
Abstract
Infigratinib (TRUSELTIQTM), a fibroblast growth factor receptor (FGFR)-specific tyrosine kinase inhibitor, is being co-developed by QED Therapeutics and Helsinn for the treatment of cholangiocarcinoma, urothelial carcinoma and other FGFR-driven conditions. Infigratinib was recently approved in the USA for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement as detected by a test approved by the US Food and Drug Administration. This article summarizes the milestones in the development of infigratinib leading to this first approval for advanced cholangiocarcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this Adis Insight Report can be found at https://doi.org/10.6084/m9.figshare.14870097. |
A tyrosine kinase inhibitor is being co-developed by QED Therapeutics and Helsinn for the treatment of cholangiocarcinoma, urothelial carcinoma and other fibroblast growth factor receptor (FGFR)-driven conditions. |
Received its first approval on 28 May 2021 in the USA. |
Approved for use in previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement. |
1 Introduction
Infigratinib (TRUSELTIQTM) is a fibroblast growth factor receptor (FGFR)-specific tyrosine kinase inhibitor being co-developed by QED Therapeutics and Helsinn for the treatment of advanced cholangiocarcinoma [1]. FGFRs play a role in cell proliferation, differentiation and angiogenesis [2]. Infigratinib received its first approval on 28 May 2021 in the USA for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement as detected by a test approved by the US Food and Drug Administration (FDA) [2, 3]. The recommended dosage of infigratinib is 125 mg orally once daily for 21 consecutive days in a 28-day cycle, continued until disease progression or unacceptable toxicity [2]. Treatment with infigratinib may cause retinal pigment epithelial detachment (RPED), hyperphosphatemia, soft tissue mineralization and embryo-foetal toxicity [2].
Infigratinib is also being developed in urothelial carcinoma (currently enrolling a phase III trial) and, at much lower doses, achondroplasia (at phase II) [1]. Development of infigratinib in haematological malignancies, malignant melanoma, solid tumours and other cancers (head and neck, squamous cell, colon, pancreatic and rectal) are not ongoing.

Key milestones in the development of infigratinib. CCA cholangiocarcinoma, NDA New Drug Application
1.1 Company Agreements
The intellectual property rights to infigratinib for the treatment of FGFR-mediated diseases were licensed from Novartis Oncology to BridgeBio Pharma in January 2018 [4]. BridgeBio Pharma is developing the drug with its subsidiary, QED Therapeutics [4]. In September 2019, a collaboration agreement was signed between the Ivy Brain Tumor Center and QED Therapeutics to investigate infigratinib for the treatment of glioblastoma, with particular focus on FGFR genetic alterations [5]. LianBio and BridgeBio Pharma entered into an agreement in August 2020 to develop and commercialise infigratinib and BBP 398 in China and other Asian territories [6]. LianBio will have commercial rights in most of these markets and preferential access to many of BridgeBio’s development candidates in the future [6]. In March 2021, QED Therapeutics entered into a global collaboration and licensing agreement with Helsinn for the development and commercialisation of infigratinib in all indications except skeletal dysplasia [7]. Under the terms of the agreement, all rights to infigratinib in skeletal dysplasia (including achondroplasia) will be retained by BridgeBio Pharma. Helsinn and BridgeBio will co-commercialise infigratinib in oncology indications in the USA, and Helsinn will have exclusive commercialisation rights outside the USA (excluding China, Hong Kong and Macau, which are covered by the agreement with LianBio) [7].

Chemical structure of infigratinib
2 Scientific Summary
2.1 Pharmacodynamics
FGFR activity can lead to the proliferation of malignant cancer cell lines with activating FGFR amplifications, fusions or mutations [2]. Infigratinib selectively binds to and inhibits FGFR1, FGFR2, FGFR3 and FGFR4 (IC50 1.1 nM, 1 nM, 2 nM and 61 nM, respectively). This inhibition of FGFR leads to reduced cell proliferation in tumours with these FGFR mutations or rearrangements. The binding affinities for the major metabolites, BHS697 and CQM157, are similar to that of infigratinib. Infigratinib demonstrated anti-tumour activity in xenograft models of cholangiocarcinoma with activating FGFR2 and FGFR3 alterations [2]. The FGFR inhibitory effect of infigratinib can also lead to increased serum phosphate levels at the approved dosage; the risk of hyperphosphataemia is higher with increased doses [8]. Infigratinib dose adjustments (reduction, temporary or permanent discontinuation) may be required [2].
