
Abstract
Fabry disease (FD) is a rare X-linked lysosomal storage disease based on a deficiency of α-galactosidase A (AGAL) caused by mutations in the α-galactosidase A gene (GLA). The lysosomal accumulation of glycosphingolipids, especially globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3, deacylated form), leads to a multisystemic disease with progressive renal failure, cardiomyopathy with potentially malignant cardiac arrhythmias, and strokes, which considerably limits the life expectancy of affected patients. Diagnostic confirmation in male patients is based on the detection of AGAL deficiency in blood leukocytes, whereas in women, due to the potentially high residual enzymatic activity, molecular genetic detection of a causal mutation is required. Current treatment options for FD include recombinant enzyme replacement therapy (ERT) with intravenous agalsidase-alfa (0.2 mg/kg body weight) or agalsidase-beta (1 mg/kg body weight) every 2 weeks and oral chaperone therapy with migalastat (123 mg every other day), which selectively and reversibly binds to the active site of AGAL, thereby correcting the misfolding of the enzyme and allowing it to traffic to the lysosome. These therapies enable cellular Gb3 clearance and improve the burden of disease. However, in about 40% of all ERT-treated men, ERT can lead to infusion-associated reactions and the formation of neutralizing antidrug antibodies, which reduces the efficacy of therapy. In chaperone therapy, there are carriers of amenable mutations that show limited clinical success. This article provides a brief overview of the clinical picture in FD patients, diagnostic confirmation, and interdisciplinary clinical management of FD. The focus is on current and future therapeutic options.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Diagnosis of Fabry disease in males includes the detection of decreased α-galactosidase A activity and in females the determination of a disease-causing mutation. |
Current treatment options of enzyme replacement therapy and chaperone therapy can improve disease burden and quality of life. |
New therapy options include modified enzyme replacement therapy, substrate reduction therapy, and gene therapy. |
1 Introduction
Fabry disease (FD) is an X-linked lysosomal storage disorder based on a deficiency in α-galactosidase A (GLA/AGAL) activity causing a progressive, life-threatening multisystemic disease due to intracellular accumulation of glycosphingolipids (mainly globotriaosylceramide [Gb3)] [1]. The aim of this article is to provide a brief overview of the clinical picture of patients with FD, the diagnostic confirmation, and interdisciplinary clinical management, with an additional special focus on current and future therapeutic options.
1.1 Overview of Epidemiology and Pathogenesis
FD affects men and women with an incidence of 1:40,000–1:117,000, although newborn screenings have pointed to higher incidences of 1:3100–1:1250 in males [2, 3]. Disease-causing mutations are detected in the GLA gene coding for AGAL, which is located on the long arm of the X chromosome (band q22), has a length of 12 Kb, and consists of 7 exons and 6 introns. Currently more than 1000 different GLA mutations are known, while most patients carry "private" (family-specific) mutations. Due to X-linked inheritance, family history is important and genetic counseling is recommended for every family with member who has FD.
Since almost all body cells accumulate Gb3, numerous organs/organ systems such as kidneys, heart, blood vessels, and the central and peripheral nervous system are affected, and patients show varying degrees of progressive functional deficits.
1.2 Clinical Picture and Diagnostics for Diagnosis Confirmation
FD patients suffer from a multisystemic disease with progressive (dialysis-dependent) renal insufficiency, cardiomyopathy with sometimes life-threatening cardiac arrhythmias, recurrent strokes, gastrointestinal pain, and neuropathic pain of the extremities, which can lead to Fabry crises (Table 1 and Fig. 1) [1, 4]. Due to the random inactivation of one of the two X chromosomes in each cell during early embryogenesis, the variability of the clinical picture is greater in women than in men [5].

Fabry disease is a multisystemic disease. TIA transient ischemic attack, WML white matter lesion
FD can be suspected in the presence of a the family history and/or from the evidence of the typical manifestations. In males, the determination of AGAL activity in blood leukocytes or from dried blood spots is the method of choice for confirmation a diagnosis. A pathologically low AGAL activity indicates the presence of FD. In females, molecular genetic testing demonstrating a disease-causing mutation in the GLA gene is necessary to confirm the diagnosis, since women with FD often present with AGAL activities within the reference range. In men with pathologically decreased enzymatic AGAL activity, molecular genetic testing should be used to detect the underlying mutation in order to select an appropriate FD-specific therapy (see below). As a marker of disease burden, a pathologically elevated globotriaosylsphingosine (lyso-Gb3) in plasma or urine can contribute to improved diagnosis and subsequent monitoring. In unclear diagnostic cases (disputed mutation, ambiguous AGAL activity and lyso-Gb3 values, comorbidities) organ biopsies may be helpful. If biopsies are performed, electron microscopic multilamellar myelin bodies (so-called "zebra bodies" or "paper roll phenomenon") can be detected, which are pathognomonic for FD, but require special sample preparations. Prenatal diagnosis can be performed by measuring AGAL activity in chorionic villi or cultured amniotic cells and, in the case of a mutation known in the family, by molecular genetic methods (see Table 2).
