
Abstract
Filgotinib (Jyseleca®) is an oral, ATP-competitive, reversible JAK1 preferential inhibitor that is being developed by Galapagos NV and Gilead Sciences for the treatment of inflammatory autoimmune diseases, including inflammatory arthritis and inflammatory bowel disease. The JAK-STAT signalling pathway has been implicated in the pathogenesis of inflammatory and autoimmune diseases, and filgotinib modulates this pathway by preventing the phosphorylation and activation of STATs. In September 2020, filgotinib received its first approvals in the EU and Japan. In the EU, filgotinib is indicated for the treatment of moderate to severe active rheumatoid arthritis (RA) in adults who have responded inadequately to, or who are intolerant to, one or more disease-modifying anti-rheumatic drugs (DMARDs). In Japan, filgotinib is indicated for the treatment of RA in patients who had an inadequate response to conventional therapies (including prevention of structural damage to joints). Clinical studies of filgotinib for the treatment of inflammatory autoimmune diseases are ongoing worldwide. This article summarizes the milestones in the development of filgotinib leading to this first approval.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
An oral, ATP-competitive, reversible JAK1 preferential inhibitor being developed by Galapagos NV and Gilead Sciences for the treatment of inflammatory autoimmune diseases |
Received its first approval on 24 September 2020 in the EU and Japan |
Approved for the treatment of RA in adults who have responded inadequately to, or who are intolerant to, one or more DMARDs |
1 Introduction
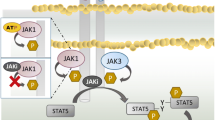
The Janus kinase (JAK)-signal transducer of activation (STAT) signalling pathway has been implicated in the pathogenesis of inflammatory and autoimmune diseases, such as rheumatoid arthritis (RA), psoriatic arthritis (PsA), ankylosing spondylitis (AS) and inflammatory bowel disease (IBD) [1]. JAKs are cytoplasmic tyrosine kinases that associate with cytokine receptors and transduce cytokine signalling via phosphorylation of STATs [2]. Four different types of JAKs (JAK1, JAK2, JAK3 and TYK2) and seven mammalian STAT family members have been identified (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6) [2]. JAK1 is important in mediating inflammatory cytokine signals, JAK2 in mediating myelopoiesis and erythropoiesis and JAK3 plays critical roles in immune homeostasis and lymphopoiesis [3]. Given the major role of the JAK-STAT pathway in the pathogenesis of inflammatory diseases, JAKs were considered logical targets for pharmacological manipulation, leading to the development of JAK inhibitors.
Filgotinib (Jyseleca®) is an oral, ATP-competitive, reversible JAK1 preferential inhibitor that is being developed by Galapagos NV (Galapagos) and Gilead Sciences for the treatment inflammatory diseases, such as RA, PsA, AS, Crohn's disease, ulcerative colitis, and non-infectious uveitis [4]. Filgotinib modulates the JAK-STAT signalling pathway by preventing the phosphorylation and activation of STATs.

Key milestones in the development of filgotinib. NDA New Drug Application, RA rheumatoid arthritis, UC ulcerative colitis
On 24 September 2020, filgotinib received its first approval in the EU [3] and Japan [5]. In the EU, filgotinib is indicated for the treatment of moderate to severe active RA in adults who have responded inadequately to, or who are intolerant to, one or more disease-modifying anti-rheumatic drugs (DMARDs) [3]. In Japan, filgotinib is indicated for the treatment of RA in patients who had an inadequate response to conventional therapies (including prevention of structural damage to joints) [6]. The recommended dose of filgotinib in adults is 200 mg once daily [3, 6]. The US FDA issued a complete response letter for filgotinib in the treatment of RA in August 2020, and have requested data from the MANTA (NCT03201445) and MANTA-Ray (NCT03926195) studies, which are evaluating the impact of filgotinib on sperm parameters, before completing its review [7]. Filgotinib is under regulatory review for use in ulcerative colitis in the EU [8]. Clinical studies of filgotinib for the treatment inflammatory diseases, including RA, PsA, AS, Crohn's disease, ulcerative colitis and non-infectious uveitis, are ongoing worldwide.
1.1 Company Agreements
In February 2012, Galapagos and Abbott (now AbbVie) entered into a global collaboration to develop and commercialise filgotinib [4, 9]. Upon successful completion of phase 2 trials for RA, AbbVie was to license the programme and assume sole responsibility for phase 3 trials and global manufacturing. Galapagos had retained co-promotion rights in Belgium, the Netherlands and Luxembourg [4, 9]. In May 2013, Galapagos and AbbVie announced an extension of their filgotinib clinical development collaboration to include Crohn's disease [10]. Galapagos was to fund and complete a phase 2 program in Crohn's disease and AbbVie was responsible for funding and performing clinical development beyond phase 2 and completing regulatory and commercialization activities [10]. In September 2015, AbbVie terminated its development and marketing agreement with Galapagos, with all rights to filgotinib retained by Galapagos [4, 11].
Galapagos and Gilead Sciences entered into a global partnership in December 2015, for the development and commercialization of filgotinib for the treatment of inflammatory diseases, including RA [4, 12]. In January 2016, the company reported closing of the deal and entry into force [13].

Chemical structure of filgotinib
The companies decided to jointly develop the product, starting with phase 3 trials for RA, with Galapagos co-funding 20% of the activities, while Gilead was to be responsible for all manufacturing and commercial activities [13]. In December 2017, Galapagos decided to opt-in on the co-promotion of filgotinib with collaboration partner Gilead Sciences in UK, Germany, France, Italy, Spain, Belgium, the Netherlands and Luxembourg, once filgotinib was approved for commercial sale [14]. The companies agreed to a lock-up and standstill arrangement, which was to expire on 31 December 2017 [14]. In February 2018, Galapagos reported that it had exercised its option to co-promote filgotinib in Europe with Gilead [15]. In July 2019, the deal was amended under the terms of which Gilead and Galapagos were to co-commercialize filgotinib in France, Germany, Italy, Spain and the United Kingdom and retain the 50/50 profit share in these countries. However, Galapagos retained exclusive rights in Belgium, the Netherlands and Luxembourg [16].
In December 2019, Gilead Sciences entered into an agreement with Eisai, for the distribution and co-promotion of filgotinib for the treatment of RA in the Japanese market. Under the terms of the agreement, Gilead was to assume marketing approval and manufacturing of filgotinib, and Eisai was responsible for product distribution in Japan for RA and other future indications. Both the companies were to jointly commercialize filgotinib if approved in Japan [17].
