
Abstract
Intravenous (IV) and subcutaneous (SC) tocilizumab (RoActemra®), an IL-6 receptor antagonist, are approved (± methotrexate) in numerous countries throughout the world, for the treatment of adults with moderate to severe active rheumatoid arthritis (RA). Extensive clinical experience has firmly established the short- and long-term efficacy and safety of tocilizumab [monotherapy or in combination with conventional synthetic DMARDs (csDMARDs)] in adults with early-stage and longer-duration established RA. In the clinical trial and real-world settings, tocilizumab monotherapy or combination therapy provided rapid and sustained improvements in clinical and radiographic outcomes and health-related quality of life. The safety profile of tocilizumab is consistent over time and, in general, is consistent with that of other immunomodulatory agents. This narrative review, written from an EU perspective, summarizes the clinical use of IV and SC tocilizumab in RA. Given its low risk of immunogenicity, the flexibility of IV and SC administration and the convenience of the once-weekly, self-administered, SC regimen, tocilizumab provides an effective treatment for severe, active and progressive RA in adults not previously treated with methotrexate and an effective biologic first- or subsequent-line treatment for moderate to severe active RA in adults who have either responded inadequately to or were intolerant of previous therapy with ≥ 1 csDMARD or TNF inhibitor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Available as IV and SC formulations; convenience of SC formulation permits once-weekly self-administration |
Well-established efficacy based on extensive experience in the clinical trial and real-world settings |
SC and IV formulations exhibit similar efficacy |
As monotherapy or combination therapy, provides rapid, sustained improvements in clinical and radiographic outcomes and HRQOL in both early-stage and established RA |
Safety profile during short- and long-term therapy is consistent over time and, in general, with that of other immunomodulatory agents; exhibits low immunogenicity |
1 Introduction
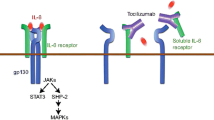
Extensive clinical experience over the past decade in the clinical trial and real-world settings has firmly established the efficacy of intravenous (IV) tocilizumab (RoActemra®) in the treatment of adult patients with rheumatoid arthritis (RA; reviewed previously in Drugs [1]). In the EU [2] and elsewhere, tocilizumab is also available as a subcutaneous (SC) formulation. The pharmacological properties of tocilizumab, a humanized monoclonal antibody that acts as an IL-6 receptor antagonist, have been reviewed in detail [1] and are summarized in Table 1. IL-6, a pleiotropic pro-inflammatory cytokine, is involved in diverse physiological processes and has been implicated in the pathogenesis of RA. This narrative review, written from an EU perspective, focuses on the clinical use of IV and SC tocilizumab, as monotherapy or in combination with conventional synthetic DMARDs (csDMARDs), in adults with moderate to severe, active RA, both in early-stage and longer-duration established disease. Tocilizumab is also approved for use in systemic juvenile idiopathic arthritis, juvenile idiopathic polyarthritis and giant cell arteritis in adults [2, 3], with discussion of these indications beyond the scope of this review.
2 Therapeutic Efficacy
2.1 Intravenous Tocilizumab
2.1.1 In Clinical Trials
The efficacy of IV tocilizumab monotherapy or combination therapy with csDMARDs in improving disease activity, structural joint damage and health-related quality of life (HRQOL) in adult patients with moderate to severe active RA was firmly established in several large (n > 300), randomized, controlled trials (RCTs) of ≥ 24 weeks’ duration [4,5,6,7,8,9,10,11,12,13,14]. Most of these trials [4,5,6,7, 9,10,11,12, 14] have been reviewed in detail [1] and are briefly summarized here. Discussion focuses on the recommended EU dosage regimen of tocilizumab 8 mg/kg once every 4 weeks (Sect. 4).