2 Therapy Goals and Treatment Recommendations
Once the diagnosis is confirmed, patients should be referred to an interdisciplinary Fabry center for initial examination, therapy planning, and therapy initiation. The following therapeutic goals should be aimed for in the context of multimodal care [6, 7]: (1) Reduction of complaints (especially pain reduction), (2) delaying/preventing the progression of organ manifestations (especially in the kidney, heart, and central nervous system), (3) improvement of quality of life, and (4) normalization of life expectancy.
Male FD patients generally have a higher risk of worse disease progression and outcome compared to females. Consequently, the international recommendations for initiating FD-specific treatment in adult male and female patients with classic or late-onset mutations reflect a personalized treatment approach that takes into account the natural history of the specific disease phenotype [6]. In an adult male with a classic Fabry mutation, FD-specific treatment should be considered regardless of the patient’s symptomatology (symptomatic or asymptomatic). Treatment decisions may be influenced by advanced patient age and severe comorbidity. In a female with a classic Fabry mutation, initiation of FD-specific treatment is warranted if she is symptomatic and symptoms/manifestations are indicative of organ involvement. In an asymptomatic woman with a classic Fabry mutation, initiation of FD-specific treatment should be considered if there is laboratory, histologic, or imaging evidence of renal, cardiac, or central nervous system damage. In males and females with a later-onset Fabry mutation (such as p.N215S), FD-specific treatment should be considered when there is laboratory, histologic, or imaging evidence of renal, cardiac, or central nervous system damage, even in the absence of typical Fabry symptoms. The abnormalities should be due to FD; this may require histologic evaluation or biochemical evidence of Gb3 accumulation. In the absence of detectable FD-related tissue pathology or clinical symptoms, FD-specific treatment may not be appropriate, especially in heterozygous female patients. These patients should be monitored regularly by a multidisciplinary care team. Patients with "benign," non-disease-causing GLA variants/polymorphisms should not be treated with any FD-specific therapy.
Therapeutic goals are not always achievable in clinical practice. Consensus recommendations [6, 7] provide guidance by establishing goals between clinicians and patients and considering treatment-related challenges to enable individualized treatment, optimize patient care, and improve quality of life. The patient-management algorithm addresses the need for early disease-specific therapy to delay or slow disease progression, as well as nonspecific adjunctive therapies that prevent or treat organ damage and stabilize quality of life [6].
3 Fabry Disease-Specific Current Treatment
Currently approved medications including enzyme replacement therapy (ERT) as well as chaperone therapy aim to reduce intracellular Gb3 accumulation by either replacing the deficient endogenous AGAL or correcting the misfolded endogenous AGAL, with subsequent improvement of trafficking and increased enzymatic activity within the lysosomes. Future therapies such as next-generation ERT or gene therapy aim to diminish present Gb3 content whereas substrate reduction therapies aim to prevent the de novo synthesis of Gb3.
3.1 Enzyme Replacement Therapy (ERT)
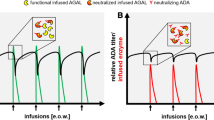
First attempts with ERT were carried out in 1973 when Brady and colleagues successfully administered purified human placental AGAL to two FD patients [8]. Both patients tolerated the infusion well and AGAL activity increased about 68% compared to pre-infusion levels [8]. Over the course of time, AGAL obtained from spleen as well as human plasma was administered to two brothers with FD. The administration of plasma-derived AGAL resulted in a Gb3 clearance of up to 70% [9]. Due to the complex purification and insufficient availability of human AGAL for infusion, clinical research progress was limited. However, improved biotechnology processes enabled the production of high amounts of recombinant AGAL protein in cell culture required for appropriate clinical studies. Since 2001, ERT is available as a causal treatment option for FD [10, 11]. The enzyme is administered every 2 weeks as an intravenous infusion (Figs. 2 and 3a). Two preparations in different dosages are currently approved for life-long therapy, including agalsidase-alfa (Replagal, Takeda) produced in a human cell line (human fibrosarcoma cells HT-1080), with an approved dosage of 0.2 mg/kg body weight leading to an infusion duration of approximately 40 min and agalsidase-beta (Fabrazyme, Sanofi Genzyme), which is produced in Chinese hamster ovary (CHO) cells with a recommended dose of 1.0 mg/kg body weight and an infusion duration of approximately 240 min. Depending on individual tolerance, the infusion duration of agalsidase-beta may be progressively reduced to 90 min.