2 Scientific Summary
2.1 Pharmacodynamics
In biochemical assays, filgotinib selectively inhibited the activity of JAK1 (> 5-fold higher potency) over JAK2, JAK3 and TYK2. In human cellular assays, filgotinib preferentially inhibited JAK1/JAK3-mediated interleukin (IL)-2, IL-4 and IL-15 signalling, JAK1/2-mediated IL-6 signalling and JAK1/TYK2-mediated type I interferon signalling downstream of the heterodimeric cytokine receptors, with functional selectivity over cytokine receptors that signal via pairs of JAK2 or JAK2/TYK2 [3, 18]. In in vitro assays, the main metabolite of filgotinib, GS-829845, was approximately 10-fold less active than filgotinib, while exhibiting a similar JAK1 preferential inhibitory activity [3]. An in vivo rat model showed that the overall pharmacodynamic effect of filgotinib was largely driven by the metabolite [3]. In human whole blood assays, filgotinib dose-dependently inhibited JAK1-associated IL-6-induced STAT1 phosphorylation but had no effect on JAK2-associated GM-CSF induced STAT5 phosphorylation and demonstrated ≈ 30-fold selectivity for inhibition of JAK1- over JAK2-dependent signalling [18]. The selectivity of filgotinib for JAK1 over JAK2 was also demonstrated in gene expression analyses using JAK-dependent gene signatures in blood from healthy volunteers and patients with RA [19].
During 24 weeks’ treatment in the pivotal phase 3 FINCH 1, 2 and 3 studies, the median and interquartile ranges for serum IgG, IgM, and IgA values remained largely within the normal reference ranges [3]. Serum C-reactive protein (CRP) levels decreased as early as week 2 of treatment and were maintained over 24 weeks’ treatment with filgotinib. Filgotinib therapy was associated with a small, transient increase in mean absolute lymphocyte count that remained within normal reference ranges and gradually returned to, or close to, baseline levels over 12 weeks’ treatment. Although median platelet counts remained within the normal range, treatment with filgotinib resulted in a slight decrease in median platelet counts within the first 4 weeks of therapy and remained stable thereafter through 24 weeks’ therapy. Median haemoglobin values remained stable within the normal range during 24 weeks’ treatment in FINCH 1, 2 and 3 [3, 20].
2.2 Pharmacokinetics
Oral filgotinib is absorbed rapidly, with the median peak plasma concentration (Cmax) of filgotinib reached 2–3 h post dose and that of its main metabolite GS-829845 reached 5 h post dose following multiple dosing [3]. Filgotinib and GS-829845 exposures (area under the concentration-time curve; AUC) and Cmax were similar between healthy adult subjects and patients with RA and values were dose proportional over the therapeutic range. Steady-state concentrations of filgotinib and GS-829845 were reached in 2–3 and 4 days, respectively. Following once daily oral administration, there is negligible accumulation of filgotinib and approximately two-fold accumulation of GS-829845. There were no clinically relevant differences in exposures when filgotinib was administered with a high-fat or low-fat meal compared with the fasted state; therefore, filgotinib can be administered with or without food. Human plasma protein binding of filgotinib and GS-829845 is low (55–59% and 39–44%, respectively). The blood-to-plasma ratio of filgotinib is 0.85–1.1, indicating no preferential distribution of filgotinib and GS-829845 into blood cells. Filgotinib and GS-829845 are substrates of the P-glycoprotein transporter [3].
Filgotinib is extensively metabolized, primarily by carboxylesterase (CES) 2, and to a lesser extent by CES1, to form the main circulating metabolite GS-829845 [3]. Following oral administration, approximately 9.4% and 4.5% of a filgotinib dose was recovered as unchanged drug in urine and faeces, respectively. In a clinical pharmacology study, the majority of radioactivity in plasma was accounted for by filgotinib (2.9%) and GS-829845 (92%), with no other major metabolites identified. Following oral administration, ≈ 87% of the administered dose was eliminated in the urine (as filgotinib and its metabolites) and 15% in the faeces. GS-829845 accounted for ≈ 54% of the dose recovered in the urine and 8.9% in the faeces. The mean terminal half-lives of filgotinib and GS-829845 were approximately 7 and 19 h, respectively [3].
Bodyweight, gender, race, age, mild renal impairment [creatinine clearance (CLCR) ≥ 60 mL/min), and mild or moderate hepatic impairment (Child-Pugh A or B) did not have a clinically relevant effect on the pharmacokinetics of filgotinib or GS-829845 [3]. Exposure to filgotinib and GS-829845 was increased in patients with moderate (CLCR 30 to < 60 mL/min) or severe (CLCR 15 to < 30 mL/min) renal impairment; therefore, a filgotinib 100 mg once daily dose is recommended in these patients. The pharmacokinetics of filgotinib have not been assessed in patients with end stage renal disease (CLCR < 15 mL/min) or severe hepatic impairment (Child-Pugh C) [3].
In vitro studies showed that CES2, the main enzyme involved in the metabolism of filgotinib, can be inhibited by medicinal products such as fenofibrate, carvedilol, diltiazem or simvastatin; however, the clinical relevance of this interaction is unknown [3]. Filgotinib is not a clinically relevant inhibitor or inducer of most enzymes or transporters commonly involved in drug interactions, such as cytochrome P450 enzymes and UDP-glucuronosyltransferases [21]. However, in vitro studies are inconclusive regarding the potential of filgotinib to induce CYP2B6 or to induce or inhibit CYP1A2 and the potential for GS-829845 to inhibit P-gp or BRCP. Caution is required when filgotinib is coadministered with CYP1A2, P-gp or BRCP substrates that have a narrow therapeutic index. In vitro, filgotinib and GS-829845 are inhibitors of OATP1B1 and OATP1B3. Coadministration of filgotinib with OATP1B1 and OATP1B3 substrates may increase exposure to the coadministered OATP1B1 or OATP1B3 substrate and the risk of adverse events; filgotinib coadministration with sensitive OATP1B1 and OATP1B3 substrates such as valsartan or statins is not recommended [3]. Further in vivo investigation of drug-drug interactions between filgotinib and OATP substrates is ongoing.