2.1.1.1 In Longer-Duration Established RA
As monotherapy [4, 6, 7], tocilizumab significantly improved ACR20, 50 and 70 response rates and DAS28 remission rates compared with methotrexate [4], csDMARD [7] or adalimumab [6] monotherapy (Table 2). In AMBITION, the ACR20 response rate at 24 weeks (primary endpoint) with tocilizumab was superior to that of methotrexate (Table 2), with significant between-group differences (BGDs) observed from 2 weeks onwards [4]. At 24 weeks in the ADACTA trial, tocilizumab monotherapy was superior to adalimumab monotherapy for the mean change in DAS28 score (− 3.3 vs. − 1.8; BGD − 1.5; p < 0.0001) (primary endpoint) and was more effective than adalimumab in terms of secondary outcomes (Table 2) [6].
As combination therapy with methotrexate [5, 10] or csDMARDs [11, 12], add-on tocilizumab was more effective than methotrexate or csDMARDs alone in improving clinical signs and symptoms of disease (Table 2). Several other endpoints also favoured (p < 0.05) tocilizumab combination therapy at 24 weeks, including improvements in swollen (SJC) and tender joint counts (TJC) [5, 10, 12], CRP levels and ESR [11, 12], HRQOL measures [5, 10, 12], SF-36 physical function scores [5, 10] and FACIT-fatigue (FACIT-F) scores [5, 10, 12].
Tocilizumab (+ methotrexate) therapy significantly improved radiographic outcomes compared with methotrexate alone at 52 [9] and 104 [15] weeks in the 2-year LITHE trial [9, 15], with these benefits maintained during the 3-year long-term extension (LTE) study [16]. At 52 [9] and 104 [15] weeks, tocilizumab recipients experienced significantly less radiographic progression of structural joint damage, including changes from baseline in Genant-mTSS (coprimary endpoint; p < 0.01), erosion scores (p < 0.05) and joint space narrowing (JSN) scores (p < 0.05). In terms of HRQOL, tocilizumab recipients experienced significantly (p < 0.0001) greater improvements in the adjusted mean AUC for change in HAQ-DI score (coprimary endpoint) than methotrexate recipients at 52 [9] and 104 [15] weeks, with significantly (p < 0.05) more tocilizumab recipients achieving a clinically meaningful improvement (i.e. a decrease of ≥ 0.3 in HAQ-DI score) in physical function at 52 weeks [15]. The beneficial effects of tocilizumab combination therapy on radiographic outcomes and HAQ-DI scores were maintained after ≤ 5 years’ treatment [16].
The beneficial effects of long-term (≤ 5 years) tocilizumab monotherapy (n = 134) or combination therapy (n = 109) on clinical signs and symptoms of RA were maintained or improved during the LTE of AMBITION [17]. The short- and long-term (≤ 4.6 years’ exposure) efficacy of tocilizumab combination therapy was confirmed in pooled analyses of RCTs, their LTE studies and a PK study [18]. Similarly, in a meta-analysis of six Japanese RCTs, improvements in clinical signs and symptoms of RA were sustained during ≤ 9 years’ tocilizumab therapy [19].
2.1.1.2 In Early-Stage RA
Tocilizumab (+ methotrexate) therapy was also effective in improving the clinical signs and symptoms of RA in patients with early-stage disease who were methotrexate- or biologic DMARD (bDMARD)-naive (FUNCTION) [8] or methotrexate-naive (U-Act-Early) [13] (Table 2). For example, in the 2-year FUNCTION trial, clinical response rates at 24 weeks (primary timepoint for primary outcome) in terms of DAS28 remission rates (primary outcome) and ACR20, 50 and 70 response rates were all significantly higher with tocilizumab combination therapy than with methotrexate (+ placebo), as was the DAS28 remission rate with tocilizumab monotherapy versus methotrexate alone (Table 2) [8]. These clinical outcomes also significantly (p < 0.05) favoured tocilizumab combination therapy over methotrexate (+ placebo) at 52 weeks [8] and were maintained at 104 weeks [20]. With tocilizumab monotherapy, the DAS28 remission rate was significantly higher than with methotrexate monotherapy at 24 weeks, with other clinical outcomes numerically higher in the tocilizumab monotherapy group (Table 2) [8].
