
Abstract
Sepsis is a syndrome which is defined as a dysregulated host response to infection leading to organ failure. Since it remains one of the leading causes of mortality worldwide, numerous drug candidates have already been tested, and continue to be developed, as potential adjunct therapies. Despite convincing mechanisms of action and robust pre-clinical data, almost all drug candidates in the field of sepsis have failed to demonstrate clinical efficacy in the past two decades. Accordingly, the development of new sepsis drugs has markedly decreased in the past few years. Nevertheless, thanks to a better understanding of sepsis pathophysiology and pathways, new promising drug candidates are currently being developed. Instead of a unique sepsis profile as initially suspected, various phenotypes have been characterised. This has resulted in the identification of multiple targets for new drugs together with relevant biomarkers, and a better understanding of the most appropriate time to intervention. Within the entire sepsis drugs portfolio, those targeting the immune response are probably the most promising. Monoclonal antibodies targeting either cytokines or infectious agents are undoubtedly part of the potential successful therapeutic classes to come.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Identification of accurate biomarkers to better select appropriate phenotypes is essential to evaluate new drug candidates for the adjunctive treatment of sepsis |
More than 10 promising drugs are currently in clinical development in the sepsis field |
Immune response is probably one of the most appropriate targets to work on |
Several drugs, including monoclonal antibodies, are likely to be marketed in a near future |
1 Introduction
Sepsis is one of the leading causes of mortality worldwide [1]. This syndrome is defined as a dysregulated host response to infection leading to organ dysfunction and failure [2]. With the exception of a short-lived therapy, namely the administration of recombinant activated protein C for severe sepsis [3], the majority of studied drugs have failed to improve sepsis-related mortality despite numerous encouraging preclinical data. It was probably a conceptual error to consider that a single therapeutic strategy or medication would provide an equal benefit to such a heterogeneous population with multiple underlying comorbidities, variable clinical presentation and physiological reserve [4]. Negative trials that were designed to block cytokine-mediated hyperinflammation are particularly illustrative of this type of misconception [5]. Over the past few years, more attention has been directed towards the description of distinct biological phenotypes in septic patients, the characterisation of more homogenous subpopulations to explore, and the identification of more appropriate biomarkers to subsequently better select and evaluate new drug candidates for the treatment of sepsis [6, 7]. The development of new biomarkers should follow rigorous standardised methodology to help in selecting eligible patients for new therapies based on their expression, while guiding administration modalities of drug candidates according to their monitoring [8].
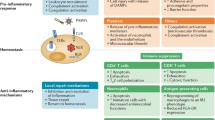
The present manuscript intends to describe the new developments and drugs currently tested to improve bacterial sepsis-induced organ dysfunction and patient-centred outcome. We did not perform a systematic review of the literature but rather chose to emphasise innovative and promising approaches in the field of sepsis, and to discuss drug candidates for which randomised, double-blinded, placebo-controlled, add-on trials (RCTs) were recently available. Both the pathophysiological approach and specific target used in sepsis pathway for the development of RCTs are described (Table 1). Drug candidates targeting pathogen-associated molecular patterns and resulting pro-inflammatory response to infectious insult, the coagulation system, the endothelium, end-organ damage, and host immune response are reviewed (Fig. 1).

Adapted from Azeredo da Silveira S, Shorr AF. Critical parameters for the development of novel therapies for severe and resistant infections-A case study on CAL02, a non-traditional broad-spectrum anti-virulence drug. Antibiotics (Basel). 2020;9(2):94
Schematic representation of the complex sepsis pathway and various targets of currently investigated drug candidates.
2 Drug Candidates Targeting Pathogen-Associated Molecular Patterns
These drug candidates include liposomal agents and antibacterial antibodies, especially directed against Staphylococcus aureus and Pseudomonas aeruginosa. Vaccine candidates have also been developed and have shown efficacy in inducing specific immunogenicity in critically ill patients [9, 10]. Nevertheless, pre-operative vaccination failed to efficiently prevent serious S. aureus infections after cardiothoracic surgery [10], and a P. aeruginosa vaccine was unsuccessful in reducing mortality or preventing invasive and respiratory tract infections due to P. aeruginosa in intensive care unit (ICU) ventilated patients [9].
2.1 Liposomal Agent
Numerous bacteria secrete toxins which damage or kill host cells in creating pores within cell membrane and plasmalemmal lipids degradation. Gram-positive bacteria like Streptococcus pneumonia and S. pyogenes secret pneumolysin and streptolysin O, respectively. Artificial liposomes have been built to sequester bacterial toxins [11]. In mice, the administration of these liposomes after S. pneumonia septicaemia improved survival and protected animals against invasive pneumococcal pneumonia. CAL02 is a novel antitoxin agent consisting of a mixture of liposomes creating large and stable liquid-ordered lipid microdomains able to trap bacterial toxins. This agent was recently tested for safety in a first-in-human RCT [12]. CAL02 has a half-life of eight hours and rapidly reaches the lung and fails to impair microcirculation due to its small size. In 19 patients admitted to an ICU for severe pneumococcal pneumonia, a low or high dose of CAL02 versus placebo was administered intravenously over 2 days in addition to standard antibiotic therapy. The drug was safe. Although the small sample size was inappropriate to detect any benefit on mortality, the reduction of organ dysfunction score was more pronounced in the intervention arms when compared to placebo. C-reactive protein and procalcitonin levels declined more rapidly in CAL02 groups. These data support the need for larger clinical trials to confirm the clinical value of the adjunct therapy by CAL02 for the treatment of severe infection [12].