Overview of the ceramide pathway and therapeutic targets of Fabry disease-specific approved and future treatments. AGAL/GLA α-galactosidase A, loss of function results in Fabry disease, A4GALT α-1,4-galactosyltransferase, B3GALNT1 β-1,3-N-acetylgalactosaminyltransferase 1, B4GALT6 β-1,4-galactosyltransferase 6, GBA glucosylceramidase beta, loss of function results in Gaucher disease, GCS glucosylceramide synthase, GLB1 galactosidase-beta 1, loss of function results in GM1 gangliosidosis, HEXA hexosaminidase-subunit alpha, loss of function results in GM2 gangliosidosis

Schematic overview of current and future treatment approaches for Fabry disease. a Enzyme replacement therapy. b Migalastat-based chaperone treatment. c Lucerastat- and Venglustat-based substrate reduction therapy. d Lentiviral-based gene correction in haematopoietic stem cells. e Adenoviral-based transduction of liver cells. rhAGAL recombinant human α-galactosidase A, Gb3 globotriaosylceramide, GCS glucosylceramide synthase, ER endoplasmic reticulum, gDNA genomic DNA, mRNA messenger RNA
ERT shows good efficacy with stabilization or even improvement of disease load (reviewed in Lenders and Brand [12]) including improvement of quality of life and reducing life-threatening events [13,14,15]. Unfortunately, females with FD are often still untreated despite organ manifestations [16], although an early initiation of therapy is particularly important for the (long-term) success of the therapy in males as well as females [17,18,19,20,21].
Depending on the patients and their manifestations, the following therapeutic effects can be achieved with ERT: Stabilization of kidney function or delay of progression to terminal kidney failure, stabilization of the thickness of the heart wall and function or reduction of left ventricular hypertrophy, relief of neuropathic pain and improvement of gastrointestinal complaints, and improvement in the ability to sweat [12, 20, 22, 23]. With respect to Fabry nephropathy, a dose-dependent reduction of Gb3 inclusions in podocytes was demonstrated in children, suggesting a dose-dependent ERT efficacy with regard to the stabilization of renal function [24,25,26,27,28].
Due to the origin (production) and application of the enzymes, ERT also has limitations resulting in challenges in everyday clinical practice. During infusion, allergic infusion reactions may occur, including cephalia, paresthesia, drop in blood pressure, fever, chills, nausea, and fatigue (especially after infusions). These symptoms can usually be alleviated by reducing the infusion rate and administration of non-steroidal anti-inflammatory drugs, antihistamines, and/or glucocorticoids. More critically, approximately 40% of all males with FD undergoing ERT produce antibodies against the infused enzyme (especially males without endogenous AGAL activity), leading to an inhibition of the infused enzyme, resulting in a worse disease course and poorer outcomes [29, 30]. These neutralizing antidrug antibodies (ADAs) already inhibit the infused enzyme during infusion [31], and impact endothelial enzyme uptake as well as intracellular enzymatic activity [32]. Depending on the titers, higher enzyme doses can compensate for ADAs, but approved dosages limit this approach [31, 33]. So far, no specific immunosuppression approaches in clinical routine have been reported, but in a collective of transplanted male FD patients a suppression of the antibody-mediated ERT inhibition was demonstrated due to immunosuppressive therapy [34]. In this respect, higher immunosuppressive doses were associated with lower antibody titers and a reduced enzyme inhibition [34]. Future studies are needed, aimed at developing ADA-specific immunosuppressive therapy protocols leading to improved disease course in patients with ADAs.
3.2 Chaperone Therapy
Missense mutations within the GLA gene often result in an unstable and misfolded protein, leading to reduced intracellular AGAL activities. Misfolded proteins will not pass the protein quality-control mechanism within the endoplasmic reticulum (ER), resulting in premature degradation before reaching the lysosomes (Fig. 3b) [35, 36]. To restore folding and stability of the protein, pharmacological chaperones can be used, binding reversibly to the active center of the protein [37].
The small molecule 1-deoxygalactonojirimycin (DGJ) is an iminosugar, initially identified as a competitive inhibitor of the AGAL enzyme. At sub-inhibitory concentrations (extracellular: 20–100 µM), the binding to the enzyme’s catalytic center (wild-type and amenable mutations) promotes improved protein folding in the ER as well as accelerated maturation and trafficking to the lysosome [38, 39]. This results in an increase of enzymatic AGAL activity in healthy control cells and (more importantly) in Fabry patient-derived lymphoblasts carrying the first described responsive mutations p.R301Q and p.Q279E [38, 39].
Since May 2016, the pharmacological chaperone migalastat (Galafold; 123 mg hard capsules, every other day; Amicus Therapeutics) is approved as the first oral therapy for FD [40]. For numerous AGAL protein mutants, the addition of migalastat under cell culture conditions (HEK293T cells) leads to a significantly increased enzymatic activity [41]. According to current knowledge, this is based on a reversible binding of migalastat to the active center of the AGAL and, thus, as a consequence of the resulting stabilization, facilitates improved transport to the lysosomes. After uptake of the chaperone enzyme complex into the lysosomes, the small molecule dissociates due to the acidic pH and the lysosomal AGAL concentration increases. As a result of more effective transport, a higher enzyme activity can be achieved in the lysosomes, leading to an improved catabolism of Gb3 (Fig. 2). AGAL mutants showing an increase in enzyme activity after migalastat administration are described as "amenable." The small molecule compound is available for the long-term treatment of adults and adolescents aged 16 years and older with confirmed FD diagnosis and an amenable mutation. The website www.galafoldamenabilitytable.com allows specialists to find out which mutation responds to migalastat in vitro (status as at May 2020: 1384 amenable mutations).
