
Abstract
Statins are a group of lipid-lowering drugs that inhibit cholesterol biosynthesis and have anti-inflammatory, anti-tumor, and immunomodulatory properties. Several lines of evidence indicate that statins regulate multiple proteins associated with the regulation of differing cellular pathways. The 5′-adenosine monophosphate-activated protein kinase (AMPK) pathway plays an important role in metabolism homeostasis with effects on cellular processes including apoptosis and the inflammatory responses through several pathways. Recently, it has been shown that statins can affect the AMPK pathway in differing physiological and pathological ways, resulting in anti-cancer, cardio-protective, neuro-protective, and anti-tubercular effects; additionally, they have therapeutic effects on non-alcoholic fatty liver disease and diabetes mellitus-associated complications. Statins activate AMPK as an energy sensor that inhibits cell proliferation and induces apoptosis in cancer cells, whilst exerting its cardio-protective effects through inhibition of inflammation and fibrosis, and promotion of angiogenesis. Furthermore, statin-associated AMPK activation leads to decreased lipid accumulation and decreased amyloid beta deposition in the liver and brain, respectively, and may have therapeutic effects on the liver and neurons. In this review, we summarize the results of studies of AMPK-associated therapeutic effects of statins in different pathological conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Statins are lipid-lowering agents that inhibit cholesterol synthesis |
AMPK is a biologic sensor for cell energy and is involved in homeostasis and maintenance |
Activation of AMPK by statins could exert therapeutic effects in a number of pathological conditions |
1 AMP-Activated Protein Kinase
5′-Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a highly adaptive complex in eukaryotes that regulates cellular metabolism and energy hemostasis. It detects low levels of cellular adenosine triphosphate (ATP) as a biologic sensor and becomes active in response to the elevated AMP/ATP ratio (reduced energy supply index) via attachment to adenosine diphosphate and/or AMP [1,2,3,4]. The AMPK structure is that of a heterotrimeric complex including a catalytic α-subunit (the major and key component with a kinase domain and an important residue that is phosphorylated by upstream kinases) and regulatory β (includes the carbohydrate binding module that is essential for glycogen attachment) and γ (that allows the AMPK to reacts to different levels of AMP, adenosine diphosphate, and ATP) subunits. In humans, each subunit has several distinct isoforms encoded by different genes and comprises 12 different AMPKs [5,6,7]. Following its activation, AMPK directs metabolism to increase catabolism or decrease anabolism via phosphorylation of key proteins in various pathways including protein synthesis, lipid homeostasis, glucose metabolism, and mitochondria hemostasis and is associated with growth, apoptosis, and inflammation [8,9,10].
AMPK regulates cell growth through the inhibition of the mammalian target of the rapamycin complex (mTOR) via direct phosphorylation of the tuberous sclerosis complex [11, 12]. Other reported targets of AMPK are p53 tumor suppressor, cyclin-dependent kinase inhibitors, p21WAF1, and p27CIP1 that are related to cell-cycle arrest [13, 14]. In addition, AMPK is the upstream kinase of key metabolic enzymes involved in cholesterol and fatty acid synthesis including acetyl CoA carboxylase and 3-hydroxy-3-methylglutaryl-coenzyme A reductase [4]. Moreover, glucose uptake is regulated by AMPK through modulatory effects on glucose transporter type 4 trafficking in specialized tissues such as muscle and fat [15]. Furthermore, AMPK inhibits glycolysis and downregulates gluconeogenesis enzymes including phosphoenolpyruvate carboxykinase and glucose-6-phosphatase that control the hepatic production of glucose [16, 17]. It has been reported that in energy deprivation conditions, AMPK stimulates the unc-51-like kinase 1 complex that induces auto-phagosome formation as a result of effects on Beclin-1 and Vps34 [18].
Studies have shown that dysregulation in AMPK and associated pathways is involved in cancers, inflammatory disorders, obesity, diabetes mellitus, atherosclerosis, and cardio-metabolic diseases. Hence, this pathway could be considered as a potential molecular target for new therapeutics in those disease conditions [19,20,21,22,23,24].