Anti-tumour activity has been seen in a phase I trial of infigratinib in patients with advanced urothelial carcinoma with FGFR3 mutations [9], leading to an expanded phase I trial in this cohort [10]. Hyperphosphataemia was associated with increasing infigratinib exposure and enhanced efficacy in these patients [11]. Animal model data also suggest the potential of infigratinib (at much lower doses) in treating children with achondroplasia [12].
2.2 Pharmacokinetics
Following administration with the approved dosage of infigratinib in patients with cholangiocarcinoma, the mean steady-state maximum drug concentration (Cmax) and area under the time-concentration curve from time zero to 24 h (AUC0−24) were 282.5 ng/mL and 3780 ng·h/mL, respectively [2]. Cmax and AUC0−24 increased more than dose proportionally over a dose range of 5–150 mg, with a mean accumulation ratio of 8- and 5-fold, respectively. In healthy subjects, mean Cmax and AUCinfinity were both increased following administration of infigratinib with a high-fat and high-calorie meal (by 60–80% and 80–120%, respectively). However, coadministration of infigratinib with a low-calorie meal only increased the mean AUCinfinity by 70% [2].
Steady state was reached within 15 days in patients with cholangiocarcinoma who were administered infigratinib [2]. At steady state, the median time to reach Cmax (tmax) was 6 h, with a mean apparent volume of distribution of 1600 L. Infigratinib binds dose-dependently to protein (mean 96.8%), mainly to lipoprotein. The geometric mean total apparent clearance was 33.1 L/h and terminal half-life was 33.5 h [2].
In vitro infigratinib metabolism was predominantly CYP3A4-mediated (94%) [2]. The two major metabolites (both > 10% of dose) BHS697 and CQM157 accounted for ≈ 16–33% and ≈ 9–12%, respectively, of the overall pharmacological activity of infigratinib. In plasma, 38% of the infigratinib dose was unchanged infigratinib [2]. In healthy subjects, a single dose of radiolabeled infigratinib resulted in ≈ 77% excretion in faeces and 7.2% in urine (3.4% and 1.9% as unchanged infigratinib) [13].
The relative potency adjusted steady-state AUC for infigratinib (and its metabolites) were increased in patients with abnormal kidney or liver function, therefore the infigratinib dosage should be reduced in patients with mild to moderate kidney or liver dysfunction [2]. In vitro studies show infigratinib did not induce CYP1A2, CYP2B6, CYP2C9 or CYP3A4; infigratinib and its metabolites did not inhibit major CYP450 enzymes. Infigratinib inhibited MATE1 and BCRP, and is a substrate for P-glycoprotein (P-gp) and BCRP. At clinically relevant concentrations, infigratinib and its metabolites have a low potential to inhibit OATP1B1, OATP1B3, P-gp or BCRP [2].
2.3 Therapeutic Trials
The first approval of infigratinib for the treatment of cholangiocarcinoma was granted following the results from a multicentre, open-label, single-arm, phase II trial (NCT02150967) [2]. Patients with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement received oral infigratinib at the approved dosage [2]. In a primary analysis (data cutoff 30 June 2016) of 61 patients [48 patients (78.7%) had FGFR2 fusions], the primary endpoint of overall response rate, as assessed by local site radiographic review, was achieved by 14.8% of patients [14]. Median progression-free survival (PFS) was estimated at 5.8 months (95% CI 4.3–7.6 months) and the disease control rate was 75.4% (95% CI 62.7–85.5%) [14].
