
Abstract
Casirivimab/imdevimab (Ronapreve™; REGEN-COV™) is a co-packaged combination of two neutralizing immunoglobulin gamma 1 (IgG1) human monoclonal antibodies (casirivimab and imdevimab) against the spike protein of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19). Casirivimab/imdevimab received its first emergency use authorization for the treatment of COVID-19 in November 2020 in the USA, with similar authorizations subsequently granted in various other countries, including India, Canada, and Switzerland. In February 2021, casirivimab/imdevimab was granted a positive scientific opinion in the EU for the treatment of COVID-19. In July 2021, casirivimab/imdevimab received its first approval in Japan for the treatment of mild or moderate COVID-19, followed in August 2021 by its conditional approval for the prophylaxis and treatment of acute COVID-19 infection in the UK. The combination was also granted provisional determination in Australia in August 2021, indicating its eligibility to be considered for provisional registration for COVID-19 treatment and prevention. This article summarizes the milestones in the development of casirivimab/imdevimab leading to these first approvals for COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.16689520. |
Co-packaged combination of two neutralizing IgG1 human monoclonal antibodies against SARS-CoV-2 for co-administration, being developed by Regeneron Pharmaceuticals for the prevention and treatment of COVID-19 |
Received its first emergency use authorization for the treatment of COVID-19 on 21 November 2020 in the USA |
Received its first approval on 19 July 2021 in Japan (treatment of mild or moderate Covid-19 infection) and on 20 August 2021 in the UK (conditional approval for the prophylaxis and treatment of acute COVID-19 infection) |
1 Introduction
A novel highly transmissible coronavirus, designated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in China late in 2019 and subsequently spread rapidly throughout the world, reaching pandemic status in March 2020 [1]. SARS-CoV-2 gains entry into host cells, such as nasal/bronchial epithelial cells and pneumocytes, by binding via its spike protein to the angiotensin-converting enzyme 2 (ACE2) receptor on their surface [2]. SARS-CoV-2 infections can be asymptomatic or associated with an acute respiratory disease, known as coronavirus disease 2019 (COVID-19), manifestations of which can vary in severity from mild to fatal [1, 2]

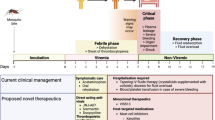
Key milestones in the development of casirivimab/imdevimab for the treatment and prevention of COVID-19. CMA conditional marketing authorization, COVID-19 coronavirus disease 2019, EUA emergency use authorization, IOA interim order authorization
.
Casirivimab/imdevimab is a co-packaged combination of two neutralizing immunoglobulin gamma 1 (IgG1) recombinant human monoclonal antibodies against the SARS-CoV-2 spike protein for co-administration: casirivimab and imdevimab. Casirivimab/imdevimab is being developed by Regeneron Pharmaceuticals (and is licensed to Roche outside the USA) for the prevention and treatment of COVID-19. Casirivimab/imdevimab received its first emergency use authorization for the treatment of COVID-19 in November 2020 in the USA [3], with similar authorizations subsequently granted in a number other countries, including India, Canada and Switzerland [4]. In February 2021, casirivimab/imdevimab was granted a positive scientific opinion in the EU for the treatment of confirmed COVID-19 in patients who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19 [5]. In July 2021, casirivimab/imdevimab received its first approval in Japan for the treatment of mild or moderate COVID-19 [6, 7], followed in August 2021 by its conditional approval in the UK for the prophylaxis and treatment of acute COVID-19 infection [8, 9]. Casirivimab/imdevimab was also granted provisional determination in Australia in August 2021, indicating its eligibility to be considered for provisional registration in the setting of COVID-19 treatment and prevention [10].
Casirivimab and imdevimab must be administered together in a single intravenous infusion [7, 9] or, alternatively, administered consecutively by subcutaneous injection [9]. For the treatment of COVID-19, the recommended dose is casirivimab 600 mg plus imdevimab 600 mg [7, 9]. For the prevention of COVID-19, the recommended single-dose regimen is casirivimab 600 mg plus imdevimab 600 mg [9]. For subjects who require ongoing prevention, a repeat-dose regimen is recommended comprising a single dose of casirivimab 600 mg plus imdevimab 600 mg followed by subsequent doses of 300 mg plus 300 mg once every 4 weeks [9]. Hereafter, doses of casirivimab plus imdevimab refer to the sum dose of the two antibodies in equal-dose combination (i.e. a 1200 mg dose refers to casirivimab 600 mg plus imdevimab 600 mg) and casirivimab plus imdevimab is referred to as casirivimab/imdevimab for simplicity. Co-packaging may differ between countries and there are regional differences in registered doses of casirivimab/imdevimab; please see local prescribing information for dosage and administration details.