2 Statins
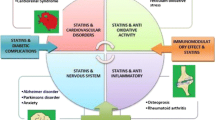
During the last 40 years, statins have revolutionized the prevention of atherosclerotic coronary artery disorders. Lovastatin, the first statin family member, was introduced as a drug agent to decrease low-density lipoprotein cholesterol [25]. Statins are 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors that have a similar structure to 3-hydroxy-3-methylglutaryl-coenzyme A and inhibit the L-mevalonate synthesis and subsequently decrease the hepatocellular cholesterol production [26,27,28]. The statin family includes naturally occurring members (lovastatin, mevastatin, pravastatin, pitavastatin, simvastatin) that are fungal derivatives and synthetic agents (atorvastatin, fluvastatin, rosuvastatin) that bind to the target enzyme in nano-molar concentrations and act as competitive inhibitors of substrates [29]. Several studies have confirmed that statins have pleiotropic effects that are not limited to lowering cholesterol levels, with a wide range of anti-inflammatory, anti-tumor, immunomodulatory, and anti-thrombotic effects [26, 30,31,32,33,34] (Fig. 1; Table 1). To provide the rationale for ongoing clinical trials in the areas of immunology, oncology, and pharmacology, this review focusses on the mechanisms that statins use to exert their effects in an AMPK-associated manner.

5′-Adenosine monophosphate-activated protein kinase (AMPK)-associated therapeutic effects of statins. DMED diabetes mellitus-induced erectile dysfunction, eNOS endothelial nitric oxide synthase, ER endoplasmic reticulum, FOXO3a Forkhead box O3, HIF-1a hypoxia-inducible factor-1 alpha, LKB1 liver kinase B1, mTOR mammalian target of the rapamycin complex, mTORC1 mechanistic target of rapamycin complex-1, SREBP1 sterol regulatory element binding protein‐1, TFEB transcription factor EB
3 AMPK-Associated Therapeutic Effects of Statins
3.1 Antitumor Effects
Potential therapeutic effects of statins in different types of cancers including head and neck squamous cell carcinoma, lung, prostate, colorectal, and breast cancer were demonstrated through the induction of apoptosis and cell-cycle arrest, and therefore as potential adjunct anti-cancer agents [35,36,37,38,39]. Statins activate AMPK as a cellular energy sensor to induce the survival of metabolic homeostasis under stress conditions, which leads to apoptosis induction and inhibition of the viability of cancer cells [40, 41]. From the data accrued, whilst statin therapy appears to have an antitumor effect it is not clear whether healthy cells may also be damaged in some circumstances.
3.1.1 Myeloid Leukemia
Mojjarad and Pazhang showed that rosuvastatin and orlistat combination therapy on the K562 human myeloid leukemia cell line in vitro synergistically inhibited cell proliferation via the AMPK/Akt/c-Myc axis [42].
3.1.2 Glioblastoma
Misirkic et al., who evaluated the effects of simvastatin on U251 and C6 glioma cell lines, reported that simvastatin induced AMPK activation, downregulated Akt, and inhibited mTOR and its substrate, p70 S6 kinase 1, which results in autophagolysosome-like intracytoplasmic acidic vesicle formation. In addition, autophagy inhibition via 3-methyladenine, bafilomycin A1, LC3β shRNA as well as Compound C and AMPK siRNA significantly increased apoptotic death in simvastatin-treated U251 cells, which demonstrated AMPK-associated statin antitumor effects [43].
3.1.3 Renal Cancer
Vorinostat is an antitumor agent that is a histone deacetylase inhibitor that phosphorylates the S6 ribosomal protein and activates the intrinsic mTOR pathway that is associated with decreased anticancer potency and the efficacy of vorinostat. Okubo and colleagues evaluated the effects of vorinostat in combination with fluvastatin in renal cancer cells and showed that fluvastatin activated AMPK as an mTOR inhibitor, preventing tumor protection in vitro (human renal cancer cells ACHN, A498, and murine renal cancer Renca) and in vivo (Renca murine model) [44].
3.1.4 Bladder Cancer
In another study, Okubo et al. demonstrated that the simvastatin and romidespin combination enhanced apoptosis and death in bladder cancer cells (UMUC-3, T-24, J-82, MBT-2) as a result of simvastatin-induced AMPK activation that led to histone acetylation and endoplasmic reticulum stress induction, and inhibitory effects of romidespin on histone deacetylase [45].
3.1.5 Squamous Cell Carcinoma
Ma et al. reported that lovastatin activated the liver kinase B1 (LKB1)/AMPK pathway in SCC9 and SCC25 squamous cell carcinoma cell lines as a result of mitochondrial dysfunction induction and decreased the cellular adenosine diphosphate/ATP ratio [46].