2.2 Antibacterial Antibodies
Hospital-acquired pneumonia (HAP) and ventilator-associated pneumonia (VAP) are associated with significant morbidity and mortality [13]. They constitute the second leading type of nosocomial infection and the leading cause of death from nosocomial infection in the USA [14]. S. aureus and P. aeruginosa remain the primary cause of nosocomial pneumonia.
Despite the development of new antibiotics against most frequent pathogens, the emergence of bacterial resistance requires new therapeutic approaches. Monoclonal antibodies (mAbs) targeting virulence factors of causative bacteria represent probably one of the most promising therapeutic innovation in the ICU setting, when used either preventively or adjunctively to antibiotic therapy. For the pre-emptive approach, an early documentation of the pulmonary colonisation by a specific pathogen is required in order to dose the patient prior to the invasion phase. Accordingly, real-time polymerase chain reactions targeting single or multiple pathogens are performed serially on tracheobronchial samples. In certain clinical settings such as Clostridium difficile infection, mAbs (e.g. bezlotoxumab) could be administered as a single primary treatment [15]. Nevertheless, VAP being frequently multi-bacterial with numerous virulence factors involved, the actual mAbs portfolio is not large enough to support such a therapeutic approach.
2.2.1 Anti-Staphylococcus aureus Antibodies
MEDI4893 (suvratoxumab) is a human immunoglobulin G1 kappa mAb with an extended half-life that binds S. aureus alpha toxin with a high affinity, and effectively blocks alpha toxin-induced pore formation in target cell membranes [16]. Alpha toxin is expressed by 90% of S. aureus clinical isolates. In different murine infection models such as pneumonia, lethal bacteraemia or sepsis, and dermonecrosis, the prophylactic administration of this type of mAb reduces disease severity [17]. No adverse effect was observed in cynomolgus monkeys following the intravenous infusion of MEDI4893 at doses exceeding expected human therapeutic dosage.
In a Phase 1 RCT dose-escalation study to evaluate the safety, tolerability, and pharmacokinetics, MEDI4893 was administered as a single IV dose of 225, 750, 2250, or 5000 mg compared with placebo in 33 healthy adult subjects [18]. No serious adverse events or new-onset chronic diseases were reported. The MEDI4893 terminal half-life was estimated between 70.1 and 84.6 days. A recent Phase-2 RCT has tested a pre-emptive use of suvratoxumab in ICU mechanically ventilated patients colonised with S. aureus susceptible or resistant to methicillin. A single intravenous dose of suvratoxumab produced a trend toward a reduced incidence of S. aureus pneumonia in these high-risk ICU patients compared with placebo (26% in the placebo group vs 17.7% in the treatment group with a relative risk reduction of 31.9%), with acceptable safety [19].
AR-301 (KBSA301, Salvecin®) is a human immunoglobulin G1 monoclonal antibody also specific for the alpha-toxin of S. aureus. AR-301 binds to an N-terminal epitope of alpha-toxin, thereby preventing functional toxin pore oligomerisation. AR-301 is developed for passive immunotherapy in the treatment of S. aureus pneumonia as an adjunctive therapy to standard antibiotic treatment. AR-301 is expected to neutralise S. aureus alpha-toxin in situ, thereby reducing the ability of the exotoxin to damage the surrounding tissues and immune cells, irrespective of methicillin susceptibility. A Phase 2a dose escalation study in patients with severe S. aureus HAP, VAP, or community-acquired pneumonia (CAP), showed that AR-301 was safe to administer at doses of 1, 3, 10 and 20 mg/kg and had a pharmacokinetic profile consistent with that of IgG1 immunoglobulins [20]. In the subgroup of patients with VAP (n = 25), a trend towards shorter duration of ventilation (16.8 ± 8.4 days in the placebo group vs 9.5 ± 7.6 days in treatment groups) suggesting potentially faster resolution of pneumonia was observed. In this first small-size trial, there was no discernible difference in mortality rates (14.0% in treatment groups vs 6.3% in the placebo group: p = 0.40) or clinical cure rates between treatment and control groups (71.0% vs 87.5%: p = 0.39). A pivotal Phase 3 trial has just recently started, still using an adjunctive approach.
2.2.2 Anti-Pseudomonas aeruginosa Antibodies
MEDI3902 is a bivalent, bispecific human immunoglobulin G1 kappa mAb that selectively binds to both the PcrV protein and Psl exopolysaccharide on the surface of P. aeruginosa. It operates through three mechanisms of action. MEDI3902 prevents type 3 secretion injectisome-mediated cytotoxicity and damage to host cells, mediates opsonophagocytic killing of P. aeruginosa by host effector cells, and inhibits the bacterium’s attachment to host epithelial cells [21]. When administered prophylactically, MEDI3902 provided protection in four murine infection models (immunocompetent pneumonia, immunocompromised pneumonia, bacteraemia, and thermal injury). MEDI3902 provided synergistic enhancement of antibiotic therapy against P. aeruginosa pneumonia, without adverse effect on safety pharmacology parameters.
A Phase 1 dose escalation, single-centre, first-time in human RCT evaluated the safety and tolerability of MEDI3902 administered as a single IV dose of 250, 750, 1500, or 3000 mg compared with placebo in 56 healthy adult subjects [22]. A Phase 2 dose-ranging, proof-of-concept RCT evaluating MEDI3902 in mechanically ventilated patients colonised with P. aeruginosa in the lower respiratory tract in the ICU who are at high risk for VAP or HAP has recently been completed and results are expected soon.