1.1 Company Agreements and Patent Information
In December 2020, Chugai Pharmaceutical and Roche entered into a licensing agreement for casirivimab/imdevimab, in which Chugai Pharmaceutical was granted development and exclusive marketing rights for the antibody cocktail for COVID-2019 in Japan [11].
In August 2020, Regeneron Pharmaceuticals and Roche entered into an agreement to develop, manufacture and distribute casirivimab/imdevimab globally [12]. Each company committed to dedicate a certain manufacturing capacity to casirivimab/imdevimab each year and to bear its own distribution expenses in its designated territory (Regeneron Pharmaceuticals in the USA; Roche outside the USA). Together the companies will fund and execute the ongoing phase III prevention and phase I healthy volunteer safety studies, and any extra global studies to further assess casirivimab/imdevimab in the treatment or prevention of COVID-19. Roche will mainly be responsible for securing regulatory approvals (and conducting any additional studies required) outside the USA, following the initial approval by the European Medicines Agency [12].
In July 2020, Regeneron entered into an agreement with the Biomedical Advanced Research and Development Authority and the US Department of Defense to manufacture and supply casirivimab/imdevimab, so that it could be available in the USA immediately upon emergency use authorization or approval being granted by the US Food and Drug Administration [13]. The agreement covered a certain number of bulk lots aimed for completion in the autumn of 2020, in addition to fill/finish and storage activities [13].
Prior to 31 December 2020, Regeneron Pharmaceuticals was granted a composition of matter patent covering casirivimab and imdevimab in the USA, which is valid until June 2040 [14].
2 Scientific Summary
2.1 Pharmacodynamics
Casirivimab and imdevimab bind with high affinity (e.g. Kd 45.8 and 46.7 pmol/L [9]) to distinct non-overlapping epitopes of the SARS-CoV-2 spike protein receptor binding domain (RBD) [9, 15]. Each antibody almost completely (≥ 95%) blocks the SARS-COV2 spike protein RBD from binding to the human ACE2 receptor in vitro [15]. Casirivimab and imdevimab block this binding with half-maximal inhibitory concentrations of 56.4 and 165 pmol/L individually and 81.8 pmol/L in combination [9]. Casirivimab/imdevimab displayed therapeutic potential when used prophylactically or as a treatment for SAR-CoV-2 infection in animal studies, limiting airway viral load and viral-induced lung pathology in monkeys and loss of bodyweight in hamsters [16].
In patients with COVID-19, treatment with casirivimab/imdevimab did not appear to alter the evolution of SARS-CoV-2 or select for resistant variants [17]. Similarly, in a SARS-CoV-2 hamster challenge model, resistant variants emerged in 18 of the 40 animals who received casirivimab or imdevimab alone versus none of the 20 animals who received the antibodies in combination [17]. Indeed, in vitro, casirivimab/imdevimab lessened the likelihood of SAR-CoV-2 resistance emerging relative to either antibody alone [17].
On the basis of in vitro studies, the neutralization potency of casirivimab/imdevimab is anticipated to be retained against a number of SARS-CoV-2 variants of concern/interest, including B.1.1.7 (Alpha/UK origin) [9, 18], B.1.351 (Beta/South Africa origin), P.1 (Gamma/Brazil origin), B.1.617.2 (Delta/India origin), AY.1/AY.2 (Delta/India origin), B.1.427/B.1.429 (Epsilon/California origin) [9], B.1.526 (Iota/New York origin) [9, 19], B.1.617.1/B.1.617.3 (Kappa/India origin) and C.37 (Lambda/Peru origin) [9]. Notably, one study found casirivimab/imdevimab to be 9.1-fold less potent against B.1.351 than an earlier variant in vitro [18]. Whether clinical outcomes correlate with in vitro neutralization potency is unknown [9].
Anti-casirivimab and anti-imdevimab antibodies have developed in some subjects who have received casirivimab/imdevimab via intravenous infusion or subcutaneous injection (0.8% and 1.7%) [9]. Among subjects who received subcutaneous casirivimab/imdevimab 1200 mg every 4 weeks (n = 707), treatment-emergent anti-casirivimab and anti-imdevimab antibodies occurred with an incidence of 0.1% and 2.0% [9]. Antibody titers were low in subjects receiving repeat-dose casirivimab/imdevimab, with no evidence of altered pharmacokinetic profiles of casirivimab or imdevimab.