3.1.6 Breast Cancer
Huang et al. demonstrated that lovastatin increased phosphorylation of LKB1 and AMPK in the MCF-1 breast cancer cell line in a time-dependent manner, and myc-tagged AMPK dominant negative mutant cell transfection significantly inhibited lovastatin-induced p53 and p38MAPK phosphorylation. In addition, LKB1 knockdown with LKB1 siRNA dramatically decreased AMPK, P38MAPK, and p53 phosphorylation in lovastatin-treated MCF-7 cells. These results confirmed that lovastatin may activate the LKB1-AMPK-p38MAPK-p53 signaling cascade and lead to reduced survival and increased death of MCF-7 and MDA-MB-231 cell lines [47]. In a further study, Wang et al. evaluated anti-angiogenesis effects of simvastatin on the 4T1 mice breast cancer cell line and reported that simvastatin lead to post-translational down-regulation of hypoxia-inducible factor-1 alpha, as a pro-angiogenic factor, via AMPK activation. They demonstrated that this effect was abrogated by compound C that inhibited AMPK phosphorylation [48].
3.1.7 Hypo-Pharyngeal Carcinoma
In another study, Yen et al. demonstrated that the antitumor effects of lovastatin in FaDu hypo-pharyngeal carcinoma cells are the result of AMPK-p38MAPK-p63-survivin cascade activation. They showed that lovastatin induced phosphorylation and activation of AMPK, p38MAPK, and p63 transcription factor as well as p63 acetylation, which increases its affinity to the surviving promoter and leads to cell-cycle arrest and apoptosis induction in vitro and inhibits subcutaneous FaDu xenograft proliferation in a mice model in vivo [39].
3.1.8 Hepatocellular Carcinoma
Wang et al. demonstrated that simvastatin-induced G0/G1 cell-cycle arrest in hepatocellular carcinoma was due to p21 and p27 accumulation in HepG2 and Hep3B cells. Simvastatin activated AMPK and lead to p21 up-regulation and increased p27 expression as a result of STAT3-Skp2 inhibition. In HepG2 xenograft mice, simvastatin reduced the expression of Skp2 and induced AMPK activation and lead to p21 and p27 over-expression in tumor sections [41].
3.1.9 Osteocarcoma
Kamel et al. evaluated the anti-tumor effects of simvastatin in a murine model of osteosarcoma and showed that simvastatin inhibited the proliferation and migration of osteosarcoma cells as well as apoptosis induction. While it has been shown that AMPK and p38MAPK inhibitors may reduce simvastatin-induced apoptosis, metformin enhances apoptosis via AMPK induction [40].
FOXO3a protein (member of forkhead box class O subfamily) is a transcription factor that regulates multiple physiologic and pathologic pathways including proliferation, cell-cycle progression, cell survival, DNA damage, and apoptosis. FOXO3a phosphorylation by AMPK and MST1 facilitates its nuclear translocation and mediates multiple cellular processes as a result of transcription induction of target genes [49]. Lee et al. showed that pitavastatin had a higher inhibitory potential in OSCC15 compared with OSCC4, as human oral squamous cell carcinoma cell lines. Higher sensitivity of OSCC15 may be as a result of higher expression of FOXO3a and upstream kinases including Akt and AMPK, though pitavastatin-induced AMPK and Akt modulation patterns could be another explanation. Pitavastatin lead to reduced Akt phosphorylation and increased AMPK and FOXO3a phosphorylation as well as FOXO3a expression in SCC15 though not SCC4 cells. The anti-tumor effects of statins are exerted via Akt inhibition and AMPK activation that leads to FOXO3a translocation and apoptosis induction via the p53 upregulated modulator of apoptosis [50]. AMPK phosphorylated FOXO3a in six residues and induced gene transcription that lead to cell-cycle arrest and apoptosis in breast cancer cells and hepatoma cancer cells [51, 52].
