from baseline in PSAAD total score at week 12 (also a key secondary endpoint) were not considered to be statistically significant. Each abrocitinib dosage was associated with improvements (based on 95% CIs) in patient-reported AD symptoms/severity and quality of life versus placebo at 12 weeks, as assessed by LSM changes from baseline in POEM and CDLQI [18].
2.3.1.5 JADE COMPARE
The efficacy of efficacy of abrocitinib and dupilumab was compared with that of placebo. in adults (aged ≥ 18 years) with moderate-to-severe AD receiving background topical therapy in a randomized, double-blind, placebo-controlled, multicentre, phase 3 trial (NCT03720470; JADE COMPARE) [19]. Eligibility criteria in this double-dummy trial included an inadequate response to ≥ 4 weeks of topical medication or a need for systemic therapy for AD control, during the last 6 months. Patients were randomized to receive abrocitinib 100 mg (n = 238) or 200 mg (n = 226) once daily, subcutaneous dupilumab 300 mg every other week (subsequent to a 600 mg loading dose) [n = 242] or placebo (n = 131) for 16 weeks in combination with a topical medication (a low- or medium-potency corticosteroid, calcineurin inhibitor and/or phosphodiesterase 4 inhibitor) plus emollients. A significantly (p < 0.001) greater proportion of patients treated with abrocitinib 100 or 200 mg/day than with placebo achieved an IGA response (36.6% and 48.4% vs 14.0%) and an EASI-75 response (58.7% and 70.3% vs 27.1%) at week 12 (co-primary endpoints); 36.5% and 58.1% of patients achieved these outcomes with dupilumab [19].
Each abrocitinib dosage provided significant (p < 0.001) benefit over placebo for the three key secondary endpoints: rate of PP-NRS4 response at week 2, IGA response at week 16 and EASI-75 response at week 16 [19]. In addition, significantly (p < 0.001) more abrocitinib 200 mg/day than dupilumab recipients achieved a PP-NRS4 response at week 2 (49.1% vs 26.4%), whereas abrocitinib 100 mg/day did not significantly differ from dupilumab in this regard (31.8% vs 26.4%) [key secondary comparison]. While not pre-defined analyses, other key secondary endpoints generally did not significantly differ between the abrocitinib and dupilumab groups, with the exception of the rate of IGA response at week 16, which significantly favoured abrocitinib 200 mg/day (based on 95% CIs) [19].
2.3.1.6 JADE DARE
When taken in combination with background topical therapy in adults with moderate-to-severe AD, abrocitinib (200 mg once daily, n = 362) was superior to subcutaneous dupilumab (600 mg loading dose then 300 mg every other week, n = 365) for each efficacy measure evaluated in a randomized, double-blind, double-dummy, phase 3 trial (NCT04345367; JADE DARE) [20]. The co-primary endpoints of this 26-week multicentre trial were the proportion of patients with a PP-NRS4 response at week 2 and the proportion with a 90% improvement from baseline at week 4 in the EASI (EASI-90). A significantly (p < 0.001) greater proportion of abrocitinib recipients than dupilumab recipients achieved a PP-NRS4 response at week 2 (48.2% vs 25.5%) and an EASI-90 response at week 4 (28.5% vs 14.6%) [co-primary endpoints]. The higher EASI-90 response seen with abrocitinib versus dupilumab was sustained at week 16 (54.3% vs 41.9%, p < 0.001; key secondary endpoint) [20].
2.3.1.7 JADE EXTEND
The longer-term efficacy of abrocitinib, used with or without medicated topical therapy, as a treatment for moderate-to-severe AD was demonstrated in the ongoing extension study, JADE EXTEND (NCT03422822) [3,4,5]. To be eligible for this trial, patients must have completed the full treatment period of the parent trial (JADE MONO-1, MONO-2, COMPARE, TEEN or REGIMEN [3,4,5]; patients in REGIMEN [17] who did not meet the responder criteria at the end of the open-label induction period had the option to enter the extension study). Those who had been randomized to abrocitinib 100 or 200 mg once daily in one of these parent studies continued to receive abrocitinib at the same dosage (with maintenance of blinding) in JADE EXTEND. Up to 71% of patients who had achieved a response after 12 weeks of treatment with abrocitinib 100 or 200 mg/day in a parent trial maintained their response after receiving abrocitinib at the same dosage for a total of 48 weeks, with response being an IGA response (53% and 57%), EASI-75 response (69% and 71%) or PP-NRS4 response (52% and 69%) [using non-responder imputation]. In addition, some patients who had not achieved a response with abrocitinib 100 or 200 mg/day by week 12 in a parent trial achieved an IGA response (22% and 27%) or an EASI-75 response (45% and 54%) by week 24 of continued treatment (i.e. a late-onset response) [3,4,5].