Features and properties of filgotinib
Alternative names | Filgotinib hydrochloride; G-146034; G-146034-101; GLPG-0634; GS-6034; Jyseleca |
Class | 2 ring heterocyclic compounds; amides; anti-inflammatories; antirheumatics; cyclopropanes; pyridines; skin disorder therapies; small molecules; thiamorpholines; triazoles; urologics |
Mechanism of Action | Modulates the JAK-STAT pathway by preventing the phosphorylation and activation of STATs |
Route of Administration | Oral |
Pharmacodynamics | Preferentially inhibits the activity of JAK1 over JAK2, JAK3 and TYK2 |
Inhibited JAK1/JAK3-mediated IL-2, IL-4 and IL-15 signalling, JAK1/2-mediated IL-6 signalling and JAK1/TYK2-mediated type I interferon signalling in human cellular assays | |
Dose-dependently inhibited IL-6-induced STAT1 phosphorylation in human whole blood assays | |
Pharmacokinetics | Peak plasma concentration reached in 2–3 h |
Filgotinib and GS-829845 (main metabolite) steady state reached in 2–3 and 4 days, respectively | |
Mean terminal half-lives of filgotinib and GS-829845 approximately 7 and 19 h, respectively | |
Most frequent ARs in patients with RA | Nausea, upper respiratory tract infection, urinary tract infection, dizziness |
ATC codes | |
WHO ATC code | L04A-A45 (filgotinib) |
EphMRA ATC code | L4 (immunosuppressants) |
Chemical Name | N-(5-{4-[(1,1-dioxo-λ6-thiomorpholin-4-yl)methyl]phenyl}[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide |
2.3 Therapeutic Trials
2.3.1 Rheumatoid Arthritis
2.3.1.1 FINCH 1
The 52-week randomized, double-blind, placebo- and active-controlled study phase 3 FINCH 1 trial (NCT02889796) showed that filgotinib significantly improved signs and symptoms of disease and physical function and slowed radiographic progression in patients with RA who had an inadequate response to ≥ 12 weeks’ methotrexate therapy (n = 1755) [3, 22]. Patients were randomized to receive filgotinib 100 mg or 200 mg once daily (n = 480 and 475), adalimumab 40 mg every 2 weeks (n = 325) or placebo (n = 475) in addition to stable background treatment with methotrexate. At week 24, patients receiving placebo were re-randomised to filgotinib 100 mg or 200 mg once daily up to week 52. The primary endpoint was the proportion of subjects who achieved an ACR20 response at Week 12 [3, 22].
At week 12, significantly (p ≤ 0.001) more patients in the filgotinib 100 mg and 200 mg groups than in the placebo group achieved American College of Rheumatology 20 (ACR20; 70% and 77% vs. 50%; primary endpoint). ACR50 (36% and 47% vs. 20%) and ACR 70 (19% and 26% vs. 7%) responses were also more frequent in the filgotinib 100 mg and 200 mg groups than in the placebo group (nominal p ≤ 0.001) [3, 22]. The proportions of patients achieving Disease Activity Score 28 C-reactive protein (DAS28-CRP) low disease activity (score ≤ 3.2) [39% and 50% vs. 23%; p < 0.001] and DAS28-CRP remission (score < 2.6) [34% and 24% vs. 9%; nominal p < 0.001) at 12 weeks were also higher with filgotinib 100 mg and 200 mg than with placebo. In addition, noninferiority of filgotinib 200 mg and adalimumab was demonstrated for DAS28-CRP low disease activity at 12 weeks (50% vs. 43%; p ≤ 0.001 for noninferiority). At week 24, filgotinib 100 mg or 200 mg recipients experienced significantly less radiographic progression (assessed by the modified Total Sharp Score (mTSS)] than placebo recipients (mean change from baseline 0.17 and 0.13 vs. 0.37; p ≤ 0.001) [3]. Patients receiving filgotinib 100 mg or 200 mg had significant (p < 0.001) improvements in physical function [assessed by the Health Assessment Questionnaire-Disability Index (HAQ-DI)] than placebo recipients (mean change from baseline of -0.56 and -0.69 vs -0.42) [3, 22].
The efficacy of filgotinib 100 mg or 200 mg in terms of ACR20, ACR50 and ACR70 and DAS28-CRP low disease activity was sustained during 52 weeks’ treatment [23].
2.3.1.2 FINCH 2
The 24-week randomized, double-blind, placebo-controlled phase 3 FINCH 2 trial (NCT02873936) showed that filgotinib significantly improved clinical responses and physical function in patients with active RA who had an inadequate response or intolerance to ≥ 1 biologic DMARD (n = 448) [3, 24]. Patients received filgotinib 100 mg or 200 mg once daily (n = 153 and 147) or placebo (n = 148), in addition to a stable background dose of conventional synthetic DMARDs. At week 12, significantly more patients receiving filgotinib 100 mg or 200 mg than those receiving placebo achieved ACR20 (58% and 66% vs. 31%; p < 0.001; primary endpoint). ACR50 (32% and 43% vs. 15%; nominal p ≤ 0.001) and ACR70 (14% and 22% vs. 7%; nominal p ≤ 0.05) responses were also more frequent in patients receiving filgotinib 100 mg or 200 mg than in those receiving placebo. DAS28-CRP low disease activity (37% and 41% vs. 16%; p < 0.001) and DAS28-CRP remission (26% and 22% vs. 8.1%; nominal p < 0.001) at week 12 were evident in more filgotinib 100 mg or 200 mg than placebo recipients. Improvements in HAQ-DI scores with filgotinib 100 mg or 200 mg were significantly greater than with placebo at week 12 (mean change from baseline − 0.48 and − 0.55 vs − 0.23; p < 0.001) and more filgotinib 100 mg or 200 mg than placebo recipients had a HAQ-DI reduction of ≥ 0.22 points (66% and 67% vs. 44%; nominal p < 0.001). Efficacy improvements with filgotinib 100 mg or 200 mg were sustained at week 24 versus placebo (nominal p ≤ 0.05) [3, 24].
2.3.1.3 FINCH 3
The 52-week randomized, double-blind, active-controlled phase 3 FINCH 3 trial (NCT02886728) showed that filgotinib significantly improved signs and symptoms of disease, physical function and HRQOL, and slowed radiographic progression in methotrexate-naïve patients with moderately to severely active RA [3, 25]. Patients received filgotinib 100 mg or 200 mg once daily plus methotrexate (n = 207 and 416), filgotinib 200 mg monotherapy (n = 210) or methotrexate monotherapy (n = 416). The primary endpoint was the proportion of patients who achieved an ACR20 response at week 24.