3.2 Cardio-Protective Effects
Statin-induced AMPK activation appears to have a cardio-protective effect via activating endothelial nitric oxide synthase (eNOS), inducing angiogenesis, and exerting anti-inflammatory, anti-fibrotic, and anti-hypertrophic effects. Sun et al. reported that atorvastatin phosphorylated and activated AMPK and increased eNOS levels downstream both in vivo (murine) and in vitro (human umbilical vein endothelial cells) [53]. Pravastatin induced AMPK and eNOS activation is reported to be associated with angiogenesis progression in a murine model of ischemia-induced angiogenesis, and compound C abrogated these effects as an AMPK inhibitor [54]. In another study, Rossoni et al. demonstrated that simvastatin activated AMPKα and eNOS, while having no effect on Akt expression, and AMPK phosphorylation led to significant modulation of arterial resistance via AMPK/phospho-eNOS (Ser1177)/NO-associated pathways [55].
Investigation into the anti-hypertrophic effects of atorvastatin showed that statin-associated AMPK activation was the main mechanism. In a rat model of myocardium hypertrophy, atorvastatin significantly inhibited the heart weight gain and halted destruction of cardiac tissue. In addition, it increased viability and inhibited apoptosis in H9c2 cells. Molecular analysis demonstrated that these effects were due to miR-143-3p inhibition as a result of AMPK and FOXO activation [56].
Following the cardio-protective effects of statins, multiple studies evaluated AMPK-associated anti-fibrotic effects of statins in different animal models. They showed that rosuvastatin inhibited pro-fibrotic markers such as procollagen type II, its carboxy-terminal proteinase, and lysy oxidase in a murine model of metabolic syndrome, while using AMPKα1/α2 siRNA transfected fibroblasts abrogated these effects [57]. Increased adiponectin secretion with significantly reduced expression of type I and III collagens, together with inhibition of angiotensin II-induced cardiac fibrosis was shown as a result of atorvastatin-associated AMPK activation in the angiotensin II-induced cardiac fibrosis model in adiponectin-knock-out mice [58].
In the case of acute myocardial infarction, dramatically reduced fibrosis and inflammation levels as well as a reduction in cell death and reduced infarct size in the primary phase of ischemia were demonstrated in rat, pig, and mice models. They showed that these effects are associated with AMPK activation and mTOR signaling pathways, and those effects were reversed with compound C [59, 60].
3.3 NAFLD
Studies have reported that statins reduce hepatic lipid accumulation and may have therapeutic utility in non-alcoholic fatty liver disease (NAFLD), although the exact mechanism of action needs clarification. AMPK activation could inhibit acetyl CoA carboxylase as a key enzyme in lipogenesis as well as through the regulation of hepatic fatty acid metabolism via total mitochondrial content and function modulation [61]. AMPK acts as an energy sensor to cell hemostasis maintenance and increases fatty acid oxidation via peroxisome proliferator-activated receptor-alpha and proliferator-activated receptor-gamma coactivator 1-alpha activation [62]. Zhang et al. reported that atorvastatin significantly inhibited NAFLD progression via AMPK signaling pathway promotion in the golden hamster model of fat diet-NAFLD. In addition, they showed that AMPK inhibition by compound C counteracted the inhibitory effect of atorvastatin on fat accumulation in the HepG2 cell line [63]. In another study, Kim and colleagues who evaluated the effects of fluvastatin on cholesterol hemostasis regulation in the HepG2 cell line, demonstrated that fluvastatin induced AMPK and LKB phosphorylation and activation as a result of Sirtuin 6 activation with downstream phosphorylated sterol regulatory element binding protein‐1 that prevented lipid accumulation in hepatocytes [64].
3.4 Neuro-Protective Effects
Researchers have shown that a combination of statins (atorvastatin, fluvastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin) may prevent neuroinflammation-mediated neurodegeneration. The mechanism may be through statin downregulated autophagy to provide neuroprotective effects [65].
Amyloid β (Aβ) deposition leads to Tau protein hyper-phosphorylation and neuronal death. It can also cause neuronal insulin resistance, Akt downstream blockage, and glycogen synthase kinase-3β activation, which further results in neuron apoptosis. Statins could alleviate insulin resistance, upregulate Akt, and inhibit glycogen synthase kinase-3β-induced Tau hyper-phosphorylation and impede Aβ deposition-induced neuron apoptosis [66]. Kornelius et al., who evaluated the neuroprotective effects of mevastatin against Aβ deposition-induced neurotoxicity in SK-N-Mc human neuroblastoma cells, reported that this protection was associated with AMPK activation that may increase insulin sensitivity. They demonstrated that mevastatin reduced insulin resistance and Aβ deposition-induced neurotoxicity as a result of AMPK activation [67]. In another study, Singh et al. examined the efficacy of lovastatin and 5-aminoimidazole-4-carboxamide ribonucleotide, an AMPK activator, in experimental autoimmune encephalomyelitis in the murine model of multiple sclerosis, and showed that combination therapy provided more protection for mitochondria/peroxisome and myelin/axons than treatment with either alone. In the spinal cord, inhibition of RhoA by lovastatin and activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleotide increased the expression of transcription factors and regulators (such as proliferator-activated receptor-alpha/beta, Sirtuin 1, nuclear respiratory factor 1, and mitochondrial transcription factor A), which are required for mitochondrial biogenesis and function (such as oxidative phosphorylation and manganese superoxide dismutase) and peroxisome (such as peroxisomal membrane protein 70 and catalase) [68].