3 Drug Candidates Targeting the Coagulation System
3.1 Thrombomodulin
ART-123 is a recombinant human soluble thrombomodulin, a glycoprotein with a molecular weight of approximately 64,000 Da. Experimental data suggest that ART-123 exhibits both an anti-coagulation and anti-inflammatory effect [23]. When binding to thrombin, ART-123 changes thrombin substrate specificity from pro-coagulant to anti-coagulant activity through the activation of protein C [24, 25]. ART-123 improves disseminated intravascular coagulation (DIC) parameters and exhibits inhibitory effects on pro-inflammatory cytokines in animal models of sepsis. Animal toxicology studies have shown no side effects beyond those expected for an anticoagulant.
Two Phase 1 studies performed in 20 healthy volunteers showed that ART-123 has a terminal half-life of 25–28 h when injected intravenously as a bolus, and is predominantly cleared by the kidney [26, 27]. In DIC patients with severe renal impairment, the decomposition of ART-123 may increase and compensate for reduced elimination by the kidneys. Accordingly, the clearance of ART-123 in patients with severe renal impairment appears to be similar to that of patients without such kidney failure [28]. A Phase 2b trial conducted in 371 septic patients with suspected DIC showed non-significant reduced mortality in patients allocated to ART123 versus placebo (28-day mortality: 17.8% vs 21.6%: p = 0.273) [29]. Post hoc analysis indicated that the reduction of mortality obtained with ART-123 was best predicted in the presence of at least one organ dysfunction, and sepsis-associated coagulopathy defined by an INR greater than 1.4 at baseline. The safety profile was consistent with the incidence rate of complications expected in such population [29].
A Phase 3 RCT designed to confirm the efficacy and safety of 0.06 mg/kg of ART-123 in patients with severe sepsis and coagulopathy has recently enrolled 800 patients [30]. The 28-day all-cause mortality was not significantly different between the ART-123 and the placebo group (106/395 [26.8%] vs 119/405 patients [29.4%]: p = 0.318). The absolute risk reduction of death in patients treated with ART-123 was 2.55% (95% CI − 3.68%/+ 8.77%). Post hoc analysis revealed a greater signal in the subset of patients who met sepsis-induced coagulopathy criteria at baseline, with an absolute risk reduction of death reaching 5.40% (95% CI − 1.68%/+ 12.48%). The incidence of major bleeding events was 5.8% in the ART-123 group and 4.0% in the placebo group [30].
After numerous pre-clinical studies, 14 clinical trials enrolled approximately 1300 patients exposed to ART-123. In 2008, ART-123 was approved for the treatment of DIC in Japan and approximately 180,000 patients have received the drug (Recomodulin™). Overall, ART-123 has been shown to be safe at effective doses. Currently, despite the recent negative Phase 3 trial, ART-123 remains approved and largely used in Japan for sepsis-associated coagulopathy.
4 Drug Candidates Targeting the Endothelium
4.1 Adrenomedullin-Targeted Therapy
Sepsis is associated with endothelial dysfunction [31]. This results in vascular leak with associated tissue edema, microvascular thrombosis, and ultimately participates in the development of sepsis-induced organ failure [32]. Adrenomedullin (ADM) is a short half-life free circulating peptide mainly expressed by endothelial and vascular smooth muscle cells [33]. High levels of ADM induce vasodilatation and hypotension [33], while reinforcing the endothelial barrier, thus reducing sepsis-induced vascular leakage [34]. Patients with septic shock exhibit higher ADM levels than those without shock [35], and plasma ADM concentration is associated with organ failure and mortality in sepsis [36, 37].
Adrecizumab is a recombinant monoclonal anti-ADM antibody that protects endogenous ADM from proteolytic decay and hence increases its plasma half-life in a dose-dependent manner [38]. Proposed mechanism of action of adrecizumab relies on the modulation of ADM equilibrium between the blood compartment and interstitium [38]. Adrecizumab is restricted to the blood compartment, whereas ADM freely diffuses across the endothelium in the extravascular space [39]. Accordingly, adrecizumab may induce a shift of ADM from the interstitium to the circulation; hence, reducing ADM-mediated vasodilatation [38]. In addition, the increased and prolonged ADM-adrecuzimab plasma concentration increases its endothelial barrier stabilising effects [40]. These beneficial effects are anticipated to be even more pronounced in the presence of elevated levels of circulating ADM, suggesting high associated concentration of ADM within the interstitial space.
Intravenous injection of adrecizumab induces dose-dependent peak concentration, significantly increases circulating concentration of ADM, and is well tolerated across non-human species [41]. Adrecuzimab also showed an excellent safety profile in Phase 1 human studies, both in the absence and presence of systemic inflammation [42]. A proof-of-concept and dose-finding Phase 2 RCT to investigate the safety, tolerability and efficacy of adrecizumab in patients with septic shock and elevated ADM levels (plasma concentration > 70 pg/mL) has recently been completed. The AdrenOSS-2 trial that enrolled 300 patients randomised to receive adrecizumab (2 and 4 mg/kg) or placebo in a 1:1:2 ratio has been recently completed [43].
4.2 Selepressin
Vascular responsiveness to vasoconstrictor agents is blunted in septic shock, and a high dose of vasopressors is usually required to maintain adequate blood pressure [39]. Selepressin is highly selective for the V1a- versus V2-receptor when compared to vasopressin [44]. In addition, it appears to prevent microvascular leak and associated positive fluid balance in ovine models of sepsis [45, 46]. For these reasons, selepressin may be preferable to vasopressin in septic shock [47]. In experimental studies, selepressin has been shown to more efficiently increase and stabilise mean arterial pressure, at lower doses than vasopressin [48], and to increase survival rates [49].