Similarly, most patients (≥ 76.5%) who had responded to dupilumab in JADE COMPARE (n = 29–90, depending on the response definition) had their response (IGA, EASI-75 and PP-NRS4) maintained 12 weeks after switching to abrocitinib 100 or 200 mg/day in JADE EXTEND [21]. Moreover, among dupilumab recipients who were non-responders for IGA, EASI-75 and PP-NRS4 in JADE COMPARE (n = 106, 51 and 66, respectively), after switching to abrocitinib 100 or 200 mg/day in JADE EXTEND, response rates were achieved at 12 weeks for IGA (34.3% and 47.2% for 100 and 200 mg/day abrocitinib, respectively), EASI-75 (67.7% and 80.0%) and PP-NRS4 (37.8% and 81.0%) [22].
2.3.2 Phase 2 Study
Abrocitinib showed promise as a treatment for adults (aged 18–75 years) with moderate-to-severe AD in a randomized, double-blind, placebo-controlled, phase 2b study (NCT02780167) [23]. Eligible patients had had an inadequate response to ≥ 4 weeks of topical medication or had not been able to receive topical treatment in the last 12 months as it was medically inadvisable. A total of 267 patients were randomized and treated once daily with abrocitinib 10, 30, 100, 200 mg (n = 49–56 per group) or placebo (n = 56) for 12 weeks (use of oral antihistamines and non-medicated emollients was permitted). The proportion of patients who achieved an IGA response at 12 weeks (primary endpoint) was 43.8%, 29.6%, 8.9% and 10.9% with abrocitinib 200, 100, 30 and 10 mg/day, respectively, and 5.8% with placebo, with the difference from placebo being significant (p < 0.001) for the abrocitinib 200 and 100 mg/day groups. Similarly, abrocitinib 100 and 200 mg/day (but not 30 or 10 mg/day) was associated with significantly (p < 0.01) more favourable LSM changes from baseline in EASI than placebo at week 12 (key secondary endpoint) [23]. Patient-reported AD symptoms/severity and quality of life also significantly (p < 0.05) improved with abrocitinib 100 and 200 mg/day versus placebo at most/all timepoints evaluated during treatment, as assessed by LSM changes from baseline in pruritus NRS score, PSAAD total score and POEM total score and the proportion of patients with an improvement in PtGA [24].
2.3.3 Other Analyses
The efficacy of abrocitinib monotherapy in the treatment of moderate-to-severe AD in adults and adolescents is supported by a pooled analysis (n = 942) of the phase 3 JADE MONO-1 and MONO-2 trials and the phase 2b study (NCT02780167) [25, 26]. For instance, the proportion of patients who achieved a PP-NRS4 response was numerically greater with abrocitinib 100 mg/day (n = 369) and 200 mg/day (n = 363) than with placebo (n = 210) at all timepoints evaluated from week 2 (24.9% and 44.2% vs 5.8%) to week 12 (42.9% and 57.3% vs 16.5%) [25]. This clinically meaningful improvement in itch occurred partially independently of overall disease improvement, as measured by IGA response [25].
2.4 Adverse Events
Abrocitinib is generally well tolerated in patients with AD, based on pooled data from across four (n = 1,540) [27] or five (n = 1,825) [3,4,5] placebo-controlled trials, from placebo-controlled studies plus JADE REGIMEN (open-label phase) and JADE EXTEND (n = 2,856) [27] and from all patients treated with abrocitinib in AD clinical studies, including JADE EXTEND (n = 3,128) [3,4,5].