2.2 Pharmacokinetics
Following a single intravenous dose of casirivimab/imdevimab 300–8000 mg, the pharmacokinetic profile of each of the antibodies is linear and dose-proportional [9]. After a single 1200 mg intravenous dose of casirivimab/imdevimab, the respective agents have mean maximum serum concentrations (Cmax) of 182.7 and 181.7 mg/L and mean concentrations 28 days after administration (i.e. on day 29) [C28] of 37.9 and 31.0 mg/L [9]. After subcutaneous administration of a single 1200 mg dose of casirivimab/imdevimab, the mean Cmax values of the respective agents are 52.2 and 49.2 mg/L and the mean C28 values are 30.5 and 25.9 mg/L. Intravenous and subcutaneous repeat-dose regimens of casirivimab/imdevimab (i.e. a loading dose followed by maintenance doses once every 4 weeks) are predicted to have serum trough concentrations similar to the mean C28 values seen after a single 1200 mg subcutaneous dose [9].
The estimated total volume of distribution of casirivimab is 7.16 L and that of imdevimab is 7.43 L [9]. It is expected that both antibodies are degraded into small peptides and amino acids as per endogenous IgG and will not be metabolized by CYP450 enzymes or be excreted renally or hepatically to any significant extent. Following a single intravenous dose of casirivimab/imdevimab 1200 mg, the respective agents have mean half-lives of 31.2 and 27.3 days according to population pharmacokinetic analysis [9]. Similarly, in single-dose studies of subcutaneous casirivimab/imdevimab 1200 mg, casirivimab had mean half-lives of 30.2 [20] and 32.4 [21] days and imdevimab of 26.5 [20] and 27.0 [21] days.
Patient characteristics, including age, sex, bodyweight, race, albumin level, renal impairment or mild hepatic impairment, do not appear to impact casirivimab or imdevimab exposure to any clinically relevant extent [9]. It is considered unlikely that casirivimab or imdevimab will interact with medications that are substrates, inhibitors or inducers of CYP450 enzymes or that are renally excreted [9].
Features and properties of casirivimab/imdevimab
Alternative names | Casirivimab and imdevimab; casirivimab + imdevimab; Ronapreve™; REGEN-COV™; REGEN-COV; REGEN-COV2; REGN COV2; REGN-10933 + REGN-10987; REGN10933-10987; RG 6413/RG 6412; SARS-CoV-2 virus multi-antibody therapy; anti-spike (S) SARS-CoV-2 monoclonal antibodies |
Class | Antibodies, antivirals, monoclonal antibodies |
Mechanism of action | Virus internalization inhibitor |
Route of administration | IV; SC |
Pharmacodynamics | Casirivimab and imdevimab bind with high affinity to non-overlapping epitopes of the SARS-CoV-2 spike protein RBD; neutralization potency of the antibodies in combination is anticipated to be retained against a number of SARS-CoV-2 variants of concern/interest |
Pharmacokinetics | Casirivimab and imdevimab have Cmax values of 183 and 182 mg/L after a 1200 mg IV dose and 52 and 49 mg/L after a 1200 mg SC dose; the half-life for each antibody is broadly similar after a single IV (31 and 27 days) or SC (30–32 and 27 days) dose |
Adverse reactions | |
Common | Injection-site reactions (after SC administration) |
Uncommon | Dizziness, nausea, rash, chills, infusion-related reactions (after IV administration); lymphadenopathy, dizziness (after SC administration) |
Rare/very rare | Flushing, urticaria, anaphylaxis (after IV administration); pruritus (after SC administration) |
ATC codes | |
WHO ATC code | J05A-X (other antivirals) |
EphMRA ATC code | J5B9 (antivirals, others) |
2.3 Clinical Trials
2.3.1 Treatment of COVID-19
2.3.1.1 Non-Hospitalized Patients
Casirivimab/imdevimab reduced hospitalizations and death in non-hospitalized adults with COVID-19 in the phase 3 portion [9] of an ongoing randomized, double-blind, placebo-controlled, phase 1–3 trial (NCT04425629) [22]. Patients were aged ≥ 18 years, had tested positive for SARS-CoV-2 infection ≤ 3 days before randomization and had symptom onset ≤ 1 week prior to randomization [22]. In phase 3 of this multicentre [22] study, patients with one or more risk factors for severe COVID-19 were randomized to receive a single intravenous dose of casirivimab/imdevimab 1200 mg (n = 838), 2400 mg (n = 1529) or 8000 mg (n = 700) or placebo (n = 1500) [9]. Only 2400 mg and 8000 mg were originally included; however, on the basis of the generally similar findings with these doses in the phase 1/2 portion of the trial (discussed later) [22], the phase 3 protocol was amended to compare 2400 mg and 1200 mg with placebo [9].