Significantly more patients receiving filgotinib 100 mg or 200 mg plus methotrexate combination therapy than those receiving methotrexate monotherapy achieved an ACR20 response at week 24 (80% and 81% vs. 71%; both p < 0.05); there was no significant difference between filgotinib 200 mg monotherapy and methotrexate recipients in the ACR20 response rate (78% vs. 71%) [3, 25]. At 24 weeks, patients receiving filgotinib 100 mg or 200 mg plus methotrexate or filgotinib 200 mg monotherapy had higher (nominal p < 0.01) ACR50 (57%, 62% and 58% vs. 46%) and ACR70 (40%, 44% and 40% vs. 26%) response rates, and DAS28-CRP low disease activity (63%, 69% and 60% vs. 46%) and significantly higher DAS28-CRP remission (43%, 54% and 42% vs. 29%; p ≤ 0.001) rates than patients receiving methotrexate [3, 25]. Patients receiving filgotinib 200 mg monotherapy had less radiographic progression than patients receiving methotrexate at week 24 (mean change from baseline in mTSS − 0.04 vs. 0.51; nominal p < 0.01); there were no significant differences between patients receiving filgotinib 100 mg or 200 mg plus methotrexate and those receiving methotrexate for this outcome (0.22 and 0.21 vs. 0.52). The mean change from baseline in HAQ-DI at week 24 was significantly (p ≤ 0.01) greater with filgotinib 100 mg or 200 mg plus methotrexate than with methotrexate (− 0.90 and − 0.94 vs − 0.79) [3, 25].
Filgotinib efficacy was sustained up to week 52, with higher ACR20, ACR50 and ACR70 response rates and DAS28-CRP low disease activity and remission rates in the filgotinib 100 mg and 200 mg combination therapy and filgotinib 200 mg monotherapy groups than in the methotrexate group (nominal p < 0.05) [3, 26]. Patients receiving filgotinib 100 mg or 200 mg combination therapy or filgotinib 200 mg monotherapy had less radiographic disease progression than patients receiving methotrexate (0.27, 0.21 and 0.23 vs. 0.74; nominal p < 0.05). The mean change from baseline in HAQ-DI scores was greater with filgotinib 200 mg combination therapy and filgotinib 200 mg monotherapy than with methotrexate(nominal p < 0.05), while there was no significant difference between the filgotinib 100 mg combination therapy and methotrexate groups [26].
2.3.1.4 Phase 2 Trials
The 24-week, randomised, double-blind, placebo-controlled, phase 2b, dose-finding DARWIN 1 trial (NCT01888874) of filgotinib 50–200 mg once or twice daily as add-on treatment to methotrexate showed that filgotinib as add-on to MTX improved the signs and symptoms of disease in patients with moderate-to-severe active RA (n = 594), and was associated with a rapid onset of action [27]. At week 12, significantly (p < 0.05) more patients achieved ACR20 responses (primary endpoint) in the filgotinib 100 mg once daily [64% (54/85)], 200 mg once daily [69% (59/86)] and 100 mg twice-daily [79% (66/84)] groups compared with placebo [44% (38/86)], with an onset of response as early as week 2 of treatment with filgotinib 100 mg twice daily and 200 mg once daily [27]. Patient-reported outcomes withy filgotinib, including physical function (HAQ‐DI), pain (VAS), fatigue (FACIT‐F) and SF-36 PCS, also improved rapidly (weeks 2–4), with benefits sustained at week 12 [28].
The 24-week, randomised, double-blind, phase 2b, dose-finding DARWIN 2 trial (NCT01894516) of filgotinib 50–200 mg once daily monotherapy showed that filgotinib was effective in treating the signs and symptoms of disease in patients with moderately to severely active RA (n = 283), with a rapid onset of action [29]. At week 12, significantly (p < 0.0001) more patients achieved ACR20 responses in all filgotinib groups [50 mg: 67% (48/72); 100 mg: 66% (46/70); 200 mg: 73% (50/69)] versus placebo [29% (21/72)] (primary endpoint), with an onset of response as early as week 1 of treatment with filgotinib 200 mg group [29].
During up to 24 weeks treatment in DARWIN 1 and DARWIN 2, filgotinib as add-on therapy to methotrexate or as monotherapy demonstrated improvements in health-related quality of life and functional status in patients with active RA, with some measures improving as early as week 1–4 of treatment and improvements sustained up to week 24 [30].
Patients who had completed 24 weeks’ treatment in DARWIN 1 or DARWIN 2 were eligible to enter the ongoing, open-label, long-term extension study DARWIN 3 (NCT02065700), during which all patients received filgotinib 200 mg/day (apart from 15 males who received a 100 mg/day dosage) [31]. An interim analysis of DARWIN 3 showed that clinical efficacy of filgotinib is maintained up to week 156, as assessed by ACR20, ACR50 and ACR70 response rates (87%/72%/46% in patients switching from combination therapy and 90%/63%/40% in patients continuing monotherapy), and DAS28-CRP low disease activity and remission rates (69%/53% in patients switching from combination therapy and 65%/46% in patients from continuing monotherapy) [31].
2.3.2 Psoriatic Arthritis
Filgotinib was effective in improving signs and symptoms of disease in patients with active moderate-to-severe PsA who were participating in the 16-week, randomized, double-blind, placebo-controlled phase 2 EQUATOR trial (NCT03101670) [32]. At week 16, significantly more patients receiving filgotinib 200 mg once daily (n = 65) than those receiving placebo (n = 66) achieved ACR20 (80% vs. 33%; p < 0.0001; primary endpoint), ACR50 (48% vs. 15%; p < 0.0001) and ACR70 (23% vs. 6%; p < 0.005) responses [32]. The onset of response to filgotinib was early, with a median time to first ACR20 response of 4.1 weeks in the filgotinib group compared with 12.3 weeks in the placebo group (p < 0.0001) [33]. The effect of filgotinib on key efficacy measure, including ACR20, ACR50 and ACR70 responses, were generally consistent across subgroups, regardless of patient, disease and treatment characteristics [34]. Patients receiving filgotinib reported significantly greater and clinically meaningful improvements in patient-reported outcomes at week 16, including improvements in the PsA Impact of Disease 9 questionnaire (PsAID9) [35], Patient’s Global Assessment of Disease Activity, pain, HAQ-DI, FACIT-F and SF-36 PCS (all p < 0.01), but not in the SF-36 Mental Summary (MCS) [36].
Patients completing EQUATOR could join an ongoing 304-week open-label safety extension (EQUATOR 2; NCT03320876). A prespecified interim analysis at week 52 showed sustained efficacy in patients continuing treatment with filgotinib 200 mg once daily (n = 54); of the patients who achieved ACR20, ACR50 and ACR70 responses at week 16, 85%, 93% and 77% maintained these at week 52 [37].