At 52 weeks, tocilizumab (+ methotrexate) recipients had significantly greater improvements in radiographic outcomes than methotrexate recipients in terms of van der Heijde mTSS (mean change 0.08 vs. 1.14; p = 0.0001) and erosion scores (0.05 vs. 0.63; p = 0.0006), with no significant BGD in JSN scores (0.03 vs. 0.51) [8]. Improvements in radiographic outcomes were numerically higher in the tocilizumab monotherapy group than in the methotrexate group [8]. The beneficial effects of tocilizumab combination therapy on radiographic outcomes were maintained at 104 weeks [20].
2.1.1.3 Adding Versus Switching to Tocilizumab
In the 3-year ACT-RAY trial, there were no significant BGDs at week 24 between adding (+ methotrexate) or switching to tocilizumab for DAS28 remission rates (primary endpoint) or secondary outcomes, including ACR response rates (Table 2) and radiographic progression [14]. The clinically meaningful improvements in clinical and radiographic responses achieved at 24 weeks were maintained at 52 weeks with both regimens [21]. At 2 years, there were generally no significant BGDs in clinical and radiographic outcomes, with efficacy maintained in both treatment groups [22]. Patients who achieved sustained remission (i.e. DAS28 < 2.6 at two consecutive 12-week visits) between weeks 52 to 104 discontinued tocilizumab treatment, and if remission was maintained, csDMARDs and then methotrexate were discontinued, with 76% of patients completing 2 years’ treatment. More tocilizumab recipients achieved drug-free remission in the add-on than switch arm (8.6% of 243 patients vs. 3.1% of 229 patients; p = 0.01). In these respective arms, 53.1 and 47.6% of patients achieved tocilizumab-free remission; of whom, 82.5 and 88.5% experienced flare within 52 weeks, with the majority of patients responding rapidly to tocilizumab retreatment [22].
2.1.2 In the Real-World Setting
Extensive evidence from several large, prospective, post-marketing studies (n = 557–1681) [23,24,25,26,27,28,29], including ACT-UP (a multinational, umbrella project involving 16 multicentre, observational studies sharing a set of design elements, patient selection criteria and core data) [23], and registry databases (n = 1491–7901) (French REGATE [30]; TOCERRA collaboration (9 EU registries) [31, 32]; Japanese post-marketing surveillance [33]; BSRBR-RA (UK) [34]; Germany [35]; US CORRONA Registry [36]), have firmly established the efficacy of IV tocilizumab in the clinical practice setting.
In ACT-UP, the efficacy of tocilizumab monotherapy (n = 506) or combination therapy (+ csDMARD; n = 830) was generally similar after 6 months’ treatment, with most patients continuing tocilizumab treatment throughout the study (primary endpoint) [80 vs. 87%; log-rank p < 0.001] [23]. At 6 months, mean changes from baseline for DAS28 scores with tocilizumab monotherapy and combination therapy were − 2.9 and − 3.2 (n = 178 and 365) and mean changes from baseline in CDAI scores were − 20.3 and − 22.3 (n = 186 and 416), with 94.4 and 92.1% of patients achieving a EULAR good or moderate response (n = 178 and 365). The tocilizumab dosing regimen was based on local label recommendations, with ≥ 94.5% of patients in both groups initiating tocilizumab treatment at a dose of 8 mg/kg [23].
In the global ACT-iON study in RA patients with an inadequate response to csDMARDs and initiating biologic therapy, patients initiating tocilizumab 8 mg/kg once every 4 weeks experienced significantly (p < 0.001) greater improvements in DAS28 scores at week 24 (primary endpoint; adjusted mean BGD − 0.831) and 52 (adjusted mean BGD − 0.910) than those initiating a TNF inhibitor (TNFi) [27]. With the exception of changes in TJC scores, adjusted mean changes from baseline for secondary endpoints all favoured tocilizumab over TNFi therapy at 24 weeks (p < 0.05), including improvements in ESR, CRP levels, and scores for SJC, CDAI, SDAI, HAQ-DI, FACIT-F and pain VAS. The benefits of tocilizumab over TNFi therapy persisted at 52 weeks for mean adjusted changes in ESR and scores for SJC, CDAI, SDAI, HAQ-DI and pain VAS (p < 0.05), with TJC scores also favouring tocilizumab at 52 weeks (p = 0.004). At 52 weeks, tocilizumab recipients were less likely to discontinue treatment than TNFi recipients (cumulative probability of drug discontinuation 15 vs. 27%; p < 0.001) [27].