Among patients whose SARS-CoV-2 infection was confirmed by reverse transcription quantitative PCR (RT-qPCR) [modified full analysis set; mFAS], the proportion who had at least one COVID-19-related hospitalization or died of any cause through day 29 (primary endpoint) was 1% with casirivimab/imdevimab 1200 mg versus 3% with concurrent placebo, a significant (p = 0.0024) relative risk reduction of 70% [9]. There did not appear to be a dose effect with casirivimab/imdevimab as, overall, findings were generally similar for 1200 mg versus 2400 mg. The number of patients who died did not significantly differ between casirivimab/imdevimab 1200 mg and placebo (one per group) or 2400 mg and placebo (one vs three). Moreover, the median time to symptom resolution was shorter (p ≤ 0.0001) with casirivimab/imdevimab 1200 mg and 2400 mg than with placebo (10 vs 14 days for each comparison). Casirivimab/imdevimab also significantly (p < 0.0001) reduced the least-squares mean (LSM) viral load from baseline to day 7 versus placebo, with the difference being − 0.71 log10 copies/mL with 1200 mg and − 0.86 log10 copies/mL with 2400 mg [9].
In the phase 1–2 portion of this trial, patients were randomized to receive a single intravenous dose of casirivimab/imdevimab 2400 mg (n = 92) or 8000 mg (n = 90) or placebo (n = 93) [22]. Data are from an interim analysis of the first 275 patients enrolled (FAS), 228 of whom were confirmed to be SARS-CoV-2 positive by RT-PCR at baseline (mFAS). At baseline, 123 (45%) patients were seropositive for antibodies against SARS-CoV-2, 113 (41%) were seronegative and the serum antibody status of 39 (14%) was unknown. The proportion of patients who had at least one COVID-19-related medically-attended visit within 29 days was numerically lower with casirivimab/imdevimab 2400 mg or 8000 mg than with placebo in the FAS (difference vs placebo − 3% and − 3%) and among patients who were seronegative at baseline (− 10% and − 8%) [prespecified key clinical endpoint analyses] [22]. For the respective casirivimab/imdevimab doses, the LSM differences from placebo in the time-weighted average (TWA) change from baseline in viral load from baseline through day 7 were − 0.52 and − 0.60 log10 copies/mL in mFAS patients who were seronegative at baseline (n = 28–35 per group) [prespecified key virological endpoint analysis] and − 0.25 and − 0.56 log10 copies/mL in the overall mFAS (n = 70–78 per group). Differences versus placebo were significant (based on 95% CIs) for casirivimab/imdevimab 8000 mg for the virological endpoint in both populations [22].
Casirivimab/imdevimab reduced viral load in non-hospitalized patients with SARS-COV-2 infection in an ongoing, randomized, double-blind, placebo-controlled, phase 2 study (NCT04666441) [9]. A total of 803 adults received a single intravenous or subcutaneous dose of casirivimab/imdevimab within 3 days of testing positive for SARS-CoV-2 infection. In patients randomized to an intravenous dose of 1200 mg (n = 116), the TWA daily change from baseline in viral load from day 1 to 7 in those who were positive for SARS-CoV-2 via RT-qPCR and seronegative at baseline (i.e. the seronegative mFAS) was − 0.56 log10 copies/mL with casirivimab/imdevimab versus placebo (p < 0.0007; primary endpoint). Mean changes from baseline in viral load on day 7 in seronegative mFAS patients who received an intravenous or subcutaneous dose of casirivimab/imdevimab 1200 mg were − 3.8 and − 4.1 versus − 3.5 log10 copies/mL with placebo (values estimated from a graph) [9].
2.3.1.2 Hospitalized Patients
Casirivimab/imdevimab improved survival in hospitalized seronegative patients with severe COVID-19 in a randomized, multicentre, phase 2/3 trial (NCT04381936; RECOVERY) [23]. A total of 9785 patients were randomized to receive either casirivimab/imdevimab 8000 mg via intravenous infusion in addition to usual care (e.g. corticosteroids and remdesivir) or usual care alone. Among seronegative patients (n = 3153; primary analysis population), 24% of those who received casirivimab/imdevimab in addition to usual care had died of any cause by day 28 compared with 30% of those who received usual care alone, a significant 20% reduction in all-cause mortality (rate ratio 0.80; 95% CI 0.70–0.91; p = 0.001) [primary endpoint] [23]. There was also a 17% lower relative risk of progressing to invasive mechanical ventilation or death (composite endpoint) with casirivimab/imdevimab plus usual care than with usual care alone among seronegative patients not on such ventilation at baseline (30% vs 37% of patients; relative risk ratio 0.83; 95% CI 0.75–0.92). Moreover, in the respective treatment groups, the median duration of hospital stay was 13 versus 17 days and the proportion of patients who were discharged alive by day 28 was 64% versus 58% (rate ratio 1.19; 95% CI 1.08–1.30). Notably, the benefits with regard to these endpoints were not seen with casirivimab/imdevimab in the overall trial population, which included patients who were seronegative, seropositive (n = 5272) or of unknown serum antibody status (n = 1360) [23].