2.3.3 Ankylosing Spondylitis
Filgotinib reduced disease activity and signs and symptoms in patients with active AS who were participating in the 12-week, randomized, double-blind, placebo-controlled, phase 2 TORTUGA trial (NCT03117270) [38]. At week 12, the mean change from baseline in the AS disease activity score (ASDAS) was significantly greater in patients receiving filgotinib 200 mg once daily than in those receiving placebo (− 1.47 vs. − 0.57; p < 0.0001) [38]. Filgotinib significantly reduced inflammation of the sacroiliac joint and spine, as indicated by significant (p < 0.05) reductions in the Spondyloarthritis Research Consortium of Canada (SPARCC) spine, SPARCC sacroiliac joint, erosion and backfill scores [38, 39]. Filgotinib significantly decreased levels of circulating biomarkers associated with active AS disease, including proinflammatory cytokines and chemokines, cell adhesion molecules, and markers of matrix remodelling [40].
2.3.4 Ulcerative Colitis
Filgotinib was effective as an induction treatment in biologic-naïve and biologic-experienced patients with moderately to severely active ulcerative colitis [41] and as maintenance treatment in those who had achieved clinical response with induction therapy [42] in the 58-week phase 2b/3 SELECTION study (NCT02914522). SELECTION is a randomized, double-blind study that evaluated once-daily filgotinib 100 mg or 200 mg versus placebo as (a) induction therapy in patients with moderately to severely active ulcerative colitis who were biologic naïve but failed conventional therapy (Induction Study A; n = 659) or were biologic experienced (Induction Study B; n = 689) and (b) maintenance treatment for patients with moderately to severely active ulcerative colitis who had achieved either clinical remission or Mayo Clinic Score (MCS) response with induction treatment in any of the treatment arms (n = 664).
Clinical remission at week 10 (primary endpoint in the induction study) was achieved in significantly higher proportions of patients treated with filgotinib 200 mg than placebo in both biologic-naïve (26.1% vs 15.3%; p = 0.0157) and biologic experienced (11.5% vs 4.2%; p = 0.0103) patients. Additionally, among biologic-naïve patients, significantly more filgotinib 200 mg than placebo recipients achieved MCS remission (p = 0.0053), endoscopic remission (p = 0.0047) and Geboes histologic remission (p < 0.0001) [key secondary endpoints]. [41].
Patients entering the maintenance study who had received filgotinib induction therapy were re-randomized to receive either their induction filgotinib dose or placebo; patients who had received placebo in the induction study continued placebo in the maintenance study. In the group of patients who had previously received filgotinib in the induction study (n = 558), clinical remission at week 58 (primary endpoint in the maintenance study) was achieved in a significantly higher proportion of patients treated with filgotinib 200 mg (37.2% vs 11.2%; p < 0.025) or filgotinib 100 mg (23.8% vs 13.5%; p < 0.05) than placebo. Significantly higher proportions of patients receiving filgotinib 200 mg maintenance therapy also achieved 6-month corticosteroid-free clinical remission, sustained clinical remission, MCS remission, endoscopic remission and histologic remission at week 58 compared with placebo (p < 0.025) [42].
Key clinical trials of filgotinib
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor |
|---|---|---|---|---|---|---|
Filgotinib, adalimumab, placebo, methotrexate | RA | 3 | Completed | International | FINCH 1; NCT02889796; GS-US-417-0301; 2016-000568-41 | Gilead Sciences |
Filgotinib, placebo, csDMARDs | RA | 3 | Completed | International | FINCH 2; NCT02873936; GS-US-417-0302; 2016-000569-21 | Gilead Sciences |
Filgotinib, placebo, methotrexate | RA | 3 | Completed | International | FINCH 3; NCT02886728; GS-US-417-0303; 2016-000570-37 | Gilead Sciences |
Filgotinib, placebo | RA | 3 | Ongoing | International | FINCH 4; NCT03025308; GS-US-417-0304; 2016-003630-25 | Gilead Sciences |
Filgotinib, placebo | RA | 2b | Completed | International | DARWIN 1; NCT01888874; GLPG0634-CL-203; 2012-003635-31 | Galapagos NV |
Filgotinib, placebo | RA | 2 | Completed | International | DARWIN 2; NCT01894516; GLPG0634-CL-204 | Galapagos NV |
Filgotinib | RA | 2 | Ongoing | International | DARWIN 3; NCT02065700; GLPG0634-CL-205; 2012-003655-11 | Gilead Sciences |
Filgotinib, placebo | RA | 2 | Completed | International | NCT01668641; GLPG0634-CL-202 | Galapagos NV |
Filgotinib | RA | 2 | Completed | Republic of Moldova | NCT01384422; GLPG0634-CL-201 | Galapagos NV |
Filgotinib, adalimumab, placebo | PsA | 3 | Recruiting | International | PENGUIN 1; NCT04115748; GS-US-431-4566; 2019-001996-35; JapicCTI-205202 | Gilead Sciences |
Filgotinib, placebo | PsA | 3 | Recruiting | International | PENGUIN 2; NCT04115839; GS-US-431-4567; 2019-002021-29; JapicCTI-205201 | Gilead Sciences |
Filgotinib, placebo | PsA | 2 | Completed | International | EQUATOR; NCT03101670; GLPG0634-CL-224 | Galapagos NV |
Filgotinib | PsA | 2 | Ongoing | International | EQUATOR 2; NCT03320876; GLPG0634-CL-225; 2017-000545-52 | Galapagos NV |
Filgotinib, placebo | AS | 2 | Completed | International | TORTUGA; GLPG0634-CL-223 | Galapagos NV |
Filgotinib, placebo, SOC | RA, PsA, AS, AxSp | 2 | Ongoing | International | MANTA-Ray; NCT03926195; GLPG0634-CL-227; 2018-003933-14 | Galapagos NV |
Filgotinib, placebo | CD | 3 | Recruiting | International | DIVERSITY; NCT02914561; GS-US-419-3895; 2016-001367-36 | Gilead Sciences |
Filgotinib, placebo | CD | 3 | Enrolling by invitation | International | DIVERSITYLTE; NCT02914600; GS-US-419-3896; 2016-002763-34 | Gilead Sciences |
Filgotinib, placebo | CD | 2 | Completed | International | NCT02048618; FITZROY; GLPG0634-CL-211; 2013-002857-32 | Galapagos NV |
Filgotinib, placebo | CD | 2 | Completed | International | Divergence 1; NCT03046056; GS-US-419-4015; 2016-003179-23 | Gilead Sciences |
Filgotinib, placebo | CD | 2 | Ongoing | International | Divergence 2; NCT03077412; GS-US-419-4016; 2016-003153-15 | Gilead Sciences |
Filgotinib, placebo | UC | 3 | Completed | International | SELECTION; NCT02914522; GS-US-418-3898; 2016-001392-78 | Gilead Sciences |
Filgotinib, placebo | UC | 3 | Ongoing | International | SELECTIONLTE; NCT02914535; GS-US-418-3899; 2016-002765-58 | Gilead Sciences |
Filgotinib, placebo, SOC | IBD | 2 | Ongoing | International | MANTA; NCT03201445; GS-US-418-4279; 2017-000402-38 | Gilead Sciences |
2.3.5 Crohn’s Disease
In the randomized, double-blind, placebo-controlled phase 2 FITZROY trial (NCT02048618) in patients with moderate-to-severe Crohn’s disease (n = 174), a significantly greater proportion of patients receiving filgotinib 200 mg once daily than those receiving placebo achieved clinical remission (Crohn’s Disease Activity Index < 150) at week 10 (47% vs. 23%; p = 0.0077; primary endpoint) [43]. A post hoc analysis showed that filgotinib 200 mg induced clinical remission regardless of the disease duration (< 5, 5–10 or > 10 years) or disease location (ileocolonic, ileal or colonic disease) [44]. Filgotinib treatment led to early (at week 2) and significant reductions (versus placebo) in markers of systemic and mucosal inflammation (faecal calprotectin, vascular endothelial growth factor A and CRP), with significant association observed between the decrease in both systemic (serum CRP and IL-6) and mucosal (faecal calprotectin) biomarkers and endoscopic response [45].