In the largest pooled analysis of placebo-controlled trials, in which patients received abrocitinib 200 mg (n = 684) or 100 mg (n = 703) or placebo (n = 438) once daily for up to 16 weeks, the most common (incidence ≥ 2 %) adverse reactions with abrocitinib (either dosage) included nausea (15.1% and 6.3% vs 1.8%), headache (7.9% with 200 mg/day), acne (4.8% and 1.8% vs 0.2%), herpes simplex (4.2% and 2.8% vs 1.4%), blood creatine phosphokinase increased > 5 × upper limit of normal (3.8% and 1.8% vs 1.8%), vomiting (3.5% with 200 mg/day), dizziness (3.4% with 200 mg/day) and upper abdominal pain (2.2% with 200 mg/day) [quantitative data for some treatment groups were not reported] [3,4,5]. The frequency of herpes zoster with abrocitinib 200 mg or 100 mg was 1.2% and 0.6% versus 0% for placebo; the incidence rate (IR) of herpes zoster was numerically higher in patients who had severe rather than moderate AD at baseline (4.93 vs 2.49 per 100 patient-years [PY]) [3,4,5].
In this analysis, infections occurred with an incidence of 34.8% and 34.9% with abrocitinib 200 or 100 mg/day versus 27.4% with placebo, although were generally mild or moderate; the IR of serious infections in the respective groups was 1.12, 3.32 and 1.81 per 100 PY [3,4,5]. Among all patients treated with abrocitinib in clinical trials, including JADE EXTEND, the IR of serious infections was 2.11 and 2.18 per 100 PY with abrocitinib 200 and 100 mg/day, with the most common being herpes simplex, herpes zoster and pneumonia [3,4,5].
As discussed earlier, lipid elevations can occur with abrocitinib [3,4,5]. However, in the largest pooled analysis of placebo-controlled trials, few patients had events related to hyperlipidaemia with abrocitinib 200 or 100 mg/day (0.6% and 0.4% vs 0% with placebo). Among all patients treated with abrocitinib 200 or 100 mg/day in clinical trials, including JADE EXTEND, both deep vein thrombosis and pulmonary embolism occurred with an IR of 0.23 and 0 per 100 PY in the respective groups [3,4,5].
Confirmed platelet counts < 50 × 103/mm3 and absolute lymphocyte counts (ALC) < 0.5 × 103/mm3 were uncommon with abrocitinib 200 or 100 mg/day in the largest pooled analysis of placebo-controlled trials (incidence ≤ 0.3% with either dosage vs 0% with placebo) and among all patients who received abrocitinib 200 or 100 mg/day in clinical trials, including JADE EXTEND (≤ 0.3% with either dosage; quantitative data for proportion of patients with platelet counts < 50 × 103/mm3 in the 100 mg/day group were not reported) [3,4,5]. Malignancies have occurred with abrocitinib in clinical studies [27], although data are not sufficient to determine a relationship between malignancy development and abrocitinib exposure [3,4,5].
The safety profile of abrocitinib in patients aged ≥ 65 years was generally similar to that seen overall in adult patients in clinical trials, although patients aged ≥ 65 years were more likely to discontinue treatment, experience serious adverse events or develop low platelet or ALC levels compared with younger patients, and the incidence of herpes zoster in patients aged ≥ 65 years was higher than in younger patients [3,4,5]. Indeed, patients aged ≥ 65 years had the highest IR of herpes zoster among the age groups assessed in the largest pooled analysis of placebo-controlled trials (7.40 vs 3.44 per 100 PY in patients aged 18 to < 65 years and 2.12 per 100 PY in patients aged < 18 years) [3,4,5].