2.3.2 Prevention of COVID-19
An ongoing randomized, double-blind, placebo-controlled, phase 3 trial (NCT04452318) is assessing the efficacy of subcutaneous casirivimab/imdevimab in preventing SARS-CoV-2 infection and subsequent symptomatic COVID-19 in uninfected household contacts of infected individuals (part A) and preventing symptomatic COVID-19 in household contacts recently infected with SARS-CoV-2 but asymptomatic (part B) [9, 20, 21]. The trial enrolled individuals aged ≥ 12 years living with a SARS-CoV-2-infected patient. Subjects were randomized to receive a single subcutaneous dose of casirivimab/imdevimab 1200 mg or placebo within 96 h of the infected patient’s positive SARS-CoV-2 test sample being collected. Subjects whose SARS-CoV-2 RT-qPCR test was negative entered cohort A and those whose SARS-CoV-2 RT-qPCR test was positive entered cohort B [9].
In cohort A, casirivimab/imdevimab was effective in preventing symptomatic and asymptomatic SARS-CoV-2 infection [21]. Of the 2067 subjects in this cohort, 1505 were seronegative at baseline and were the primary population for efficacy analyses. The proportion of subjects in this population who developed symptomatic SARS-CoV-2 infection (confirmed by RT-qPCR) during the 28-day assessment period (primary endpoint) was 1.5% with casirivimab/imdevimab (n = 753) and 7.8% with placebo (n = 752), a relative risk reduction of 81.4% (odds ratio [OR] 0.17; 95% CI 0.09–0.33; p < 0.001). In the respective groups, the mean duration of symptoms/symptomatic infection was 1.2 and 3.2 weeks [21]. Moreover, 4.8% of casirivimab/imdevimab recipients versus 14.2% of placebo recipients developed asymptomatic or symptomatic SARS-CoV-2 infection (confirmed by RT-qPCR), a 66.4% relative risk reduction (OR 0.31; 95% CI 0.21–0.46; p < 0.001). The mean duration of infection in the respective groups was 1.1 versus 2.2 weeks. Casirivimab/imdevimab also significantly (p < 0.001) reduced the likelihood of high viral load (> 104 copies/mL) [1.6% vs 11.3% of placebo recipients] and the total duration of high viral load (18.8 vs 181.6 weeks per 1000 participants) [21].
In cohort B, early treatment with casirivimab/imdevimab was effective in preventing asymptomatic SARS-CoV-2 infection from progressing to symptomatic COVID-19 [20]. Of the 314 subjects randomized in this cohort, 207 were seronegative at baseline and 204 of these were assessed for efficacy (three seronegative subjects were determined post-randomization to be symptomatic at baseline and excluded). Signs or symptoms of COVID-19 developed within 14 days of a baseline positive RT-qPCR test or during the 28-day efficacy assessment period in 29.0% of casirivimab/imdevimab recipients (n = 100) versus 42.3% of placebo recipients (n = 104) in this population, representing a 31.5% relative risk reduction (OR 0.54; 95% CI 0.30–0.97; p = 0.0380) [primary endpoint] [20]. The mean duration of symptoms per symptomatic patient was 21.7 and 27.3 days in the respective groups. Mean viral load appeared to decline more rapidly with casirivimab/imdevimab than with placebo, with an adjusted mean difference of − 1.5 log10 copies/mL at day 8; the mean number of days with high viral load (> 4 log10 copies/mL) was 3.4 versus 5.7. No casirivimab/imdevimab recipients had a COVID-19-related hospitalization or emergency room visit compared with six placebo recipients [20].
Exploratory efficacy data from a randomized, double-blind, placebo-controlled, phase 1 trial (NCT04519437) [9] support the cohort A findings of the phase 3 study [21]. Adult subjects who tested negative for SARS-CoV-2 at baseline received subcutaneous casirivimab/imdevimab 1200 mg (n = 729) or placebo (n = 240) every 4 weeks for 24 weeks. Clinically-diagnosed COVID-19 occurred in 0.4% of casirivimab/imdevimab recipients versus 5.0% of placebo recipients (OR 0.08; 95% CI 0.01–0.30; nominal p < 0.0001) [9].