In a Phase 2a RCT, selepressin was administered at different doses in 53 patients with early septic shock [50]. Selepressin was well tolerated, allowed reducing the doses of norepinephrine, and appeared to diminish fluid balance and shorten the duration of mechanical ventilation [50]. Accordingly, a Phase 2b/3 RCT was conducted, evaluating up to four selepressin dosing strategies [51]. An adaptive trial design was used to (1) determine the efficacy of multiple dosing regimens of selepressin (part 1, dose-finding), and (2) to confirm the efficacy of a dosing regimen in the treatment of septic shock (part 2, confirmation). Patients were assigned to receive one of three dosing regimens of selepressin (starting infusion rates of 1.7, 2.5 and 3.5 ng/kg/min; n = 585), or placebo (n = 283) as continuous infusions titrated according to hemodynamic monitoring [52]. Ventilator- and vasopressor-free days did not differ significantly between combined selepressin and placebo groups (mean 15.0 vs 14.5 days: p = 0.30). Over the first 6 h, the selepressin group had a significantly higher mean arterial pressure (mean 74 vs 70 mmHg: p > 0.001) and lower norepinephrine requirement (mean 0.29 µg/kg/min vs 0.48 µg/kg/min: p < 0.001). During the first 24 h, patients receiving selepressin had a significantly lower net fluid balance than those who received placebo (81 vs 107 mL/h: p < 0.001), but the groups were similar thereafter [52].
5 Drug Candidates Targeting End-Organ Damage
5.1 Alkaline Phosphatase
Sepsis-induced acute kidney injury significantly increases mortality and is more commonly associated with long-term end-stage kidney disease. Sepsis-associated inflammatory process alters the renal microcirculation by a leukocyte infiltration, induces tubular lesions and renal cell apoptosis. Alkaline phosphatase is an endogenous enzyme exerting detoxifying effects and protecting against renal inflammation through dephosphorylation of bacterial lipopolysaccharide but also proinflammatory mediators including adenosine triphosphate [53].
A first clinical trial performed in patients with sepsis and septic shock demonstrated an improvement of renal function in the population treated with alkaline phosphatase [54]. A lower nitric oxide production with subsequent lesser renal damage was the likely mechanism associated with this benefit. A second Phase 2 study including patients with sepsis-induced acute kidney injury supported the potential benefits of alkaline phosphatase on renal function [55]. Based on these results, together with preclinical data, a human recombinant form of alkaline phosphatase was developed and secondarily tested in a larger adaptive Phase 2a/2b study that included 301 patients admitted to ICU with sepsis and acute kidney injury [56]. The primary endpoint was the time-corrected area under the curve of the endogenous creatinine clearance for the first 7 days after randomisation. Human recombinant alkaline phosphatase did not improve short-term kidney function. However, on Days 21 and 28, the endogenous creatinine clearance was significantly higher in the alkaline phosphatase group compared with placebo. In addition, a significant benefit on the 90-day all-cause mortality was observed (17.1% in recombinant alkaline phosphatase group compared with 29% in the placebo group) and may be explained by the longer-term benefit on the kidney function [56]. A large RCT comparing human recombinant alkaline phosphatase with placebo in patients with sepsis-induced acute kidney injury will target improved survival as the primary end point in the near future.
6 Drug Candidates Targeting Host Immune Response
6.1 Soluble TREM-1 and Nangibotide
The Triggering Receptor Expressed on Myeloid cells-1 (TREM-1) is an immunomodulatory receptor expressed on innate immune cells such as mature monocytes and neutrophils [57]. While TREM-1 is expressed at a low level in physiologic conditions, it amplifies systemic inflammation with exuberant and hyperactivated immune state in response to an acute infectious aggression [58, 59]. TREM-1 pathway is one of the most upregulated pathways [60, 61]. Accordingly, it plays a central role in the pathogenesis of sepsis, in linking innate to adaptive immunity. After activation by toll-like receptor agonists, TREM-1 expressing cells such as monocytes have higher ability to recruit and induce proliferation of B and T cells [62, 63]. TREM-1 also exists in a soluble form (sTREM-1) in the blood following cleavage of membrane-bound TREM-1 by metalloproteinases after TREM-1 receptor activation [64]. sTREM-1 levels correlate with the severity of organ dysfunction assessed by the sepsis-related organ failure assessment (SOFA) score [65], and with 28-day mortality [66].
Nangibotide is a synthetic TREM-1 antagonistic peptide that inhibits the activation of the TREM-1 receptor, thus decreasing leukocyte activation and innate immune response [67]. Accordingly, nangibotide represents a new class of drug that controls innate immunity and appears as a novel mechanism-based treatment for sepsis. In experimental models of sepsis, the administration of nangibotide was associated with reduced inflammatory response, and protective effects on the cardiovascular system and survival [68].
Study MOT-C-104 was the first administration of nangibotide in healthy volunteers to assess its safety, tolerability and pharmacokinetics [69]. Nangibotide was safe and well tolerated up to the highest dose tested (5 mg/kg bolus followed by 6.0 mg/kg/h continuous infusion for 7.75 h). No drug-related adverse events were observed and no anti-drug antibodies were detected. No pharmacodynamic effects of MOT-C-104 were observed since the TREM-1 signalling pathway is only active in an activated immune system. Nangibotide had a short half-life (≈ 3 min) and a clearance rate compatible with extensive enzymatic metabolism in blood [69].
Study MOT-C-201 was the first administration of nagibotide in septic shock patients through a multicentre RCT [70]. Patients received either a continuous infusion of nangibotide (0.3, 1.0, or 3.0 mg/kg/h, after a 5 mg/kg loading dose) or placebo. Treatment began within 24 h of shock onset and continued until 12 h after cessation of vasopressors or up to 5 days. Baseline characteristics and both the frequency and types of adverse events were comparable between groups. No patient developed anti-drug antibodies. In patients with high baseline sTREM-1 levels, nangibotide tended to reduce organ dysfunction, ICU length of stay and organ support requirement when compared to placebo. Decrease in SOFA score from baseline to Day 5 in pooled nangibotide groups versus placebo was − 0.7 ± 0.85 in the randomised population and − 1.5 ± 1.12 in patients with high baseline plasma sTREM-1 concentrations (non-significant). In this Phase 2 trial, nangibotide was safe and well tolerated [71]. Accordingly, it appears as a promising drug, particularly in patients with elevated baseline sTREM-1 levels and a larger Phase 2b aiming to enrol 400 patients is ongoing.