Two phase III studies for treatment with migalastat were conducted. The FACETS study is a placebo-controlled study including treatment-naïve patients (N = 50; 22 placebo vs. 28 migalastat) [42]. The ATTRACT study analyzed the efficacy and safety of migalastat compared to ERT (agalsidase-beta, agalsidase-alfa) in 57 patients (36 migalastat vs. 21 ERT males and females) [43]. The data were promising, since renal function remained stable under a maximum 18-month treatment period with migalastat, a significant reduction of the left ventricular mass index was observed, and plasma lyso-Gb3, as a marker of disease burden, remained low and stable when switching from ERT to migalastat (ATTRACT [43]). An analysis of clinical events related to kidney, heart, and cerebrovascular events and death revealed a frequency of events observed in the migalastat group of 29% and within the ERT group of 44% (p = 0.36) [43]. Overall, migalastat therapy showed good tolerability, with side effects (including ≥ 1/10 headache) not occurring more frequently than with ERT (ATTRACT study) [43]. Although study populations were relatively small and the study duration was limited, the data are of clinical relevance since migalastat does not appear to be inferior to the previous gold standard of ERT. Both phase III studies reported a significant reduction of LVMi (− 7.7 g/m2 and − 6.6 g/m2, respectively) after 24 and 18 months of migalastat-treatment [42, 43]. These data were subsequently confirmed by appropriate “real-world” studies including two single-center studies [44, 45] and one multicenter study [46]. Interestingly, 30 months of data from the open-label extension of the randomized, phase III ATTRACT study demonstrated only a significant reduction of left ventricular mass index (− 10.0 g/m2) in those patients with left ventricular hypertrophy at baseline [47].
Oral therapy with migalastat seems to be a convenient alternative to intravenous ERT for patients with amenable mutations. However, patients with different amenable mutations will show a different clinical response, partly due to the individual increase in AGAL activity, severity of the disease (at treatment initiation), possible comorbidities, and therapy adherence. Recent studies demonstrated that in vitro amenability does not always necessarily correspond to clinical amenability [44, 46, 48]. Some amenable AGAL mutations with low residual activity within the good laboratory practice (GLP)-HEK assay failed to show a biochemical response in appropriate migalastat-treated patients carrying these mutations [46, 48]. For critical mutations in patients lacking clinical amenability, future in-depth analyses including the establishment of CRISPR/Cas9-knockout HEK cells (without endogenous AGAL activity) and/or the investigation of patient-specific cell models will allow a better understanding of the different clinical effectiveness of chaperone therapy of certain mutations [46, 48].
4 Future Treatment
4.1 Next-Generation ERTs
Pegunigalsidase alfa (PRX-102, Protalix Biotherapeutics / Chiesi) is a novel PEGylated (PEG, polyethylene glycol) and covalently cross-linked form of AGAL, developed as ERT for FD and produced in tobacco cells (tobacco plant cell-based ProCellEx System) (Fig. 3a). Pegunigalsidase seems to be equivalent in functionality to the currently available ERTs, with prolonged in vitro stability and a tenfold increase in half-life in male Fabry mice (581 min) compared to approved drugs [49]. A 1-year phase I/II clinical trial (dose-finding study) revealed increased efficacy due to prolonged plasma half-life (80 h) and reduced immunogenicity [50]. The improved half-life (80 h) due to PEGylation, which stabilizes the AGAL homodimer, might allow the infusion interval (monthly intravenous administration) to be extended [51]. Three phase III studies are currently underway (BALANCE [NCT02795676], BRIDGE, and BRIGHT [NCT03180840]). BALANCE is a multicenter, randomized, actively controlled, direct comparison study (head-to-head) to evaluate the safety and efficacy of pegunigalsidase alfa (1 mg/kg) compared to the approved dose of agalsidase beta (1 mg/kg) with a special focus on renal function. BRIDGE is an open switch-over study to assess the safety and efficacy of the switch from an approved dose of agalsidase alfa (0.2 mg/kg) to pegunigalsidase alfa (1 mg/kg). BRIGHT is an open switch-over study to evaluate the safety and efficacy of the switch from an approved dose of agalsidase alfa (0.2 mg/kg) or agalsidase beta (1 mg/kg) every 2 weeks to a higher dose of pegunigalsidase alfa (2.0 mg/kg) every 4 weeks.