At a median follow-up of 10.6 months (data cutoff 31 March 2020), treatment with infigratinib in 108 patients (81% had in-frame FGFR2 fusions, 19% had other FGFR2 rearrangements) resulted in an overall objective response rate (as assessed by blinded independent central review) of 23.1% (95% CI 15.6–32.2%; primary endpoint) [2], and 34.0% and 13.8%, respectively, in patients with ≤ 1 or ≥ 2 prior lines of treatment [15]. The median duration of response was 5.0 months (range 3.7–9.3 months) [2]. One patient (0.9%) has achieved complete response, with partial response in 22% of patients. Duration of response exceeding 6 months has been observed in 32.0% of patients [2]. The disease control rate (composite endpoint of complete and partial response) was 84.3% (95% CI 76.0-90.6%) [15].
The efficacy of oral infigratinib (125 mg/day for 21 days per 28-day cycle) was also evaluated in a prospective, open-label, single-arm, phase II trial in patients with previously treated, advanced urothelial carcinoma with FGFR3 alterations (n = 67) [10]. At primary analysis (data cutoff 30 January 2017), the primary endpoint of overall response rate (included complete and partial responses) was 25.4% of patients. In these patients, the median duration of response was an estimated 5.1 months (95% CI 3.9–7.4 months). Disease control (a composite endpoint of complete response, partial response and stable disease) was observed in 64.2% of patients. Estimated median PFS and median overall survival were 3.8 months (95% CI 3.1–5.4 months) and 7.8 months (95% CI 5.7–11.6 months), respectively [10].
2.4 Adverse Events
The tolerability of infigratinib, when administered at the approved dosage, was analysed in a pooled safety population (n = 351) of patients with cholangiocarcinoma (from study NCT02150967) and patients with other advanced solid tumours or haematological malignancies [2]. Adverse reactions of special interest from this pooled population included RPED, hyperphosphataemia and soft tissue mineralization. RPED was reported in 11% of patients, leading to dose interruption or reduction in 3.4% of patients and permanent discontinuation in 0.6% of patients. Hyperphosphataemia, which can lead to soft tissue mineralization, cutaneous calcinosis and other adverse reactions, was reported in 82% of patients, with a median time to onset of 8 days (range 1–349 days) [2].
Features and properties of infigratinib
Alternative names | BBP-831; BGJ-398; BGJ-398 phosphate; Infigratinib phosphate; NVP-BGJ398; TRUSELTIQ |
Class | Aniline compounds, Antineoplastics, Chlorobenzenes, Methylurea compounds, Phenyl ethers, Piperazines, Pyrimidines, Small molecules |
Mechanism of action | Fibroblast growth factor receptor (FGFR)-specific tyrosine kinase inhibitor that inhibits tumour angiogenesis and proliferation, leading to tumour cell death |
Route of administration | Oral |
Pharmacodynamics | Inhibits FGFRs, which are involved in malignant cell proliferation in tumours with FGFR alterations, thereby reducing tumour growth; exhibited anti-tumour activity in xenograft models of cholangiocarcinoma |
Pharmacokinetics | Exhibits non-linear pharmacokinetics in patients with cholangiocarcinoma; drug exposure increased following food; tmax 6 h; mainly protein-bound; terminal t1/2 33.5 h; mainly metabolised by CYP3A4 |
Adverse reactions | |
Most frequent (> 40%) | Nail toxicity, stomatitis, dry eye, fatigue |
Laboratory abnormalities (> 50%) | Increased: creatinine, phosphate, alkaline phosphate, alanine aminotransferase Decreased: phosphate, haemoglobin |
Special warnings and precautions | Retinal pigment epithelial detachment, hyperphosphatemia and soft tissue mineralization, embryo-foetal toxicity |
ATC codes | |
WHO ATC code | L01X-E (Protein kinase inhibitors) |
EphMRA ATC code | L1H (Protein Kinase Inhibitor Antineoplastics) |