6.2 Interleukin-7
One of the most promising new adjuvants to counteract sepsis-induced immunosuppression is interleukin-7 (IL-7), a pluripotent cytokine produced by bone marrow and thymic stromal cells [71, 72]. The current glycosylated form of recombinant human IL‑7 (RhIL-7) used in clinical trials is CYT-107. IL-7 can reverse sepsis-induced lymphopenia, while avoiding excessive inflammatory response [73, 74]. IL-7 harbours anti-apoptotic properties [75], induces proliferation of naïve and memory T-cells [76], restores the functional capacity of hyporesponsive or exhausted T-cells, increases the expression of cell adhesion molecules [77], and reverses sepsis-induced depression of interferon gamma (IFN-γ) [75].
In an animal model using a P. aeruginosa pneumonia as a second infectious hit after a treated cecal ligation and puncture peritonitis, Shindo et al. [78] showed that RhIL-7 was effective in preventing death. In septic patient, RhIL-7 substantially improved immune function with an increase in T-lymphocyte proliferation, IFN-γ production, and B Cell lymphoma 2 (BCL2) production, which is a protein that enhances lymphocytes survival [79].
In a first Phase 2 trial conducted in patients surviving a vasopressor-dependent sepsis, the intramuscular injection of two regimen doses from five (low doses) to eight (high doses) injections of 10 μg/kg CYT-107 over a maximum of four weeks restored absolute lymphocyte count (ALC). Compared to baseline, ALC had a 3.5-fold increase (0.99 × 103 lymphocytes/μL; 95% CI 0.14–2.13) in patients receiving low-dose regimen to a 4.5-fold increase (1.30 × 103 lymphocytes/μL; 95% CI 0.20–2.40) in those patients with high-dose regimen [80]. CYT-107 was well tolerated without evidence of induced cytokine storm, worsened inflammation, or organ dysfunction. The 3.5- to 4.5-fold increase in ALC and in circulating CD4 and CD8 T-cells persisted at least four weeks after the last drug administration. CYT-107 also increased T-cell proliferation and activation. The only adverse effect of CYT-107 was a grade 2–3 injection site reaction that occurred in 6 of 17 patients treated with the drug [80]. To avoid this site reaction, a second Phase 2b (IRIS-7 CD ClinicalTrials.gov: NCT03821038) is currently investigating both the efficacy and tolerance of the intravenous administration of CYT-107 in a similar sepsis population.
6.3 Antibodies Against Programmed Cell Death Protein 1 and Programmed Cell Death 1 Ligand 1
Nivolumab is a fully human monoclonal antibody directed against programmed cell death-1 (PD-1), a CD28 family receptor. PD-1 is a negative regulatory molecule highly expressed on activated T and B cells. Two ligands specific for PD-1 have been identified: programmed death-ligands 1 (PD-L1) and PD-L2. Both have been shown to down-regulate T-cell activation upon binding to PD-1 in mice and humans [81]. PD-1/PD-L1 pathway is considered as an immune checkpoint inhibitor. Nivolumab inhibits PD-1/PD-L1 interaction resulting in enhanced interferon-gamma production and promotes immune responses and antigen-specific T-cell responses [82].
Guignant et al. [83] showed that patients suffering from sepsis had increased PD-1 and PD-L1 expression on immune cells. This increase was associated with the occurrence of secondary infections and mortality [83]. Moreover, blocking PD-1/PD-L1 pathway with anti-PD-1 or anti-PD-L1 antibodies in septic patients decreased apoptosis and improved immune T-cell function [84]. The capacity of such antibodies to improve survival in animal models of sepsis and to restore T-cell function in septic patient is also related to the restoration of neutrophil and monocyte functions [85].
In a Phase 1b RCT evaluating the safety of increased doses of nivolumab (from 10 to 900 mg), Hotchkiss et al. [86] showed that PD-1/PD-L1 pathway inhibition was safe in patients with sepsis-induced immunosuppression. After high doses of nivolumab (300 and 900 mg), mHLA-DR expression remained high until Day 30. No relation was evidenced between ALC and cytokine level in the nivolumab group, irrespective of the dose administered [86]. Recently, a Phase 1/2 study evaluated two regimen of a single dose (480 mg or 960 mg) of intravenous nivolumab in patients with vasopressor-dependent sepsis and a low ALC (≤ 1100/μL) [87]. In this small study population of 13 patients, nivolumab seemed to improve ALC and mHLA-DR expression [87]. Combination immunotherapy associating nivolumab and interferon-gamma may also be considered as rescue therapy in refractory fungal infection, but its clinical value remains to be confirmed [88].
6.4 Stem Cells
The potential roles and benefits of stem cells in the treatment of sepsis have been explored mainly in preclinical models. Cx611 is an intravenously administered product of allogenic adipose-derived stem cells (eASCs) in development for sepsis and early rheumatoid arthritis. Stem cells are potent regulators of the immune response, able to inhibit T cell activation, B cell function and dendritic cell maturation. Several studies in animal models of sepsis have showed the efficacy of eASCs on mortality by a combination of reduced inflammation, release of anti-microbial effectors and an increased phagocytosis [89]. In a mice model of sepsis, the same cells decreased the mortality to 40% compared with 100% in the untreated septic group, inhibited splenocytes apoptosis, decreased pro-inflammatory cytokines and increased IL-10 [90]. In a model of pneumonia, mice treated with mesenchymal stem cells (MSCs) exhibited a reduced bacterial count, probably secondary to the release of the anti-microbial peptide LL-37 by the cells [91].