Key clinical trials of casirivimab/imdevimab
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor (collaborator) |
|---|---|---|---|---|---|---|
Casirivimab/imdevimab | Treatment of COVID-19 (non-severe) | 4 | Not yet recruiting | Japan | UMIN000045358; R000051784 | National Hospital Organization Tochigi Medical Center |
Casirivimab/imdevimab | Treatment of COVID-19 [surveillance study] | 4 | Not yet recruiting | Japan | UMIN000044927, R000051317 | Chugai Pharmaceutical Co. Ltd |
Casirivimab/imdevimab; bamlanivimab + etesevimab; bamlanivimab; sotrovimaba | Treatment of COVID-19 | 3 | Recruiting | USA | NCT04790786; OPTIMISE-C19 | University of Pittsburgh Medical Center; Berry Consultants |
Casirivimab/imdevimab; bamlanivimab + etesevimab; placebo | Treatment of COVID-19 (non-hospitalized) | 3 | Recruiting | Italy | EudraCT2021-002612-31; MANTICO | Azienda Ospedaliera Universitaria Integrata Verona |
Casirivimab/imdevimab; bamlanivimab; SAB-185; AZD7442; SNG001; camostat; BRII-196/BRII-198; BMS-986414 + BMS-986413; placeboa | Treatment of COVID-19 (non-hospitalized) | 2/3 | Recruiting | Multinational | NCT04518410; ACTIV-2 | National Institute of Allergy and Infectious Diseases; Eli Lilly and Co.; AIDS Clinical Trials Group; Brii Biosciences Ltd; Sagent Pharmaceuticals; Synairgen Research Ltd; AstraZeneca; Bristol-Myers Squibb; SAb Biotherapeutics Inc |
Casirivimab/imdevimab; placebo | Treatment of COVID-19 (non-hospitalized) | 1/2/3 | Recruiting | Multinational | NCT04425629; EudraCT2020-003690-21 | Regeneron Pharmaceuticals |
Casirivimab/imdevimab; corticosteroid; convalescent plasma; lopinavir-ritonavir; hydroxychloroquine; azithromycin; tocilizumab; immunoglobulin; aspirin; colchicine; baricitinib; anakinra; dimethyl fumarate; empagliflozin | Treatment of COVID-19 (hospitalized) | 2/3 | Recruiting | Multinational | NCT04381936; EudraCT2020-001113-21; RECOVERY | University of Oxford; UK Research and Innovation; NIHR, UK; Wellcome; Bill and Melinda Gates Foundation; Department for International Development, UK; Health Data Research UK; Medical Research Council Population Health Research Unit; NIHR Clinical Trials Unit Support Funding; NIHR Health Protection Research Unit in Emerging and Zoonotic Infections |
Casirivimab/imdevimab; bamlanivimab | Treatment of COVID-19 (non-hospitalized) | 2 | Recruiting | USA | NCT04840459 | Sohail Rao |
Casirivimab/imdevimab; placebo | Treatment of COVID-19 (non-hospitalized) | 2 | Ongoing | USA | NCT04666441 | Regeneron Pharmaceuticals |
Casirivimab/imdevimab; placebo | Treatment of COVID-19 (hospitalized) | 1/2 | Completed | Multinational | NCT04426695; EudraCT2020-002537-15 | Regeneron Pharmaceuticals |
Casirivimab/imdevimab; placebo | Prevention of COVID-19 | 3 | Ongoing | Multinational | NCT04452318; EudraCT2020-003654-71 | Regeneron Pharmaceuticals |
2.4 Adverse Events
Casirivimab/imdevimab, administered via intravenous infusion or subcutaneous injection, was generally well tolerated in clinical studies [9, 20,21,22].
2.4.1 Intravenous
In the phase 1–3 treatment trial (NCT04425629), few patients with COVID-19 who received a single intravenous dose of casirivimab/imdevimab 2400 mg (n = 258) or 8000 mg (n = 260) or placebo (n = 262) experienced serious treatment-emergent adverse events (TEAEs) [1.6% and 0.8% vs 2.3%], grade 3 or 4 TEAEs (1.2% and 0.8% vs 1.5%), TEAEs of special interest (namely grade ≥ 2 hypersensitivity reactions or infusion-related reactions) [0% and 1.5% vs 0.8%] or withdrew from the study due to TEAEs (0% and 0.4% vs 0%) [24]. No deaths occurred in any of the treatment groups [24]. The tolerability profile of casirivimab/imdevimab in this study was generally similar in adults aged < 65 years and those aged ≥ 65 years [9].
Across the casirivimab/imdevimab clinical development programme, anaphylaxis/anaphylactic reactions have occurred, albeit very rarely [9]. These events, which occurred within 1 h of the infusion being completed, resolved with supportive treatment (e.g. epinephrine) [9].
Infusion-related reactions have occurred with all doses of intravenous casirivimab/imdevimab evaluated in clinical studies, with the signs and symptoms of these reactions commonly including chills, dizziness/syncope, flushing, nausea, rash and urticaria [9]. In general, the reactions occurred during or within 24 h of the infusion, were mild to moderate, and resolved with usual care or without intervention [9].