3.5 Anti-hyperglycemic Effects
The anti-hyperglycemic effects of statins appear not to be manifested clinically, although there is an increasing concern regarding the diabetogenic effects of statins in patients who received statins to treat dyslipidemia. Casula et al. investigated the association between incident diabetes and statin use in a meta-analysis and suggested an increased risk of new-onset diabetes from 9 to 13% associated with statins [69]. However, clinically diabetes-induced erectile dysfunction is a recognized complication. AMPK plays an important role in autophagy-associated pathways and upregulates autophagy levels in poor nutrient conditions and may be considered as a potential therapeutic target for this diabetes-associated complication [70,71,72,73]. Recent studies have shown that simvastatin lead to cell-cycle arrest via AMPK activation and STAT3/SKP2 axis inhibition [41, 74]. Ding et al. demonstrated that simvastatin could activate the AMPK-SKP2-CARM1 pathway, upregulate protective autophagy, and inhibit corpus cavernosum fibrosis and improved erectile function in diabetes-induced erectile dysfunction rats [75].
3.6 Anti-tubercular Effects
Statins exert anti-tubercular effects through different pathways, and in the case of AMPK-associated effects, Bruiners et al. evaluated the effects of statins on the in vitro model of mycobacterial infection in human macrophages and showed that simvastatin inhibits cholesterol biosynthesis and influences the AMPK-mechanistic target of rapamycin complex-1-transcription factor EB-autophagy axis that lead to autophagy induction and anti-mycobacterial effects. It is noteworthy that the addition of exogenous cholesterol to the cells reversed these effects [76].
4 Conclusions
This review has shown that statins have pleiotropic effects in addition to their lipid-lowering effects through an AMPK-related mechanism that may have therapeutic potential as a molecular target. Activation of the AMPK pathway by statins could exert anti-tumor, cardio-protective, neuroprotective, hepato-protective, and anti-hyperglycemic effects, and indicates further research on statin regulation of AMPK signaling needs to be undertaken to develop new treatment modalities. However, whilst these studies have shown the potential therapeutic utility of AMPK regulation by statins, more work needs to be undertaken to elucidate the specific signaling pathways to understand their exact mechanisms for their use in clinical practice.
References
Wu S, Zou M-H. AMPK, mitochondrial function, and cardiovascular disease. Int J Mol Sci. 2020;21(14):4987.
Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr. 2011;93(4):891S-S896.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Yan Y, Zhou XE, Xu HE, Melcher K. Structure and physiological regulation of AMPK. Int J Mol Sci. 2018;19(11):3534.
Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. 2016;283(16):2987–3001.
Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449(7161):496–500.
Bobba A, Casalino E, Amadoro G, Petragallo V, Atlante A. AMPK is activated early in cerebellar granule cells undergoing apoptosis and influences VADC1 phosphorylation status and activity. Apoptosis. 2017;22(9):1069–78.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26.
O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–55.
Bodmer D, Levano-Huaman S. Sesn2/AMPK/mTOR signaling mediates balance between survival and apoptosis in sensory hair cells under stress. Cell Death Dis. 2017;8(10):e3068.
Liu X, Hu X, Kuang Y, Yan P, Li L, Li C, et al. BCLB, methylated in hepatocellular carcinoma, is a starvation stress sensor that induces apoptosis and autophagy through the AMPK-mTOR signaling cascade. Cancer Lett. 2017;395:63–71.
Björklund MA, Vaahtomeri K, Peltonen K, Viollet B, Mäkelä TP, Band AM, et al. Non-CDK-bound p27 (p27NCDK) is a marker for cell stress and is regulated through the Akt/PKB and AMPK-kinase pathways. Exp Cell Res. 2010;316(5):762–74.