A Phase 1 study in healthy volunteers has explored the safety, tolerability and effects of Cx611 after an LPS challenge [92]. Three different doses (250,000 cells/kg, 1 and 4 million cells/kg) were compared to placebo. The safety of this administration of eASCs was confirmed and few adverse events were reported. However, vital signs and symptoms were not influenced by eASCs except for the febrile response in the high dose group. The laboratory measurements revealed enhanced levels of pro-inflammatory interleukins IL-6, IL-8 and anti-inflammatory IL-10 and tumour growth factor β (TGF-β) in the 4 million cells/kg compared with placebo. The limited effect of eASCs after a low dose of LPS may be potentially explained by the too brief and limited immune response in this model unable to detect the activity of Cx611. A Phase 2b/2a study assessing the safety of two doses of Cx611 on Day 1 and Day 3 (160 million cells per infusion) in patients with severe CAP has been completed recently (ref. NCT03158727). The secondary objective of this study is to explore the efficacy of these cells to treat severe CAP. Immunological follow-up of pro- and anti-inflammatory cytokines will be performed and help for a better understanding of the mode of action of eACSs. The results of the study should be presented shortly.
7 Conclusion
Various drug candidates for the treatment of sepsis are currently developed and tested in add-on RCTs. Through different targets, these potential adjunct therapies aim at reducing organ dysfunction, hence improving patient-centred outcome. Better characterisation of distinct sepsis phenotypes to allow a more individualised approach, identification of relevant biomarkers and determination of the most appropriate time for intervention promise to better address the complexity of sepsis, and to ultimately improve the prognosis of this highly lethal prevalent condition. We can speculate that in the near future, variable associations of drug interventions according to septic patients’ phenotype will be proposed in a timely fashion, similar to the currently developed therapeutic approach in cancer patients.
References
Fleischmann C, Scherag A, Adhikari NK, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–72.
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–10.
Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709.
Opal SM, Dellinger RP, Vincent JL, Masur H, Angus DC. The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C?*. Crit Care Med. 2014;42(7):1714–21.
Hotchkiss RS, Opal SM. Activating immunity to fight a foe—a new path. New Engl J Med. 2020;382(13):1270–2.
Reinhart K, Bauer M, Riedemann NC, Hartog CS. New approaches to sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev. 2012;25(4):609–34.
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–20.
Pierrakos C, Velissaris D, Bisdorff M, Marshall JC, Vincent JL. Biomarkers of sepsis: time for a reappraisal. Crit Care. 2020;24(1):287.
Adlbrecht C, Wurm R, Depuydt P, et al. Efficacy, immunogenicity, and safety of IC43 recombinant Pseudomonas aeruginosa vaccine in mechanically ventilated intensive care patients-a randomized clinical trial. Crit Care. 2020;24(1):74.
Fowler VG, Allen KB, Moreira ED, et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA. 2013;309(13):1368–78.
Henry BD, Neill DR, Becker KA, et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat Biotechnol. 2015;33(1):81–8.
Laterre PF, Colin G, Dequin PF, et al. CAL02, a novel antitoxin liposomal agent, in severe pneumococcal pneumonia: a first-in-human, double-blind, placebo-controlled, randomised trial. Lancet Infect Dis. 2019;19(6):620–30.
Bassi GL, Ferrer M, Marti JD, Comaru T, Torres A. Ventilator-associated pneumonia. Semin Respir Crit Care Med. 2014;35(4):469–81.
Infectious Diseases Society of America; American College of Chest Physicians; American Thoracic Society; Society of Critical Care Medicine, Spellberg B, Talbot G. Recommended design features of future clinical trials of antibacterial agents for hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis. 2010;51 Suppl 1(Suppl 1):S150–70.
Lee Y, Lim WI, Bloom CI, Moore S, Chung E, Marzella N. Bezlotoxumab (Zinplava) for clostridium difficile infection: the first monoclonal antibody approved to prevent the recurrence of a bacterial infection. Pharm Ther. 2017;42(12):735–8.
Tkaczyk C, Hua L, Varkey R, et al. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin Vaccine Immunol. 2012;19(3):377–85.
Hua L, Hilliard JJ, Shi Y, et al. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob Agents Chemother. 2014;58(2):1108–17.
Yu XQ, Robbie GJ, Wu Y, et al. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob Agents Chemother. 2016;61(1):e01020–e1116.
Francois B, Sánchez García M, Eggimann P, et al. Efficacy and safety results of the SAATELLITE phase 2 study of Suvratoxumab, a Staphylococcus aureus alpha toxin-neutralizing human monoclonal antibody in mechanically ventilated patients in the Intensive Care Units. Intensive Care Med Exp. 2019;7(Suppl 3):000478.
François B, Mercier E, Gonzalez C, et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med. 2018;44(11):1787–96.
DiGiandomenico A, Warrener P, Hamilton M, et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Ps1 by phenotypic screening. J Exp Med. 2012;209(7):1273–87.
Ali SO, Yu XQ, Robbie GJ, et al. Phase 1 study of MEDI3902, an investigational anti-Pseudomonas aeruginosa PcrV and Psl bispecific human monoclonal antibody, in healthy adults. Clin Microbiol Infect. 2019;25(5):629.e1–.e6.
Abeyama K, Stern MD, Ito Y, et al. The N- terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267–74.