2.4 Adverse Events
Filgotinib was generally well tolerated in clinical trials in patients with RA [3, 46] or inflammatory bowel disease [41,42,43]. The most common adverse reactions with filgotinib 200 mg once daily in the placebo-controlled safety analysis set (up to week 12; n = 777) in trials in patients with RA were nausea (3.5%), upper respiratory tract infection (URTI; 3.3%), urinary tract infection (UTI; 1.7%) and dizziness (1.2%) [3, 20]. Nausea was generally transient and occurred during the first 24 weeks of treatment [3].
In a pooled analysis of placebo-controlled studies (FINCH 1 and 2 and DARWIN 1 and 2), the incidence of infection during 12 weeks’ treatment with filgotinib 200 mg was 18.1% compared with 13.3% with placebo; no opportunistic infections (excluding tuberculosis) were reported during this period [3]. During 12 weeks’ treatment, the incidences of infectious adverse drug reactions in filgotinib 200 mg and placebo groups were 3.3% and 1.8% for URTI, 1.7% and 0.9% for UTI, 0.6% and 0.4% for pneumonia, and 0.1% and 0.3% herpes zoster. During 12 weeks’ treatment, the incidence of serious infections was 1.0% and 0.6% with filgotinib 200 mg and placebo, respectively. In FINCH 3, involving methotrexate-naïve patients, the incidence of infections over 24 weeks was 25.2%, 23.1% and 24.5% in patients receiving filgotinib 200 mg monotherapy, filgotinib 200 mg plus methotrexate and methotrexate, respectively, and the incidence of serious infections was 1.4%, 1.0% and 1.0%, respectively. The most common serious infection with filgotinib was pneumonia. Limited data indicate that serious infections occur at a higher incidence in patients aged ≥ 75 years. Absolute neutrophil counts < 1 × 109 cells/L and absolute lymphocyte counts < 0.5 × 109 cells/L were reported in ≤ 1% of patients in clinical studies [3]. In FINCH 1, the incidences of serious adverse events, serious infections, and adverse events of interest [herpes zoster infection, adjudicated major adverse cardiovascular events (MACE), venous thromboembolism, malignancies (other than nonmelanoma skin cancer) and nonmelanoma skin cancer] with filgotinib were comparable to those with adalimumab through 24 weeks [22] and 52 weeks [23].
Filgotinib was also generally well tolerated in an integrated safety analysis of data from seven clinical trials (FINCH 1–4 and DARWIN 1–3), which included 4057 patients with RA receiving filgotinib 200 mg or 100 mg once daily (n = 2227 and 1600, respectively, including patients switching from placebo, methotrexate, adalimumab or other doses of filgotinib) [46]. There was no dose dependent effect of filgotinib on serious treatment-emergent adverse events (TEAEs) and TEAEs leading to death, and the exposure-adjusted incidence rates per 100 patient years (EAIRs/100 PYs) of these events were comparable between patients receiving filgotinib 200 mg or 100 mg, adalimumab, methotrexate or placebo (serious TEAEs 6.5, 7.7, 7.6, 7.9 and 9.3; TEAEs leading to death 0.4, 0.4, 0.3, 0 and 0.3). In filgotinib 200 mg and 100 mg, adalimumab, methotrexate and placebo recipients, the EAIRs/100 PYs for infections were 28.9, 39.0, 44.5, 44.1 and 52.7, serious infections were 1.7, 3.3, 3.4, 2.2 and 2.3, herpes zoster virus infections were 1.7, 1.1, 0.7, 1.1 and 1.0 and opportunistic infections were 0.1, 0.3, 0.7, 0.6 and 0. The EAIRs/100 PYs of MACE, venous thromboembolism, malignancies (other than nonmelanoma skin cancer) and nonmelanoma skin cancer were low (≤ 1.1) across all treatment groups. The tolerability profile of filgotinib was consistent with its mechanism of action and no new safety concerns were identified [46].
The adverse events profile of filgotinib in patients with inflammatory bowel disease in the phase 3 SELECTION [41, 42] and phase 2 FITZROY [43] studies was consistent with that seen in patients with RA. During the induction and maintenance periods of SELECTION, the incidence of adverse events and serious adverse events with filgotinib in patients with ulcerative colitis was similar to that with placebo. Serious infection and herpes zoster were infrequent in all treatment groups [41, 42]. In FITZROY, similar proportions of placebo and filgotinib 100 mg or 200 mg recipients with Crohn’s disease experienced at least one treatment-emergent adverse event; 9% of filgotinib (14/152) and 4% of placebo (3/67) recipients experienced a serious treatment-emergent adverse event. Serious infections occurred in four filgotinib recipients and no placebo recipients [43].