Liang J, Shao SH, Xu Z-X, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1–AMPK pathway regulates p27 kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9(2):218–24.
Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295(1):E29-37.
Kim T, Davis J, Zhang AJ, He X, Mathews ST. Curcumin activates AMPK and suppresses gluconeogenic gene expression in hepatoma cells. Biochem Biophys Res Commun. 2009;388(2):377–82.
Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu C-L, et al. mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci USA. 2009;106(27):11137–42.
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42.
Greenhill C. Obesity: consequences of AMPK activation. Nat Rev Endocrinol. 2016;12(7):372.
Jalili-Nik M, Soltani A, Moussavi S, Ghayour-Mobarhan M, Ferns GA, Hassanian SM, et al. Current status and future prospective of curcumin as a potential therapeutic agent in the treatment of colorectal cancer. J Cell Physiol. 2018;233(9):6337–45.
Umezawa S, Higurashi T, Nakajima A. AMPK: therapeutic target for diabetes and cancer prevention. Curr Pharm Design. 2017;23(25):3629–44.
Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjøbsted R, Jørgensen NO, et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017;25(5):1147-59.e10.
Myers RW, Guan H-P, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357(6350):507–11.
Steneberg P, Lindahl E, Dahl U, Lidh E, Straseviciene J, Backlund F, et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight. 2018;3(12):e99114.
Dehnavi S, Sohrabi N, Sadeghi M, Lansberg P, Banach M, Al-Rasadi K, et al. Statins and autoimmunity: state-of-the-art. Pharmacol Ther. 2020;214:107614.
Hashemi M, Hoshyar R, Ande SR, Chen QM, Solomon C, Zuse A, et al. Mevalonate cascade and its regulation in cholesterol metabolism in different tissues in health and disease. Curr Mol Pharmacol. 2017;10(1):13–26.
Reiner Ž, Hatamipour M, Banach M, Pirro M, Al-Rasadi K, Jamialahmadi T, Radenkovic D, Montecucco F, Sahebkar A, et al. Statins and the Covid-19 main protease: in silico evidence on direct interaction. Arch Med Sci. 2020;16(2):490–6.
Tournadre A. Statins, myalgia, and rhabdomyolysis. Joint Bone Spine. 2020;87(1):37–42.
Ciurleo R, Bramanti P, Marino S. Role of statins in the treatment of multiple sclerosis. Pharmacol Res. 2014;87:133–43.
Chruściel P, Sahebkar A, Rembek-Wieliczko M, Serban M-C, Ursoniu S, Mikhailidis DP, et al. Impact of statin therapy on plasma adiponectin concentrations: a systematic review and meta-analysis of 43 randomized controlled trial arms. Atherosclerosis. 2016;253:194–208.
Parizadeh SM, Azarpazhooh MR, Moohebati M, Nematy M, Ghayour-Mobarhan M, Tavallaie S, et al. Simvastatin therapy reduces prooxidant-antioxidant balance: results of a placebo-controlled cross-over trial. Lipids. 2011;46(4):333–40.
Stein A, Stroobants S, Gieselmann V, D’hooge R, Matzner U. Anti-inflammatory therapy with simvastatin improves neuroinflammation and CNS function in a mouse model of metachromatic leukodystrophy. Mol Ther. 2015;23(7):1160–8.
Bagheri H, Ghasemi F, Barreto GE, Sathyapalan T, Jamialahmadi T, Sahebkar A. The effects of statins on microglial cells to protect against neurodegenerative disorders: a mechanistic review. BioFactors. 2020;46(3):309–25.
Sahebkar A, Serban C, Ursoniu S, Mikhailidis DP, Undas A, Lip GYH, et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen: systematic review and meta-analysis of randomised placebo-controlled trials. Thromb Haemost. 2016;115(3):520–32.
Bansal D, Undela K, D’Cruz S, Schifano F. Statin use and risk of prostate cancer: a meta-analysis of observational studies. PLoS ONE. 2012;7(10):e46691.
Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J Clin Oncol. 2005;23(34):8606–12.
Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, et al. Breast cancer growth prevention by statins. Cancer Res. 2006;66(17):8707–14.
Chan KK, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res. 2003;9(1):10–9.