Chemical name | 3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-[6-[4-(4-ethylpiperazin-1-yl)phenylamino]pyrimidin-4-yl]-1-methylurea phosphate (1:1) |
Key clinical trials of infigratinib
Indication | Phase | Status | Location(s) | Identifier | Sponsor(s) |
|---|---|---|---|---|---|
Cholangiocarcinoma | III | Ongoing | Global | NCT03773302, EudraCT2018-004004-19, PROOF 301 | LianBio, QED Therapeutics |
Cholangiocarcinoma | II | Ongoing | Global | NCT02150967, EudraCT2013-005085-19 | Novartis Pharmaceuticals, QED Therapeutics |
Urothelial carcinoma | III | Ongoing | Global | NCT04197986, EudraCT2019-003248-63, PROOF 302 | QED Therapeutics |
Achondroplasia | II | Ongoing | Global | NCT04265651, EudraCT2019-002954-21, PROPEL 2 | QED Therapeutics |
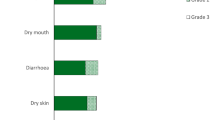
In the phase II trial (NCT02150967) in patients with advanced cholangiocarcinoma treated with infigratinib. adverse reactions with an incidence of ≥ 30% were nail toxicity (57% of patients), stomatitis (56%), dry eye (44%), fatigue (44%), alopaecia (38%), palmar-plantar erythrodysesthesia (PPE; 33%), arthralgia (32%), dysgeusia (32%) and constipation (30%) [2]. Grade 3–4 adverse reactions (incidence ≥ 5%) included stomatitis (15%), PPE (7%) and abdominal pain (5%). Laboratory abnormalities that worsened from baseline to grade 3–4 in > 10% of patients included increased urate (37%), decreased phosphate (31%), decreased sodium (20%) and increased phosphate (13%) [2].
Serious adverse reactions were reported in 32% of patients, and included infections, anaemia, pyrexia, abdominal pain, hypercalcaemia and sepsis (all ≥ 2%) [2]. One patient died due to sepsis; this was not considered treatment-related. Permanent discontinuation due to adverse reactions occurred in 15% of patients, most commonly due to increased blood creatinine, fatigue, subretinal fluid and calcinosis. Dosage interruption due to an adverse reaction occurred in 64% of infigratinib recipients and were mostly commonly (≥ 5% of patients) due to hyperphosphataemia, hypercalcaemia, PPE, stomatitis, diarrhea and increased blood creatinine. Dosage reductions due to an adverse reaction occurred in 60% of patients, most frequently (≥ 2% of patients) due to hyperphosphataemia, hypercalcaemia, PPE, stomatitis, increased blood creatinine, increased lipase and onycholysis [2].
In a phase I expansion trial of patients with urothelial carcinoma (n = 67) who received infigratinib, adverse events (AEs) with an incidence > 35% of patients included hyperphosphatemia (46.3%), increased creatinine (41.8%), fatigue (37.3%), constipation (37.3%) and anaemia (35.8%) [10]. The most frequent (incidence ≥ 7.5% of patients) grade 3–4 AEs included hyperlipasaemia (10.4%), fatigue (7.5%), anaemia (7.5%), hypophosphataemia (7.5%), and PPE (7.5%). Grade 3–4 hyperphosphataemia was reported in one patient (1.5%), which was manageable and reversible. At data cutoff, 14.9% of patients had discontinued treatment due to AEs [10].
2.5 Companion Diagnostic
The US FDA has approved the FoundationOne® CDx next-generation, sequencing-based in vitro diagnostic device for the detection of FGFR2 fusion or other rearrangement in patients with cholangiocarcinoma who may benefit from treatment with infrigatinib [3, 16].
2.6 Ongoing Clinical Trials
Infigratinib is being investigated in phase II and III trials in several indications. In addition to the ongoing phase II trial (NCT02150967) in cholangiocarcinoma (on which accelerated approval in the USA was based), the multicentre, open-label, randomized, controlled, phase III trial (NCT03773302; PROOF 301) is currently recruiting patients with advanced cholangiocarcinoma. The primary efficacy endpoint is PFS. Another phase II trial (NCT04233567) in patients with advanced or metastatic solid tumours with FGFR mutations is also at the recruitment stage, with a primary endpoint of overall response rate.