2.4.2 Subcutaneous
In subjects who did not have SARS-CoV-2 infection in the phase 3 prevention trial (NCT04452318; cohort A), subcutaneous casirivimab/imdevimab 1200 mg (n = 1311) was associated with a numerically lower incidence of TEAEs than placebo (n = 1,306) [20.2% vs 29.0%], with a higher incidence of COVID-19 with placebo accounting for this difference [21]. The incidence of non-COVID-19 TEAEs was 16.0% and 16.5% in the respective groups. Among the TEAEs that occurred most commonly (incidence ≥ 2%) across the treatment groups, only injection-site reaction occurred with a numerically greater incidence with casirivimab/imdevimab than with placebo (4.2% vs 1.5%). In the respective treatment groups, the incidence of grade ≥ 3 TEAEs (non-COVID-19 TEAEs) was 0.8% and 1.7% (0.8% and 1.3%) and the incidence of serious TEAEs (non-COVID-19 TEAEs) was 0.8% and 1.1% (0.8% and 0.8%). There were no AEs of special interest (namely grade ≥ 3 hypersensitivity and injection-site reactions) or TEAEs that lead to study drug withdrawal in either group. There were two deaths outside of the efficacy assessment period in each treatment group; none were considered to be related to COVID-19 [21].
The tolerability profile of subcutaneous casirivimab/imdevimab was generally similar in subjects with asymptomatic SARS-CoV-2 infection in this phase 3 prevention trial (cohort B) [20]. In the casirivimab/imdevimab (n = 155) and placebo (n = 156) groups, the incidence of TEAEs was 33.5% and 48.1% and that of non-COVID-19 TEAEs was 11.0% and 16.0%. The only TEAE to occur in ≥ 2% of patients (either group) and with a numerically greater incidence with casirivimab/imdevimab than with placebo was injection-site reaction (3.9% vs 0.6%). In the respective treatment groups, the incidence of grade ≥ 3 TEAEs (non-COVID-19 TEAEs) was 0.6% and 2.6% (0.6% and 0.6%) and the incidence of serious TEAEs (non-COVID-19 TEAEs) was 0.0% and 2.6% (0.0% and 0.6%). No AEs of special interest (i.e. grade ≥ 3 hypersensitivity and injection-site reactions) or TEAEs leading to withdrawal of study drug or death occurred in either group [20].
The tolerability profile of subcutaneous casirivimab/imdevimab in adolescents (aged ≥ 12 to < 18 years) was generally similar to that seen in adults in the phase 3 trial (NCT04452318; cohorts A and B) [9]. The tolerability profile of the combination was also generally similar in adults aged < 65 and ≥ 65 years across phase 3 (NCT04452318; cohorts A and B) and phase 1 (NCT04519437) studies [9].
All studies evaluating subcutaneous casirivimab/imdevimab (single or multiple doses) have reported injection-site reactions, common signs and symptoms of which include erythema, ecchymosis, pruritus, oedema, pain/tenderness and urticaria [9]. The reactions were generally mild to moderate, local and resolved either with usual care or without intervention [9].
2.5 Ongoing Clinical Trials
In addition to the ongoing trials discussed previously, a phase 3 study to determine the relative effects of monoclonal antibodies that have been granted emergency use authorization for COVID-19 in the USA (including casirivimab/imdevimab) is recruiting participants (NCT04790786; OPTIMISE-C19), as is a phase 3 trial to evaluate the efficacy of casirivimab/imdevimab and other monoclonal antibodies against SARS-CoV-2 in non-hospitalized patients with COVID-19 in Italy (EudraCT2021-002612-31; MANTICO). Recruitment is also underway in a multinational phase 2/3 trial to evaluate the safety and efficacy of various investigational agents, including casirivimab/imdevimab, that modify immune responses to SARS-CoV-2 or enhance viral control in non-hospitalized patients with COVID-19 (NCT04518410; ACTIV-2). Also recruiting is a US phase 2 trial (NCT04840459) to assess monoclonal antibodies against SARS-CoV-2 (casirivimab/imdevimab and bamlanivimab) in the treatment of non-hospitalized patients with mild to moderate COVID-19 at high risk of progressing to severe COVID-19 and/or hospitalization.
Two phase 4 trials plan to evaluate casirivimab/imdevimab use in patients with non-severe COVID-19 (UMIN000045358, R000051784) or with risk factors for severe COVID-19 (UMIN000044927, R000051317; surveillance study) in Japan. In addition, an expanded access programme is available to provide compassionate use access to casirivimab/imdevimab for adults with recently diagnosed mild-to-moderate COVID-19 who are at high risk of poor outcomes (NCT04617535).
References
Hu B, Guo H, Zhou P, et al. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19(3):141–54.
Wiersinga WJ, Rhodes A, Cheng AC, et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782–93.