Moss-aGal (ELEVA, former Greenovation) is a recombinant human AGAL expressed in Physcomitrella patens, which is a genetically modified moss (Fig. 3a). Preclinical studies suggest an improved uptake of the enzyme by mannose receptors instead of mannose-6-phosphate receptors into cells [52]. A phase I study showed good safety and tolerability of Moss-aGal in six women after a single intravenous dose of 0.2 mg/kg [53]. Phase II and III studies are in preparation.
4.2 Substrate Reduction Therapy
While ERT aims to replace the missing or defective AGAL through the infusion of a genetically engineered enzyme and migalastat increases endogenous enzyme activities of AGAL due to amenable mutations, the aim of a substrate reduction therapy (SRT) is the reduction of the substrate and, therefore, the subsequent inhibition of Gb3 accumulation in the cells (Figs. 2 and 3c). Lucerastat (Idorsia) is a low molecular weight iminosugar inhibiting the upstream-located glucosylceramide synthase (GCS) and, thus, the biosynthesis of glycosphingolipids including downstream Gb3, indicating a potential new oral treatment alternative [54, 55]. The first in vitro studies demonstrated a successful reduction of Gb3 and an amelioration of abnormal cellular membranes in FD patient-derived fibroblasts [54,55,56]. A pivotal phase III study (MODIFY [NCT03425539]) was initiated in 2018 and is still recruiting patients (last updated 24 September 2020).
In addition, Venglustat as a GCS inhibitor demonstrated a favorable safety and tolerability profile (Sanofi Genzyme; NCT01674036; NCT01710826) [57].
4.3 Gene Therapy
Gene therapy is based on the introduction of DNA carrying the genetic code for the AGAL protein into patients’ cells. Using this approach, the causal chain "mutated gene to defective enzyme with defective function to cellular Gb3 deposition in FD patients" would be reached one step earlier compared to ERT (Figs. 2 and 3). Currently, several clinical studies are evaluating the safety of gene therapy for FD, which differ in their therapeutic approaches. The first interventional, multicenter, multinational, open-label study (NCT03454893, NCT02800070, AVR-RD-01, AvroBio) is based on the lentiviral ex vivo transduction of hematopoietic stem cells (Fig. 3d). This approach aims to use transfected hematopoietic stem cell-derived cells as the production platform for functional AGAL production, which will be secreted to the plasma and subsequently internalized by AGAL-deficient cells.
Two further clinical studies (NCT04046224, NCT04040049) [58, 59] are based on adeno-associated viral (AAV) in vivo transduction of hepatocytes, using these cells as an AGAL-secreting platform instead of hematopoietic stem cell-derived cells (Fig. 3e). STAAR is a multicenter, open-label, dose-finding study for the AAV2/6 vector-based drug ST-920 (NCT04046224, Sangamo). The second clinical study on AAV-based gene therapy is also still in the recruitment phase (NCT04040049; FLT190; Freeline Therapeutics). FLT190 is a gene therapy in the test phase, based on a platform that is also developed for the potential future treatment of hemophilia A and B and Gaucher disease. FLT190 consists of a codon-optimized GLA transgene under the control of a liver-specific promoter. The construct, covered by a synthetic capsid, shows improved transduction of human hepatocytes compared to wild-type AAV serotypes [59]. A third clinical study on AAV-based gene therapy is also in the recruitment phase and uses an attenuated AAV (4D-310; 4D Molecular Therapeutics). Preclinical studies in mice demonstrated that the novel capsid 4D-C102 was especially efficient in transducing human cardiomyocytes. Since myocardial cells are hard to reach with ERT, this approach will be of interest for those patients with FD-specific cardiac manifestations.
5 Concomitant Medication
In addition to causal FD treatment, the need for concomitant medication of affected patients is strongly indicated.
5.1 Pain Management
Neuropathic pain often affects the everyday life of many FD patients, significantly lowering quality of life. However, only limited data on the symptomatic treatment of neuropathic pain in FD is available. According to current knowledge, the combination of ERT/migalastat and symptomatic analgesic drugs leads to the most effective pain relief. Even if established drugs are recommended for the treatment of neuropathic pain, some are not advisable due to additional FD-typical manifestations. This includes tricyclic antidepressants, which negatively influence the cardiac conduction system, which is often affected in FD. The effect of non-steroidal anti-inflammatory drugs on neuropathic pain is minimal, so they should be used with caution, especially due to their nephrotoxicity. Current pain-management strategies are based on clinical experience or follow national and international guidelines for the management of neuropathic pain [60,61,62,63]. Tricyclic antidepressants (cave), serotonin and norepinephrine reuptake inhibitors (e.g., duloxetine, venlafaxine), carbamazepine, gabapentin, and pregabalin are considered as first-line options. Lidocaine, topical capsaicin patches, as well as tramadol are considered second-line options; strong opioids should only be considered third-line options [61]. In this respect, the use of controlled-released opioids is preferred over short-acting opioids. Cannabinoids may also be considered as third-line treatments. Finally, methadone (with both N-methyl-d-aspartate and opioid-receptor effects), anticonvulsants with less evidence of efficacy (e.g., lamotrigine and lacosamide), tapentadol, and botulinum toxin are currently considered fourth-line treatments [64]. However, if patients suffer from neuropathic pain, a neurologist with experience in FD should be consulted.