2.5 Ongoing Clinical Trials
In addition to the ongoing phase 2 DARWIN 3 (NCT02065700) and EQUATOR 2 (NCT03320876) studies, several phase 2 and 3 studies are underway, including the phase 3 FINCH 4 (NCT03025308) long-term extension study in patients with RA, which is evaluating the long-term safety and tolerability of filgotinib in patients who had previously completed FINCH 1, 2 or 3.
Recruitment is underway for the phase 3, randomized, double-blind PENGUIN 1 trial (NCT04115748), which will compare the efficacy and safety of filgotinib versus placebo and adalimumab in patients with active PsA who are naïve to biologic DMARDs. The study consists of two parts, the main study and its long-term extension. Recruitment is also underway for the phase 3, randomized, double-blind, placebo-controlled PENGUIN 2 trial (NCT04115839), which will determine the efficacy and safety of filgotinib in patients with active PsA who have an inadequate response or are intolerant to biologic DMARDs. A phase 2, open-label long-term extension study (NCT03320876) is evaluating the long-term safety and efficacy of filgotinib in patients with moderately to severely active PsA.
The phase 3, randomized, double-blind, placebo-controlled trial SEALION2-NAÏVE (NCT04483700) plans to evaluate the efficacy and safety of filgotinib in patients with active AS who are naïve to biologic DMARDs. Also planned is the phase 3, randomized, double-blind, placebo-controlled SEALION1-IR study (NCT04483687), which will assess the efficacy and safety of filgotinib in patients with active AS who have an inadequate response to biologic DMARDs.
The phase 3, randomized, double-blind, placebo-controlled, DIVERSITY trial (NCT02914561) is recruiting patients with moderately to severely active Crohn’s disease. The study will evaluate the safety and efficacy of filgotinib during induction and maintenance treatment of patients who are biologic-naïve and biologic-experienced. Recruitment is also underway for the phase 3, long-term extension study DIVERSITY LTE (NCT02914600), which will evaluate the long-term safety of filgotinib in patients with Crohn's disease who have completed or met protocol-specified efficacy discontinuation criteria in a prior filgotinib study. In addition, the phase 2, randomized, double-blind, placebo-controlled DIVERGENCE 1 trial (NCT03046056) in patients with Crohn's disease was recently completed; the ongoing phase 2, randomized, double-blind, placebo-controlled DIVERGENCE 2 trial (NCT03077412) is assessing the efficacy and safety of filgotinib in patients with perianal fistulizing Crohn’s disease.
The ongoing phase 3, long-term extension study SELECTION LTE (NCT02914535) is evaluating the long-term safety of filgotinib in patients with ulcerative colitis who have completed or met protocol-specified efficacy discontinuation criteria in a prior filgotinib study. The phase 2 randomized, placebo-controlled HUMBOLDT study (NCT03207815) is recruiting patients with active, non-infectious uveitis to determine the efficacy and safety of filgotinib. Also underway are the phase 2 randomized, double-blind placebo-controlled MANTA (NCT03201445) and MANTA-Ray (NCT03926195) studies. MANTA will assess the testicular safety of filgotinib in adult males with moderately to severely active IBD, and MANTA-Ray will evaluate the effect of filgotinib on semen parameters in adult males with active RA, PsA, AS or non-radiographic axial spondyloarthritis.
3 Current Status
On 24 September 2020, filgotinib received its first approvals in the EU [3] and Japan [5]. In the EU, filgotinib is indicated for the treatment of moderate to severe active RA in adults who have responded inadequately to, or who are intolerant to, one or more DMARDs [3]. In Japan, filgotinib is indicated for the treatment of RA in patients who had an inadequate response to conventional therapies (including prevention of structural damage to joints) [6].
Change history
04 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01468-3
References
Banerjee S, Biehl A, Gadina M, et al. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–46.
O’Shea JJ, Schwartz DM, Villarino AV, et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28.
European Medicines Agency. Filgotinib (Jyseleca®): summary of product characteristics. 2020. https://www.ema.europa.eu/. Accessed 30 Sep 2020.
Durez P, Hoekema A, Huizinga T, et al. Treatment innovation for patients: a collaborative network in the Benelux and an inside view of 20 years of Galapagos. Acta Clin Belg. 2020. https://doi.org/10.1080/17843286.2020.1812830.
Gilead Sciences. Jyseleca® (filgotinib) approved in Japan for rheumatoid arthritis [media release]. 25 Sep 2020. https://www.gilead.com.
Gilead Sciences. Jyseleca® (filgotinib): Japanese prescribing information. 2020. https://www.pmda.go.jp/PmdaSearch/iyakuDetail/230867_39990D6F1023_1_01#HDR_Warnings. Accessed 30 Sep 2020.
Gilead Sciences. Gilead receives complete response letter for filgotinib for the treatment of moderately to severely active rheumatoid arthritis [media release]. 18 Aug 2020. https://www.gilead.com/.
Gilead Sciences. European Medicines Agency validates marketing application for filgotinib for the treatment of ulcerative colitis [media release]. 2 Nov 2020. https://www.gilead.com/.
Abbott, Galapagos. Abbott and Galapagos announce global collaboration for novel oral therapy, GLPG0634, in phase II to treat autoimmune diseases [media release]. 29 Feb 2012. https://abbott.mediaroom.com.
Galapagos. AbbVie and Galapagos extend GLPG0634 collaboration to include Crohn's disease [media release]. 17 May 2013. https://news.abbvie.com.
Galapagos. Galapagos to advance filgotinib to Phase 3 in rheumatoid arthritis [media release]. 25 Sep 2015. https://www.glpg.com
Galapagos. Galapagos and Gilead announce global partnership to develop filgotinib for the treatment of rheumatoid arthritis and other inflammatory diseases [media release]. 17 Dec 2015. https://www.glpg.com
Galapagos. Galapagos and Gilead complete closing of their global collaboration for filgotinib [media release]. 19 Jan 2016. https://www.glpg.com
Galapagos. Galapagos exercises co-promotion option for filgotinib with collaboration partner Gilead Sciences in eight European countries [media release]. 31 Dec 2017. https://www.glpg.com
Galapagos. Galapagos delivered in 2017 [media release]. 22 Feb 2018. https://www.glpg.com.
Gilead Sciences. Gilead and Galapagos enter into transformative research and development collaboration [media release]. 14 Jul 2019. https://www.gilead.com.
Gilead Sciences. Gilead and Eisai enter into agreement in Japan for the co-promotion of the investigational rheumatoid arthritis therapy filgotinib, pending regulatory approval [media release]. 24 Dec 2019. https://www.gilead.com.
Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191(7):3568–77.