Mohri M, Sugimoto E, Sata M, Asano T. The inhibitory effect of recombinant human soluble thrombomodulin on initiation and extension of coagulation—a comparison with other anticoagulants. Thromb Haemost. 1999;82(6):1687–93.
Tawara S, Sakai T, Matsuzaki O. Anti-inflammatory and anti-fibrinolytic effects of thrombomodulin alfa through carboxypeptidase B2 in the presence of thrombin. Thromb Res. 2016;147:72–9.
Nakashima M, Uematsu T, Umemura K, Maruyama I, Tsuruta K. A novel recombinant soluble human thrombomodulin, ART-123, activates the protein C pathway in healthy male volunteers. J Clin Pharmacol. 1998;38(6):540–4.
Nakashima M, Kanamaru M, Umemura K, Tsuruta K. Pharmacokinetics and safety of a novel recombinant soluble human thrombomodulin, ART-123, in healthy male volunteers. J Clin Pharmacol. 1998;38(1):40–4.
Hayakawa M, Yamamoto H, Honma T, et al. Pharmacokinetics and pharmacodynamics of recombinant soluble thrombomodulin in disseminated intravascular coagulation patients with renal impairment. Shock. 2012;37(6):569–73.
Vincent JL, Ramesh MK, Ernest D, et al. A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41(9):2069–79.
Vincent JL, Francois B, Zabolotskikh I, et al. Effect of a recombinant human soluble thrombomodulin on mortality in patients with sepsis-associated coagulopathy: the SCARLET randomized clinical trial. JAMA. 2019;321(20):1993–2002.
Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. 2010;363(7):689–91.
Reuter DA, Chappell D, Perel A. The dark sides of fluid administration in the critically ill patient. Intensive Care Med. 2018;44(7):1138–40.
Kitamura K, Kangawa K, Kawamoto M, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192(2):553–60.
Temmesfeld-Wollbrück B, Brell B, David I, et al. Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med. 2007;33(4):703–10.
Caironi P, Latini R, Struck J, et al. Circulating biologically active adrenomedullin (bio-ADM) predicts hemodynamic support requirement and mortality during sepsis. Chest. 2017;152(2):312–20.
Marino R, Struck J, Maisel AS, Magrini L, Bergmann A, Di Somma S. Plasma adrenomedullin is associated with short-term mortality and vasopressor requirement in patients admitted with sepsis. Crit Care. 2014;18(1):R34.
Mebazaa A, Geven C, Hollinger A, et al. Circulating adrenomedullin estimates survival and reversibility of organ failure in sepsis: the prospective observational multinational Adrenomedullin and outcome in sepsis and septick shock-1 (AdrenOSS-1) study. Crit Care. 2018;22(1):354.
Geven C, Bergmann A, Kox M, Pickkers P. Vascular effects of adrenomedullin and the anti-adrenomedullin antibody adrecizumab in sepsis. Shock. 2018;50(2):132–40.
Dupuis J, Caron A, Ruel N. Biodistribution, plasma kinetics and quantification of single-pass pulmonary clearance of adrenomedullin. Clin Sci. 2005;109(1):97–102.
Geven C, Pickkers P. The mechanism of action of the adrenomedullin-binding antibody adrecizumab. Crit Care. 2018;22(1):159.
Geven C, Kox M, Blet A, et al. Preclinical safety evaluation of the adrenomedullin-binding antibody Adrecizumab in reodents, dogs and non-human primates. Toxicol Appl Pharmacol. 2019;369:1–16.
Geven C, Van Lier D, Blet A, et al. Safety, tolerability and pharmacokinetics/pharmacodynamics of the adrenomedullin antibody adrecizumab in a first-in-human study and during experimental human endotoxaemia in healthy subjects. Br J Clin Pharmacol. 2018;84(9):2129–41.
Geven C, Blet A, Kox M, et al. A double-blind, placebo-controlled, randomised, multicentre, proof-of-concept and dose-finding phase II clinical trial to investigate the safety, tolerability and efficacy of Adrecizumab in patients with septic shock and elevated adrenomedullin concentration (AdrenOSS-2). BMJ Open. 2019;9(2):e024475.
Levy B, Fritz C, Jacquot A, Auchet T, Kimmoun A. Vasoplegia treatments: the past, the present, and the future. Crit Care. 2018;22(1):52.
Maybauer MO, Maybauer DM, Enkhbaatar P, et al. The selective vasopressin type 1a receptor agonist selepressin (FE 202158) blocks vascular leak in ovine severe sepsis. Crit Care Med. 2014;42(7):e525–e533533.
Rehberg S, Yamamoto Y, Sousse L, et al. Selective V(1a) agonism attenuates vascular dysfunction and fluid accumulation in ovine severe sepsis. Am J Physiol Heart Circ Physiol. 2012;303(10):H1245–H12541254.
O’Callaghan DJP, Gordon AC. What’s new in vasopressin? Intensive Care Med. 2015;41(12):2177–9.
Rehberg S, Ertmer C, Vincent JL, et al. Role of selective V1a receptor agonism in ovine septic shock. Crit Care Med. 2011;39(1):119–25.
He X, Su F, Taccone FS, et al. A selective V(1A) receptor agonist, selepressin, is superior to arginine vasopressin and to norepinephrine in ovine septic shock. Crit Care Med. 2016;44(1):23–31.
Russell JA, Vincent JL, Kjolbye AL, et al. Selepressin, a novel selective vasopressine V1A agonist, is an effective substitute for norepinephrine in a phase IIa randomized, placebo-controlled trial in septic shock patients. Crit Care. 2017;21(1):213.
Lewis RJ, Angus DC, Laterre PF, et al. Rationale and design of an adaptive phase 2b/3 clinical trial of selepressin for adults in septic shock. Ann Am Thorac Soc. 2018;15(2):250–7.