5.2 Reno-Protective Medication
Management of proteinuria is a key feature in preserving renal function in patients with FD. A consistent antiproteinuric therapy with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) was shown to decrease the progression of kidney disease in FD [19, 65]. Since a high-sodium diet can diminish the effect of ACE inhibitors and ARBs [66] and is associated with an increased risk of progression to end-stage renal disease in patients with proteinuria [67], a low-sodium diet in FD patients with proteinuria is strongly indicated [68].
5.3 Management of Gastrointestinal Symptoms
Gastrointestinal (GI) symptoms, including postprandial cramping pain, diarrhea, nausea, bloating, and vomiting are typical for patients with FD, especially in those with a classical phenotype. Some major GI symptoms in patients with FD can be treated by various symptomatically effective concomitant drugs. Patients with acute diarrhea can be treated with classical antidiarrheal medication such as loperamide. On the other hand, patients suffering from gastroparesis can benefit from treatments with pro-motility agents such as metoclopramide, which increase the contractile force and accelerate intraluminal transit [69]. Patients suffering from upper GI symptoms may benefit from proton pump inhibitors (e.g., omeprazole) or ondansetron if nausea is present [70]. Medication to combat bloating and flatulence may include the administration of simethicone, which eliminates and prevents foam formation [71, 72]. Furthermore, linaclotide, an oligo-peptide agonist of guanylate cyclase 2C, is used to treat irritable bowel syndrome with constipation and chronic constipation of unknown cause [73, 74]. However, most of these medications are not suitable for long-term use due to side effects. Hence, non-drug-management approaches including dietary modifications (including FODMAP-low diets) or oral AGAL substitution are valuable approaches [75].
6 Unmet Needs
In view of the health economic burden (cost of ERT and chaperone therapy per patient approximately €250,000/year) of a generally lifelong therapy, criteria have to be developed that consider on the one hand the therapy initiation and on the other hand the possible therapy discontinuation (hopeless clinical situation in case of FD progress or newly discovered metastatic tumor disease without foreseeable curative success). It remains to be discussed whether chaperone therapy should be favored for obese patients with an amenable mutation, as this is dosed independently of weight, in contrast to ERT, to avoid additional costs. Ethical aspects have to be considered, which complicate the daily clinical routine for every practitioner. Furthermore, the current therapy has limitations, as deaths occur despite therapy if therapy is started late. Further limitations of ERT are: (1) the short half-life, which results in short therapy intervals; (2) limited accessibility to specific tissues (does not cross the blood-brain barrier); (3) the difficult clearance of clinically relevant renal (podocytes) and cardiac cells (cardiomyocytes) compared to the endothelium; and (4) the high immunogenicity. In this respect, clinicians have not yet developed standards for the management of patients who have limited therapeutic success due to their therapy efficacy-limiting antibodies. To avoid these therapeutic limitations pharmaceutical companies are developing other therapies (gene therapy, SRTs, new ERTs).
References
Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–35.
Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40.
Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 2009;30:1397–405.
Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184–92.
Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–54.
Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123:416–27.
Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124:189–203.
Brady RO, Tallman JF, Johnson WG, Gal AE, Leahy WR, Quirk JM, et al. Replacement therapy for inherited enzyme deficiency. N Engl J Med. 1973;289:9–14.
Desnick RJ, Dean KJ, Grabowski GA, Bishop DF, Sweeley CC. Enzyme therapy XVII: Metabolic and immunologic evaluation of alpha- galactosidase A replacement in Fabry disease. Birth Defects Orig Artic Ser. 1980;16:393–413.
Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human a-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16.
Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–9.
Lenders M, Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. J Am Soc Nephrol. 2018;29:2265–78.
Watt T, Burlina AP, Cazzorla C, Schönfeld D, Banikazemi M, Hopkin RJ, et al. Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. Genet Med. 2010;12:703–12.
Hughes DA, Barba Romero MÁ, Hollak CE, Giugliani R, Deegan PB. Response of women with Fabry disease to enzyme replacement therapy: comparison with men, using data from FOS–the Fabry Outcome Survey. Mol Genet Metab. 2011;103:207–14.
Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86.
Lenders M, Hennermann JB, Kurschat C, Rolfs A, Canaan-Kühl S, Sommer C, et al. Multicenter Female Fabry Study (MFFS) - clinical survey on current treatment of females with Fabry disease. Orphanet J Rare Dis. 2016;11:88. https://doi.org/10.1186/s13023-016-0473-4.
Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119:524–9.
Germain DP, Weidemann F, Abiose A, Patel MR, Cizmarik M, Cole JA, et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-β: data from the Fabry Registry. Genet Med. 2013;15:958–65.
Warnock DG, Ortiz A, Mauer M, Linthorst GE, Oliveira JP, Serra AL, et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant. 2012;27:1042–9.
Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353–8.
Germain DP, Arad M, Burlina A, Elliott PM, Falissard B, Feldt-Rasmussen U, et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease - A systematic literature review by a European panel of experts. Mol Genet Metab. 2019;126:224–35.
Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis. 2015;10:125. https://doi.org/10.1186/s13023-015-0338-2.
Schiffmann R, Swift C, Wang X, Blankenship D, Ries M. A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J Inherit Metab Dis. 2015;38:1129–36.
Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–48.
Weidemann F, Krämer J, Duning T, Lenders M, Canaan-Kühl S, Krebs A. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. 2014;25:837–49.
Lenders M, Canaan-Kühl S, Krämer J, Duning T, Reiermann S, Sommer C, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction and switch - 2-year follow-up. J Am Soc Nephrol. 2016;27:952–62.
Skrunes R, Svarstad E, Kampevold Larsen K, Leh S, Tøndel C. Reaccumulation of globotriaosylceramide in podocytes after agalsidase dose reduction in young Fabry patients. Nephrol Dial Transplant. 2017;32:807–13.
Krämer J, Lenders M, Canaan-Kühl S, Nordbeck P, Üçeyler N, Blaschke D, et al. Fabry disease under enzyme replacement therapy - new insights in efficacy of different dosages. Nephrol Dial Transplant. 2018;33:1362–72.
Linthorst GE, Hollak CEM, Donker-Koopman WE, Strijland A, Aerts JMFG. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–95.
Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J Am Soc Nephrol. 2016;27:256–64.
Lenders M, Schmitz B, Brand SM, Foell D, Brand E. Characterization of drug-neutralizing antibodies in patients with Fabry disease during infusion. J Allergy Clin Immunol. 2018;141:2289–92.
Stappers F, Scharnetzki D, Schmitz B, Manikowski D, Brand SM, Grobe K, et al. Neutralising anti-drug antibodies in Fabry disease can inhibit endothelial enzyme uptake and activity. J Inherit Metab Dis. 2020;43:334–47.
Lenders M, Neußer LP, Rudnicki M, Nordbeck P, Canaan-Kühl S, Nowak A, et al. Dose-dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with Fabry disease. J Am Soc Nephrol. 2018;29:2879–89.
Lenders M, Oder D, Nowak A, Canaan-Kühl S, Arash-Kaps L, Drechsler C, et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J Intern Med. 2017;282:241–53.
Romeo G, D’Urso M, Pisacane A, Blum E, De Falco A, Ruffilli A. Residual activity of alpha-galactosidase A in Fabry’s disease. Biochem Genet. 1975;13:615–28.
Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005;19:12–8.
Boyd RE, Lee G, Rybczynski P, Benjamin ER, Khanna R, Wustman BA, et al. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J Med Chem. 2013;56:2705–25.
Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112–5.
Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, et al. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267:4179–86.
McCafferty EH, Scott LJ. Migalastat: A review in Fabry disease. Drugs. 2019;79:543–54.
Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19:430–8.
Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375:545–55.
Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54:288–96.
Müntze J, Gensler D, Maniuc O, Liu D, Cairns T, Oder D, et al. Oral chaperone therapy migalastat for treating Fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther. 2019;105:1224–33.
Riccio E, Zanfardino M, Ferreri L, Santoro C, Cocozza S, Capuano I, et al. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: real-life data. Eur J Hum Genet. 2020;9:1. https://doi.org/10.1038/s41431-020-0677-x (Epub ahead of print. PMID: 32647377).
Lenders M, Nordbeck P, Kurschat C, Karabul N, Kaufeld J, Hennermann JB, et al. Treatment of Fabry disease with migalastat - outcome from a prospective observational multicenter study (FAMOUS). Clin Pharmacol Ther. 2020;108:326–37.
Feldt-Rasmussen U, Hughes D, Sunder-Plassmann G, Shankar S, Nedd K, Olivotto I, et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol Genet Metab. 2020. https://doi.org/10.1016/j.ymgme.2020.07.007 (Epub ahead of print. PMID: 33012654).
Lenders M, Stappers F, Niemietz C, Schmitz B, Boutin M, Ballmaier PJ, et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J Med Genet. 2019;56:548–56.
Kizhner T, Azulay Y, Hainrichson M, Tekoah Y, Arvatz G, Shulman A, et al. Characterization of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Fabry disease. Mol Genet Metab. 2015;114:259–67.
Schiffmann R, Goker-Alpan O, Holida M, Giraldo P, Barisoni L, Colvin RB, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J Inherit Metab Dis. 2019;42:534–44.
Ruderfer I, Shulman A, Kizhner T, Azulay Y, Nataf Y, Tekoah Y, et al. Development and analytical characterization of pegunigalsidase alfa, a chemically cross-linked plant recombinant human α-galactosidase-A for treatment of Fabry disease. Bioconjug Chem. 2018;29:1630–9.