Galien R, Vayssiere B, De Vos S, et al. Analysis of the JAK1 selectivity of GLPG0634 and its main metabolite in different species, healthy volunteers and rheumatoid arthritis patients [abstract no. 478 plus poster]. Arthritis Rheum. 2013;65(Suppl 10):S209–10.
European Medicines Agency. Jyseleca: EPAR - public assessment report. 2020. https://www.ema.europa.eu/. Accessed 29 Oct 2020.
Namour F, Desrivot J, Van der Aa A, et al. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug-drug interactions. Drug Metab Lett. 2016;10(1):38–48.
Combe B, Kivitz A, Tanaka Y, et al. Efficacy and safety of filgotinib for patients with rheumatoid arthritis with inadequate response to methotrexate: FINCH 1 primary outcome results. Ann Rheum Dis. 2019;78(Suppl 2):78.
Combe B, Kivitz A, Tanaka Y, et al. Efficacy and safety of filgotinib for patients with rheumatoid arthritis with inadequate response to methotrexate: Finch 1 52-week results [abstract no. THU0198]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Genovese MC, Kalunian K, Gottenberg JE, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA. 2019;322(4):315–25.
Westhovens R, Rigby W, Van Der Heijde D, et al. Efficacy and safety of filgotinib for patients with rheumatoid arthritis Naive to methotrexate therapy: FINCH3 primary outcome results [abstract no. LB0003]. Ann Rheum Dis. 2019;78(Suppl 2):259–60.
Westhovens R, Rigby W, Van der Heijde D, et al. Efficacy and safety of filgotinib in methotrexate-naive patients with rheumatoid arthritis: finch 3 52-week results [abstract no. SAT0158]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis. 2017;76(6):998–1008.
Tanaka Y, Van Der Aa A, Jamoul C, et al. Rapid and sustained effect of filgotinib, an oral JAK1-selective inhibitor, on patient-reported outcomes: results from a phase 2b RA study [abstract no. FR120]. Int J Rheum Dis. 2018;21(Suppl 1):207.
Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. 2017;76(6):1009–19.
Genovese M, Westhovens R, Meuleners L, et al. Effect of filgotinib, a selective JAK 1 inhibitor, with and without methotrexate in patients with rheumatoid arthritis: patient-reported outcomes. Arthritis Res Ther. 2018;20(1):57.
Kavanaugh A, Westhovens R, Winthrop K, et al. Rheumatoid arthritis treatment with filgotinib: week 156 safety and efficacy data from a phase 2b open-label extension study [abstract no. 0550]. In: 83rd American College of Rheumatology and the 54th Association of Rheumatology Health Professionals Annual Scientific Meeting. 2019.
Mease P, Coates LC, Helliwell PS, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): results from a randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10162):2367–77.
Mease PJ, Gladman DD, Bosch FV, et al. Efficacy of filgotinib vs placebo in active psoriatic arthritis: patient-level data from equator, a randomized, phase 2 study [abstract no. OP0109]. Ann Rheum Dis. 2019;78(Suppl 2):128.
Helliwell P, Van den Bosch F, Coates LC, et al. Efficacy and safety of filgotinib, a selective janus kinase 1 inhibitor, in patients with active psoriatic arthritis: subgroup analyses from a randomized, placebo-controlled, phase 2 trial (Equator) [abstract no. FRI0343]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Orbai AM, Ogdie A, Gossec L, et al. Effect of filgotinib on health-related quality of life in active psoriatic arthritis: a randomized phase 2 trial (EQUATOR). Rheumatology (Oxford). 2020;59(7):1495–504.
Coates LC, Mease PJ, Gladman DD, et al. Effect of filgotinib on patient-reported outcomes in active psoriatic arthritis: results from equator, a randomized, phase 2 study [abstract no. SAT0373]. Ann Rheum Dis. 2019;78(Suppl 2):927–8.
Gladman DD, Coates LC, Van den Bosch F, et al. Long-term efficacy of the oral selective Janus Kinase 1 inhibitor filgotinib in psoriatic arthritis: week 52 response patterns in individual patients from an open-label extension (Ole) study (Equator2) [abstract no. FRI0339]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
van der Heijde D, Baraliakos X, Gensler LS, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10162):2378–87.
Maksymowych WP, stergaard M, Landewe RBM, et al. Impact of filgotinib on structural lesions in the sacroiliac joints at 12 weeks in patients with active axial spondyloarthritis: magnetic resonance imaging data from the double-blind, randomized TORTUGA trial [abstract no. THU0377]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Maksymowych WP, Tian Y, Yoon OK, et al. Filgotinib treatment results in reduction of biomarkers associated with disease in patients with ankylosing spondylitis [abstract no. FRI0285]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Feagan BG, Loftus Jr EV, Danese S, et al. Efficacy and safety of filgotinib as induction therapy for patients with moderately to severely active ulcerative colitis: results from the phase 2b/3 SELECTION study [abstract no. LB19]. In: UEG Week Online. 2020.
Peyrin-Biroulet L, Loftus Jr EV, Danese S, et al. Efficacy and safety of filgotinib as maintenance therapy for patients with moderately to severely active ulcerative colitis: results from the phase 2b/3 SELECTION study [abstract no. LB20]. In: UEG Week Online. 2020.
Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389(10066):266–75.
Leung WK, D’Haens G, Van Der Aa A, et al. Post-hoc analysis from the phase 2 FITZROY study with the selective JAK1 inhibitor filgotinib: effect of disease duration and location on clinical remission in Crohn’s disease patients [abstract no. OE-0731]. J Gastroenterol Hepatol. 2018;33(Suppl 4):63–4.
Keshav S, Roblin X, D’Haens G, et al. Filgotinib decreases inflammatory markers associated with endoscopic improvement in moderate to severely active Crohn’s disease [abstract no. PTU-003]. Gut. 2018;67(Suppl 1):A63–4.
Genovese MC, Winthrop K, Tanaka Y, et al. Integrated safety analysis of filgotinib treatment for rheumatoid arthritis from 7 clinical trials [abstract no. THU0202]. In: 21st Annual Congress of the European League Against Rheumatism. 2020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Sohita Dhillon is a contracted employee of Adis International Ltd/Springer Nature and Susan J. Keam is a salaried employee of Adis International Ltd/Springer Nature and declare no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
Enhanced material for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.13191509.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Dhillon, S., Keam, S.J. Filgotinib: First Approval. Drugs 80, 1987–1997 (2020). https://doi.org/10.1007/s40265-020-01439-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01439-0