Laterre PF, Berry SM, Blemings A, et al. Effect of selepressin vs placebo on ventilator- and vasopressor-free days in patients with septic shock: the SEPSIS-ACT randomized clinical trial. JAMA. 2019;322(15):1476–85.
Picher M, Burch LH, Hirsh AJ, Spychala J, Boucher RC. Ecto 5'-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem. 2003;278(15):13468–79.
Heemskerk S, Masereeuw R, Moesker O, et al. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit Care Med. 2009;37(2):417–23, e1.
Pickkers P, Heemskerk S, Schouten J, et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit Care. 2012;16(1):R14.
Pickkers P, Mehta RL, Murray PT, et al. Effect of human recombinant alkaline phosphatase on 7-day creatinine clearance in patients with sepsis-associated acute kidney injury: a randomized clinical trial. JAMA. 2018;320(19):1998–2009.
Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164(10):4991–5.
Gibot S, Buonsanti C, Massin F, et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun. 2006;74(5):2823–30.
Gibot S, Massin F, Marcou M, et al. TREM-1 promotes survival during septic shock in mice. Eur J Immunol. 2007;37(2):456–66.
Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032–7.
Xiao W, Mindrinos MN, Seok J, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208(13):2581–90.
Bleharski JR, Kiessler V, Buonsanti C, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol. 2003;170(7):3812–8.
Boufenzer A, Lemarié J, Simon T, et al. TREM-1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ Res. 2015;116(11):1772–82.
Gomez-Piña V, Soares-Schanoski A, Rodriguez-Rojas A, et al. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J Immunol. 2007;179(6):4065–73.
Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–10.
Su L, Liu D, Chai W, Liu D, Long Y. Role of sTREM-1 in predicting mortality of infection: a systematic review and meta-analysis. BMJ Open. 2016;6(5):e010314.
Derive M, Bouazza Y, Sennoun N, et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J Immunol. 2012;188(11):5585–92.
Derive M, Massin F, Gibot S. Effects of a TREM-like Transcript-1-derived peptide during hypodynamic shock in pigs. Shock. 2013;39(2):176–82.
Cuvier V, Lorch U, Witte S, et al. A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition. Br J Clin Pharmacol. 2018;84(10):2270–9.
François B, Wittebole X, Ferrer R, et al. Nangibotide in patients with septic shock: a Phase 2a randomized controlled clinical trial. Intensive Care Med. 2020. https://doi.org/10.1007/s00134-020-06109-z.
Sprent J, Surh CD. Interleukin 7, maestro of the immune system. Semin Immunol. 2012;24(3):149–50.
Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 2018;14(2):121–37.
Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11(5):330–42.
Hotchkiss RS, Opal S. Immunotherapy for sepsis—a new approach against an ancient foe. N Engl J Med. 2010;363(1):87–9.
Unsinger J, McGlynn M, Kasten KR, et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 2010;184(7):3768–79.
Lévy Y, Sereti I, Tambussi G, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. 2012;55(2):291–300.
Cimbro R, Vassena L, Arthos J, et al. IL-7 induces expression and activation of integrin α4β7 promoting naive T-cell homing to the intestinal mucosa. Blood. 2012;120(13):2610–9.
Shindo Y, Fuchs AG, Davis CG, et al. Interleukin 7 immunotherapy improves host immunity and survival in a two-hit model of Pseudomonas aeruginosa pneumonia. J Leukoc Biol. 2017;101(2):543–54.
Venet F, Foray AP, Villars-Méchin A, et al. IL-7 restores lymphocyte functions in septic patients. J Immunol. 2012;189(10):5073–81.
Francois B, Jeannet R, Daix T, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. 2018;3(5):e98960.
Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239–45.
Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206–10.
Guignant C, Lepape A, Huang X, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. 2011;15(2):R99.
Chang K, Svabek C, Vazquez-Guillamet C, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. 2014;18(1):R3.
Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. 2016;100(6):1239–54.
Hotchkiss RS, Colston E, Yende S, et al. Immune checkpoint inhibition in sepsis: a phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (BMS-936559). Crit Care Med. 2019;47(5):632–42.
Watanabe E, Nishida O, Kakihana Y, et al. Pharmacokinetics, Pharmacodynamics and Safety of Nivolumab in Patients With Sepsis-induced immunosuppression: A multicenter, open-label phase 1/2 study. Shock. 2020;53(6):686–94.
Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL. Nivolumab plus interferon-γ in the treatment of intractable mucormycosis. Lancet Infect Dis. 2017;17(1):18.
Lombardo E, van der Poll T, DelaRosa O, Dalemans W. Mesenchymal stem cells as a therapeutic tool to treat sepsis. World J Stem Cells. 2015;7(2):368–79.
Gonzalez-Rey E, Anderson P, González MA, Rico L, Büscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. 2009;58(7):929–39.
Krasnodembskaya A, Song Y, Fang X, et al. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;28(12):2229–38.
Perlee D, Lonneke A, Vught Van, et al. Intravenous infusion of human adipose mesenchymal stem cells modifies the host response to polysaccharide in humans: a randomized single blind prallel group placebo-controlled trial. Stem Cells. 2018;36(11):1778–88.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding.
Conflict of interest
All authors declare that they have no conflict of interest.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Author contributions
PV, PFL, TD and BF collected the data, performed the literature search, drafted the manuscript and critically reviewed it. They all read and approved the final version of the manuscript.
Rights and permissions
About this article

Cite this article
Vignon, P., Laterre, PF., Daix, T. et al. New Agents in Development for Sepsis: Any Reason for Hope?. Drugs 80, 1751–1761 (2020). https://doi.org/10.1007/s40265-020-01402-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01402-z