The ongoing multicentre, double-blind, randomized, placebo-controlled, phase III trial (NCT04197986; PROOF 302) is evaluating the efficacy and safety of infigratinib in patients with urothelial carcinoma; the primary efficacy endpoint is centrally determined disease-free survival. A phase II, open-label, dose-escalation and dose-expansion trial (NCT04265651; PROPEL 2) is recruiting paediatric patients with achondroplasia, with primary endpoints of annualized height velocity and incidence of treatment-emergent AEs.
3 Current Status
Infigratinib received its first approval on 28 May 2021 in the USA for previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement as detected by an FDA-approved test [3].
Change history
24 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01652-5
References
QED Therapeutics. QED Pipeline. 2021. https://www.qedtx.com/our-product/#pipeline. Accessed 29 Jun 2021.
QED Therapeutics. TRUSELTIQ (infigratinib) capsules, for oral use: US prescribing information. 2021. https://www.accessdata.fda.gov. Accessed 29 Jun 2021.
US Food & Drug Administration. FDA grants accelerated approval to infigratinib for metastatic cholangiocarcinoma [media release]. 28 May 2021. https://www.fda.gov.
BridgeBio Pharma. BridgeBio Pharma licenses late-stage oncology drug infigratinib to tackle FGFR-driven maladies; establishes new subsidiary QED Therapeutics with $65 million in initial financing [media release]. 30 Jan 2018. http://www.bridgebio.com.
The Ivy Brain Tumor Center. Ivy Brain Tumor Center and BridgeBio subsidiary QED Therapeutics announce collaboration to advance cancer research and treatment options [media release]. 24 Sep 2019. http://www.ivybraintumorcenter.org.
BridgeBio Pharma. BridgeBio Pharma expands reach into China and other major Asian markets through strategic collaboration with perceptive advisors-founded company, LianBio [media release]. 11 Aug 2020. http://www.bridgebio.com.
BridgeBio Pharma. BridgeBio Pharma's affiliate QED Therapeutics and Helsinn Group announce strategic collaboration to co-develop and commercialize infigratinib in oncology [media release]. 31 Mar 2021. http://www.bridgebio.com.
Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35(2):157–65.
Sequist LV, Cassier P, Varga A, et al. Phase I study of BGJ398, a selective pan-FGFR inhibitor in genetically preselected advanced solid tumors [abstract no. CT326]. Cancer Res. 2014;74(Suppl 19):CT326.
Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1–3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018;8(7):812–21.
Lyou Y, Grivas P, Rosenberg JE, et al. Hyperphosphatemia secondary to the selective fibroblast growth factor receptor 1–3 inhibitor infigratinib (BGJ398) is associated with antitumor efficacy in fibroblast growth factor receptor 3-altered advanced/metastatic urothelial carcinoma. Eur Urol. 2020;78(6):916–24.
Komla-Ebri D, Dambroise E, Kramer I, et al. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J Clin Invest. 2016;126(5):1871–84.
Reyes M, Zamora C, Andrews S, et al. A phase I, open-label, single-dose study to evaluate the excretion and mass balance, pharmacokinetics, and metabolism of [14C]-infigratinib (BGJ398) in healthy adult volunteers [abstract no. PIII-045]. Clin Pharmacol Ther. 2021;109(Suppl 1):S57.
Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276–82.
Javle MM, Roychowdhury S, Kelley RK, et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement [abstract no. 265 plus oral presentation]. In: ASCO Gastrointestinal Cancers Symposium. 2021.
QED Therapeutics, Foundation Medicine. QED Therapeutics announces a collaboration with Foundation Medicine to develop ccmpanion diagnostics for infigratinib [media release]. 10 Dec 2018. http://www.qedtx.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Connie Kang is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kang, C. Infigratinib: First Approval. Drugs 81, 1355–1360 (2021). https://doi.org/10.1007/s40265-021-01567-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01567-1