Regeneron Pharmaceuticals. Regeneron's REGEN-COV2 is first antibody cocktail for COVID-19 to receive FDA emergency use authorization [media release]. 21 Nov 2020. http://www.regeneron.com.
Regeneron Pharmaceuticals Inc. Securities and Exchange Commission: Form 10-Q. 2021. https://investor.regeneron.com/static-files/7da40791-c1e0-4d9b-9edf-3ca9768f52a8. Accessed 13 Sept 2021.
European Medicines Agency. EMA issues advice on use of REGN-COV2 antibody combination (casirivimab/imdevimab) [media release]. 26 Feb 2021. https://www.ema.europa.eu/en/news/ema-issues-advice-use-regn-cov2-antibody-combination-casirivimab-imdevimab.
Regeneron Pharmaceuticals. Japan becomes first country to approve Regeneron antibody cocktail (casirivimab and imdevimab) for the treatment of mild to moderate COVID-19 [media release]. 19 July 2021. http://www.regeneron.com.
Chugai Pharmaceutical Co Ltd. Ronapreve® for intravenous infusion set: Japanese prescribing information. 2021. https://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/450045_62505A0A1023_1_01. Accessed 13 Sept 2021.
Regeneron Pharmaceuticals. UK authorizes Regeneron antibody cocktail to prevent and treat acute COVID-19 infection [media release]. 20 Aug 2021. http://www.regeneron.com.
UK Medicines & Healthcare Products Regulatory Agency. Casirivimab/imdevimab (Ronapreve): UK summary of product characteristics. 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1012415/revised-gb-spc-ronapreve-clean-120mg-ml12aug2021docx.pdf. Accessed 13 Sept 2021.
Australian Government. TGA grants provisional determination to Roche Products Pty Ltd combination COVID-19 treatment casirivimab + imdevimab (Ronapreve) [media release]. 23 Aug 2021. https://www.tga.gov.au.
Chugai Pharmaceutical Co Ltd. Chugai in-licenses antibody cocktail for COVID-19 from Roche [media release]. 10 Dec 2020. http://www.chugai-pharm.co.jp.
Regeneron Pharmaceuticals. Regeneron and Roche collaborate to significantly increase global supply of REGN-COV2 investigational antibody cocktail for COVID-19 [media release]. 18 Aug 2020. http://www.regeneron.com.
Regeneron Pharmaceuticals. Regeneron announces manufacturing and supply agreement for BARDA and U.S. Department of Defense for REGN-COV2 anti-viral antibody cocktail [media release]. 7 July 2020. http://www.regeneron.com.
Regeneron Pharmaceuticals Inc. Securities and Exchange Commission: Form 10-K. 2021. https://www.sec.gov/ix?doc=/Archives/edgar/data/872589/000180422021000008/regn-20201231.htm. Accessed 13 Sept 2021.
Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010–4.
Baum A, Ajithdoss D, Copin R, et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370(6520):1110–5.
Copin R, Baum A, Wloga E, et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell. 2021;184(15):3949-61.e11.
Tada T, Dcosta BM, Zhou H, et al. Decreased neutralization of SARS-CoV-2 global variants by therapeutic anti-spike protein monoclonal antibodies bioRxiv. 2021. https://doi.org/10.1101/2021.02.18.431897.
Zhou H, Dcosta BM, Samanovic MI, et al. B.1.526 SARS-CoV-2 variants identified in New York City are neutralized by vaccine-elicited and therapeutic monoclonal antibodies. MBio. 2021;12(4):e01386-e1421.
O’Brien MP, Forleo-Neto E, Sarkar N, et al. Subcutaneous REGEN-COV antibody combination in early SARS-CoV-2 infection. MedRxiv. 2021. https://doi.org/10.1101/2021.06.14.21258569.
O’Brien MP, Forleo-Neto E, Musser BJ, et al. Subcutaneous REGEN-COV antibody combination to prevent Covid-19. N Engl J Med. 2021. https://doi.org/10.1056/NEJMoa2109682.
Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med. 2021;384(3):238–51.
Regeneron Pharmaceuticals. REGEN-COV™ (casirivimab and imdevimab) phase 3 RECOVERY trial meets primary outcome, improving survival in hospitalized COVID-19 patients lacking an immune response to SARS-CoV-2 [media release]. 15 June 2021. http://www.regeneron.com.
European Medicines Agency. Assessment report: procedure under Article 5(3) of Regulation (EC) No 726/2004—Regeneron Ireland DAC use of casirivimab and imdevimab for the treatment of COVID-19. 2021. https://www.ema.europa.eu. Accessed 15 Sept 2021.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Emma Deeks is a contracted employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deeks, E.D. Casirivimab/Imdevimab: First Approval. Drugs 81, 2047–2055 (2021). https://doi.org/10.1007/s40265-021-01620-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01620-z