Yen C-S, Chen J-C, Chang Y-F, Hsu Y-F, Chiu P-T, Shiue C, et al. Lovastatin causes FaDu hypopharyngeal carcinoma cell death via AMPK-p63-survivin signaling cascade. Sci Rep. 2016;6:25082.
Kamel WA, Sugihara E, Nobusue H, Yamaguchi-Iwai S, Onishi N, Maki K, et al. Simvastatin-induced apoptosis in osteosarcoma cells: a key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity. Mol Cancer Ther. 2017;16(1):182–92.
Wang S-T, Ho HJ, Lin J-T, Shieh J-J, Wu C-Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8(2):e2626.
Mojjarad B, Pazhang Y. Orlistat and rosuvastatin suppressed proliferation of K562 human myelogenous leukemia cell line through AMPK/Akt/c-Myc signaling. 2020.
Misirkic M, Janjetovic K, Vucicevic L, Tovilovic G, Ristic B, Vilimanovich U, et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol Res. 2012;65(1):111–9.
Okubo K, Isono M, Miyai K, Asano T, Sato A. Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells. Cancer Sci. 2020;111(1):112–26.
Okubo K, Miyai K, Kato K, Asano T, Sato A. Simvastatin-romidepsin combination kills bladder cancer cells synergistically. 2020.
Ma L, Niknejad N, Gorn-Hondermann I, Dayekh K, Dimitroulakos J. Lovastatin induces multiple stress pathways including LKB1/AMPK activation that regulate its cytotoxic effects in squamous cell carcinoma cells. PLoS ONE. 2012;7(9):e46055.
Huang SW, Chyuan IT, Shiue C, Yu MC, Hsu YF, Hsu MJ. Lovastatin-mediated MCF-7 cancer cell death involves LKB1-AMPK-p38MAPK-p53-survivin signalling cascade. J Cell Mol Med. 2020;24(2):1822–36.
Wang JC, Li XX, Sun X, Li GY, Sun JL, Ye YP, et al. Activation of AMPK by simvastatin inhibited breast tumor angiogenesis via impeding HIF-1α-induced pro-angiogenic factor. Cancer Sci. 2018;109(5):1627–37.
Link W, Fernandez-Marcos PJ. FOXO transcription factors at the interface of metabolism and cancer. Int J Cancer. 2017;141(12):2379–91.
Lee N, Tilija Pun N, Jang WJ, Bae JW, Jeong CH. Pitavastatin induces apoptosis in oral squamous cell carcinoma through activation of FOXO3a. J Cell Mol Med. 2020;24(12):7055–66.
Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE. 2014;9(5):e98207.
Shrestha A, Nepal S, Kim MJ, Chang JH, Kim SH, Jeong GS, et al. Critical role of AMPK/FOXO3A axis in globular adiponectin-induced cell cycle arrest and apoptosis in cancer cells. J Cell Physiol. 2016;231(2):357–69.
Sun W, Lee T-S, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114(24):2655–62.
Izumi Y, Shiota M, Kusakabe H, Hikita Y, Nakao T, Nakamura Y, et al. Pravastatin accelerates ischemia-induced angiogenesis through AMP-activated protein kinase. Hypertens Res. 2009;32(8):675–9.
Rossoni LV, Wareing M, Wenceslau CF, Al-Abri M, Cobb C, Austin C. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci. 2011;121(10):449–58.
Yu B, Liu D, Zhang H, Xie D, Nie W, Shi K, et al. Anti-hypertrophy effect of atorvastatin on myocardium depends on AMPK activation-induced miR-143-3p suppression via FOXO1. Biomed Pharmacother. 2018;106:1390–5.
Hermida N, Markl A, Hamelet J, Van Assche T, Vanderper A, Herijgers P, et al. HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP-activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovasc Res. 2013;99(1):44–54.
Choi SY, Park JS, Roh MS, Kim C-R, Kim MH, Serebruany V. Inhibition of angiotensin II-induced cardiac fibrosis by atorvastatin in adiponectin knockout mice. Lipids. 2017;52(5):415–22.
Mendieta G, Ben-Aicha S, Casani L, Badimon L, Sabate M, Vilahur G. Molecular pathways involved in the cardioprotective effects of intravenous statin administration during ischemia. Basic Res Cardiol. 2020;115(1):2.