Abrocitinib was also generally well tolerated in adolescent patients (aged 12–17 years) with AD in the 12-week JADE TEEN trial [18]. The most common treatment-emergent adverse events (TEAEs) occurring in > 5.0% of abrocitinib 200 or 100 mg/day recipients and at an incidence ≥ 2 % higher than in placebo recipients included nausea (18.1% and 7.4% vs 1.0%), dizziness (6.4% and 0% vs 1.0%), acne (5.3% and 3.2% vs 1.0%), vomiting (5.3% and 4.2% vs 0%), folliculitis (2.1% and 7.4% vs 1.0%) and pharyngitis (3.2% and 5.3% vs 3.1%). Moreover, few patients in the respective groups had serious TEAEs (1.1% and 0% vs 2.1%) or had TEAEs of special interest, including herpes zoster (0% and 1.1% vs 0%), herpes simplex (1.1% and 0% vs 0%), oral herpes (2.1% and 1.1% vs 0%), eczema herpeticum (0% and 1.1% vs 0%) or conjunctivitis (0% and 0% vs 1.0%). No patients died [18].
2.5 Ongoing Clinical Trials
In addition to the ongoing phase 3 JADE EXTEND trial (NCT03422822) discussed previously, recruitment is underway in a phase 2a trial to evaluate the mechanism of action of abrocitinib in adults with moderate-to-severe AD, by correlating efficacy outcomes with changes in key skin and blood biomarkers (NCT03915496; JADE MOA). An expanded access programme is also available in various countries, including the USA, Australia, Canada and Switzerland (NCT04564755; JADE REAL), to provide access to abrocitinib to adults and adolescents with moderate-to-severe AD who have inadequate treatment options and are otherwise ineligible to participate in abrocitinib clinical trials.
3 Current Status
Abrocitinib received its first approval on 9 September 2021 for the treatment of moderate-to-severe AD in adults and adolescents 12 years and older who are candidates for systemic therapy in the UK [3,4,5,6]. Abrocitinib was also approved in Japan for the same indication on 27 September 2021 [7]. Abrocitinib received a positive CHMP opinion in the EU on 14 October 2021 for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy [8].
Change history
11 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40265-022-01694-3
References
Ferreira S, Guttman-Yassky E, Torres T. Selective JAK1 inhibitors for the treatment of atopic dermatitis: focus on upadacitinib and abrocitinib. Am J Clin Dermatol. 2020;21(6):783–98.
He H, Guttman-Yassky E. JAK inhibitors for atopic dermatitis: an update. Am J Clin Dermatol. 2019;20(2):181–92.
Agency MHPR. Abrocitinib (CIBINQO®): 50mg film-coated tablets: summary of product characteristics. 2021. https://mhraproducts4853.blob.core.windows.net/docs/3b68809595392199c87a314cd40e80146a1fa36b. Accessed 13 Sept 2021.
Agency MHPR. Abrocitinib (CIBINQO®): 100mg film-coated tablets: summary of product characteristics. 2021. https://mhraproducts4853.blob.core.windows.net/docs/cec4db859df0798598d52dd5da18f77adb972e29. Accessed 13 Sept 2021.
Agency MHPR. Abrocitinib (CIBINQO®): 200mg film-coated tablets: summary of product characteristics. 2021. https://mhraproducts4853.blob.core.windows.net/docs/9106b0ca62531780c94245a0eb6894ba3a43a8bf. Accessed 13 Sept 2021.
Pfizer. UK’s MHRA grants marketing authorisation for Pfizer’s CIBINQO® (abrocitinib) for adults and adolescents with moderate to severe atopic dermatitis [media release]. 9 Sep 2021. https://www.pfizer.com.
Pfizer. Japan’s MHLW approves Pfizer’s CIBINQO® (abrocitinib) for adults and adolescents with moderate to severe atopic dermatitis [media release]. 30 Sep 2021. https://www.pfizer.com/news/press-release/press-release-detail/japans-mhlw-approves-pfizers-cibinqor-abrocitinib-adults.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) summary of opinion: CIBINQO (abrocitinib) [media release]. 14 Oct 2021. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-opinion-cibinqo_en.pdf.
Pfizer Inc. Pyrrolo[2,3-d]pyrimidine derivatives useful for inhibiting janus kinase. 2021. https://patents.google.com/patent/WO2016024185A1/es. Accessed 13 Sept 2021.
Vazquez ML, Kaila N, Strohbach JW, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61(3):1130–52.