Shen JS, Busch A, Day TS, Meng XL, Yu CI, Dabrowska-Schlepp P, et al. Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J Inherit Metab Dis. 2016;39:293–303.
Hennermann JB, Arash-Kaps L, Fekete G, Schaaf A, Busch A, Frischmuth T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J Inherit Metab Dis. 2019;42:527–33.
Ashe KM, Budman E, Bangari DS, Siegel CS, Nietupski JB, Wang B, et al. Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry disease. Mol Med. 2015;21:389–99.
Guérard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford RWD, et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Pharmacol Ther. 2018;103:703–11.
Brogden G, Shammas H, Maalouf K, Naim SL, Wetzel G, Amiri M, et al. Case study on the pathophysiology of Fabry disease: abnormalities of cellular membranes can be reversed by substrate reduction in vitro. 2017. Biosci Rep. https://doi.org/10.1042/BSR20160402.
Peterschmitt MJ, Crawford NPS, Gaemers SJM, Ji AJ, Sharma J, Pham TT. Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral Venglustat in healthy volunteers. Clin Pharmacol Drug Dev. 2021;10:86–98.
Hughes DA, Goker-Alpan O, Ganesh J, et al. A phase I/II multicenter gene therapy clinical study for Fabry disease. Mol Genet Metab. 2019;129:S77.
Hughes DA, Patel N, Kinch R, et al. First-in-human study of a liver-directed AAV gene therapy (FLT190) in Fabry disease. Mol Genet Metab. 2019;129:S77-78.
Zeltzer LK, Palermo T, Krane E. Ch. 62 Pain management. In: Kliegman R, Stanton B, S. Geme J, Schor N, editors. Nelson textbook of pediatrics, vol. 1. 20th ed. Philadelphia: Elsevier Inc; 2015. p. 430–46.
Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015;14:162–73.
Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17:1113-e88.
Politei JM, Bouhassira D, Germain DP, Goizet C, Guerrero-Sola A, Hilz MJ, et al. Pain in Fabry disease: practical recommendations for diagnosis and treatment. CNS Neurosci Ther. 2016;22:568–76.
Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, et al. Pharmacological management of chronic neuropathic pain: Revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19:328–35.
Warnock DG, Thomas CP, Vujkovac B, Campbell RC, Charrow J, Laney DA, et al. Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy. J Med Genet. 2015;52:860–6.
De’Oliveira JM, Price DA, Fisher ND, Allan DR, McKnight JA, Williams GH, et al. Autonomy of the renin system in type II diabetes mellitus: dietary sodium and renal hemodynamic responses to ACE inhibition. Kidney Int. 1997;52:771–7.
Vegter S, Perna A, Postma MJ, Navis G, Remuzzi G, Ruggenenti P. Sodium intake, ACE inhibition, and progression to ESRD. J Am Soc Nephrol. 2012;23:165–73.
Felis A, Whitlow M, Kraus A, Warnock DG, Wallace E. Current and investigational therapeutics for Fabry disease. Kidney Int Rep. 2019;5:407–13.
Argoff CE, Barton NW, Brady RO, Ziessman HA. Gastrointestinal symptoms and delayed gastric emptying in Fabry’s disease: response to metoclopramide. Nucl Med Commun. 1998;19:887–91.
Hilz MJ, Arbustini E, Dagna L, Gasbarrini A, Goizet C, Lacombe D, et al. Non-specific gastrointestinal features: could it be Fabry disease? Dig Liver Dis. 2018;50:429–37.
Danhof IE, Stavola JJ. Accelerated transit of intestinal gas with simethicone. Obstet Gynecol. 1974;44:148–54.
Friis H, Bod’e S, Rumessen JJ. Effect of simethicone on lactulose-induced H2 production and gastrointestinal symptoms. Digestion. 1991;49:227–30.
Bassotti G, Usai-Satta P, Bellini M. Linaclotide for the treatment of chronic constipation. Expert Opin Pharmacother. 2018;19:1261–6.
Yang Y, Fang J, Guo X, Dai N, Shen X, Yang Y, et al. Linaclotide in irritable bowel syndrome with constipation: A Phase 3 randomized trial in China and other regions. J Gastroenterol Hepatol. 2018;33:980–9.
Lenders M, Boutin M, Auray-Blais C, Brand E. Effects of orally delivered alpha-galactosidase A on gastrointestinal symptoms in patients with Fabry disease. Gastroenterology. 2020;159:1602–4.
Acknowledgements
M.L. received speaker honoraria, travel funding, and research grants from Amicus Therapeutics, Sanofi Genzyme, and Takeda and consulting honoraria from Avrobio, Inc.. E.B. received research grants and speaker/consulting honoraria from Sanofi Genzyme, Takeda, and Amicus Therapeutics and speaker/consulting honoraria from Chiesi and Greenovation/Eleva.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lenders, M., Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 81, 635–645 (2021). https://doi.org/10.1007/s40265-021-01486-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01486-1