Li Q, Dong Q-T, Yang Y-J, Tian X-Q, Jin C, Huang P-S, et al. AMPK-mediated cardioprotection of atorvastatin relates to the reduction of apoptosis and activation of autophagy in infarcted rat hearts. Am J Transl Res. 2016;8(10):4160.
Athyros VG, Boutari C, Stavropoulos K, Anagnostis P, Imprialos KP, Doumas M, et al. Statins: an under-appreciated asset for the prevention and the treatment of NAFLD or NASH and the related cardiovascular risk. Curr Vasc Pharmacol. 2018;16(3):246–53.
Lee WJ, Kim M, Park H-S, Kim HS, Jeon MJ, Oh KS, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem Biophys Res Commun. 2006;340(1):291–5.
Zhang B, Zhang C, Zhang X, Li N, Dong Z, Sun G, et al. Atorvastatin promotes AMPK signaling to protect against high fat diet-induced non-alcoholic fatty liver in golden hamsters. Exp Ther Med. 2020;19(3):2133–42.
Kim J-H, Lee JM, Kim J-H, Kim KR. Fluvastatin activates sirtuin 6 to regulate sterol regulatory element-binding proteins and AMP-activated protein kinase in HepG2 cells. Biochem Biophys Res Commun. 2018;503(3):1415–21.
McFarland A, Davey A, McDermott C, Grant G, Lewohl J, Anoopkumar-Dukie S. Differences in statin associated neuroprotection corresponds with either decreased production of IL-1β or TNF-α in an in vitro model of neuroinflammation-induced neurodegeneration. Toxicol Appl Pharmacol. 2018;344:56–73.
Li H-H, Lin C-L, Huang C-N. Neuroprotective effects of statins against amyloid β-induced neurotoxicity. Neural Regen Res. 2018;13(2):198–206.
Kornelius E, Li H-H, Peng C-H, Hsiao H-W, Yang Y-S, Huang C-N, et al. Mevastatin promotes neuronal survival against Aβ-induced neurotoxicity through AMPK activation. Metab Brain Dis. 2017;32(6):1999–2007.
Singh I, Samuvel DJ, Choi S, Saxena N, Singh AK, Won J. Combination therapy of lovastatin and AMP-activated protein kinase activator improves mitochondrial and peroxisomal functions and clinical disease in experimental autoimmune encephalomyelitis model. Immunology. 2018;154(3):434–51.
Casula M, Mozzanica F, Scotti L, Tragni E, Pirillo A, Corrao G, et al. Statin use and risk of new-onset diabetes: a meta-analysis of observational studies. Nutr Metab Cardiovasc Dis. 2017;27(5):396–406.
Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445(1):11–27.
Russo GL, Russo M, Ungaro P. AMP-activated protein kinase: a target for old drugs against diabetes and cancer. Biochem Pharmacol. 2013;86(3):339–50.
Li M-y, Zhu X-l, Zhao B-x, Shi L, Wang W, Hu W, et al. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS–AMPK–mTOR axis. Cell Death Dis. 2019;10(7):1–14.
Shin H-JR, Kim H, Oh S, Lee J-G, Kee M, Ko H-J, et al. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534(7608):553–7.
Son SM, Kang S, Choi H, Mook-Jung I. Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol Neurodegener. 2015;10(1):56.
Ding F, Shan C, Li H, Zhang Y, Guo C, Zhou Z, et al. Simvastatin alleviated diabetes mellitus-induced erectile dysfunction in rats by enhancing AMPK pathway-induced autophagy. Andrology. 2020;8(3):780–92.
Bruiners N, Dutta NK, Guerrini V, Salamon H, Yamaguchi KD, Karakousis PC, et al. The anti-tubercular activity of simvastatin is mediated by cholesterol-dependent regulation of autophagy via the AMPK-mTORC1-TFEB axis. J Lipid Res. 2020;61(12):1617–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflicts of interest
Sajad Dehnavi, Amirhossein Kiani, Mahvash Sadeghi, Ali Farhadi Biregani, Maciej Banach, Stephen L. Atkin, Tannaz Jamialahmadi, and Amirhossein Sahebkar have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
All authors were involved in the conception, design, drafting, revision, and final approval of this review.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Dehnavi, S., Kiani, A., Sadeghi, M. et al. Targeting AMPK by Statins: A Potential Therapeutic Approach. Drugs 81, 923–933 (2021). https://doi.org/10.1007/s40265-021-01510-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01510-4