Peeva E, Hodge MR, Kieras E, et al. Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: a phase 1, randomized, placebo-controlled, dose-escalation study. Br J Clin Pharmacol. 2018;84(8):1776–88.
Kieras E, Zhang W, Banfield C, et al. PF-04965842, a JAK1 inhibitor, modulates pharmacodynamic and disease biomarkers in blood of atopic dermatitis patients [abstract no. 20]. Exp Dermatol. 2018;27(Suppl 2):10.
Dowty M, Yang X, Lin J, et al. The effect of CYP2C9 and CYP2C19 genotype on the pharmacokinetics of PF 04965842, a JAK1 inhibitor in clinical development [abstract no. P190]. Drug Metabol Pharm. 2020;35(1 Suppl):S80.
Wang EQ, Le V, O’Gorman M, et al. Effects of hepatic impairment on the pharmacokinetics of abrocitinib and its metabolites. J Clin Pharmacol. 2021;61(10):1311–23.
Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–66.
Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863–73 [plus supplementary material].
Blauvelt A, Silverberg JI, Lynde CW, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate to severe atopic dermatitis: results from the JADE REGIMEN phase 3 trial. J Am Acad Dermatol. 2021. https://doi.org/10.1016/j.jaad.2021.05.
Eichenfield LF, Flohr C, Sidbury R, et al. Efficacy and safety of abrocitinib in combination with topical therapy in adolescents with moderate-to-severe atopic dermatitis: the JADE TEEN randomized clinical trial. JAMA Dermatol. 2021. https://doi.org/10.1001/jamadermatol.2021.2830 [plus supplementary material].
Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. New Engl J Med. 2021;384(12):1101–12.
Reich K, Thyssen JP, Blauvelt A, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis who received background topical therapy in a 26-week, randomized, head-to-head trial [abstract no. D3T01.2B plus presentation]. In: European Academy of Dermatology and Venereology 30th Congress. 2021.
Bhutani T, Deleuran M, Fonacier L, et al. Effective maintenance of response in atopic dermatitis patients after switching from dupilumab to abrocitinib (JADE-EXTEND) [abstract no. P551]. Ann Allergy Asthma Immunol. 2020;125(5 Suppl):S50–1.
Shi V, Bhutani T, Deleuran M, et al. Abrocitinib in the treatment of moderate-to-severe atopic dermatitis refractory to dupilumab treatment: an analysis of JADE-EXTEND, a phase 3 long-term extension study [abtsract no. 27590]. J Am Acad Dermatol. 2021;85(3 Suppl):AB36.
Gooderham MJ, Forman SB, Bissonnette R, et al. Efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155(12):1371–9.
Simpson EL, Wollenberg A, Bissonnette R, et al. Patient-reported symptoms and disease impacts in adults with moderate-to-severe atopic dermatitis: results from a phase 2b study with abrocitinib. Dermatitis. 2021. https://doi.org/10.1097/DER.0000000000000725.
Kim BS, Silverberg JI, Stander S, et al. Rapid improvement of itch associated with atopic dermatitis with abrocitinib is partially independent of overall disease improvement: results from pooled phase 2b and 3 monotherapy studies. Dermatitis. 2021. https://doi.org/10.1097/DER.0000000000000770.
Silverberg JI, Thyssen JP, Simpson EL, et al. Impact of oral abrocitinib monotherapy on patient-reported symptoms and quality of life in adolescents and adults with moderate-to-severe atopic dermatitis: a pooled analysis of patient-reported outcomes. Am J Clin Dermatol. 2021;22:541–54.
Simpson EL, Silverberg JI, Nosbaum A, et al. Integrated safety analysis of abrocitinib for the treatment of moderate-to-severe atopic dermatitis from the phase II and phase III clinical trial program. Am J Clin Dermatol. 2021;22(5):693–707.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Emma Deeks is a contracted employee of Adis International Ltd/Springer Nature and Sean Duggan is a salaried employee of Adis International Ltd/Springer Nature, and declare no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Deeks, E.D., Duggan, S. Abrocitinib: First Approval. Drugs 81, 2149–2157 (2021). https://doi.org/10.1007/s40265-021-01638-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01638-3