In the open-label, multinational, phase 3b, ACT-SURE trial, patients with active RA (i.e. DAS > 3.2) who had an inadequate response to csDMARDs or csDMARDs plus a TNFi were randomized to tocilizumab (± csDMARD) for 24 weeks [24]. Treatment with TNFi was discontinued at the start of the study, with patients switching to tocilizumab with or without a washout period (n = 976 TNFi-naive, 298 TNFi-experienced with washout, and 470 TNFi recent use with no washout). At 24 weeks, ACR20 response rates in the TNFi-naive, TNFi-experienced and TNFi-recent use groups were 71, 61 and 63%, respectively (efficacy was a secondary outcome) [24]. Rates of DAS28 remission in these respective groups were 62, 49 and 50%, with LDA achieved by 75, 61 and 62% of patients [24].
In the open-label, multicentre, phase 3b ACT-STAR trial in treatment-experienced RA patients who had an inadequate response to prior csDMARDs or bDMARDs, ACR20 response rates at 24 weeks were 40–50%, ACR50 response rates were 24–27%, DAS28 remission was achieved by 20–25% of patients and LDA by 31–46% of patients across groups randomized to tocilizumab 8 mg/kg monotherapy (n = 163), tocilizumab 4 or 8 mg/kg (+ csDMARDs) (n = 363) or tocilizumab 8 mg/kg (+ csDMARD; n = 360) [29]. Efficacy was a secondary outcome in this trial; unlike phase 3 clinical trials, eligible patients were not required to have a minimum CRP or ESR, or a washout period prior to study entry [29].
In the French 12-month ACT-SOLO study in tocilizumab-naive patients with RA (n = 577; mean RA duration 10.9 years; 98% of patients were treatment experienced), 40% of patients initiated tocilizumab as monotherapy and 60% as combination therapy (+ csDMARD) [25]. The primary objective was to describe factors influencing the use of tocilizumab as monotherapy or combination therapy. At 12 months, there was no difference in the median rate of retention in the tocilizumab monotherapy and combination therapy groups (67 vs. 71%). In multivariate analyses, after exclusion of dyslipidemia as a factor (since this was correlated with age), independent factors for monotherapy (all p < 0.05) were aged ≥ 65 years [odds ratio (OR) 1.56], no methotrexate within the previous 2 years (OR 5.74), a past history of serious infectious disease (OR 2.03) and a higher baseline DAS28 (OR 1.22). There were no BGDs in terms of efficacy outcomes at 1 year, including ACR20, 50 and 70 response rates, DAS28, SDAI and CDAI remission and low-disease activity (LDA) rates, EULAR good or moderate response rates and changes in HAQ-DI scores [25].
In the CORRONA study in tocilizumab-naive patients with RA (mean disease duration 10.5–15 years) who had prior exposure to ≥ 1 TNFi, improvements in disease activity measures at 6 months indicated that tocilizumab monotherapy was as effective as treatment with a TNFi plus methotrexate, irrespective of the methotrexate dosage (i.e. methotrexate dose ≤ 10, > 10 to ≤ 15, > 15 to ≤ 20 or > 20 mg) (abstract) [36]. For the primary outcome of the mean change in CDAI score at 6 months, improvements were similar between the tocilizumab monotherapy group and all TNFi combination therapy groups, as was the likelihood of achieving LDA (i.e. CDAI score ≤ 10) [36].
Tocilizumab significantly improved markers of anaemia [i.e. haemoglobin (Hb) and haematocrit (Hct) levels] during 2 years’ treatment in RA patients (n = 3732), irrespective of baseline anaemia status, in a real-world, longitudinal cohort study utilizing US CMER (n = 153,788) [37]. In tocilizumab recipients, adjusted mean increases in Hb levels at 24 months in the overall population and in those with anaemia at baseline were 0.23 and 0.72 g/dL, with respective improvements in Hct levels of 0.96 and 2.06%. There was an 86% increase (OR 1.86; 95% CI 1.43–2.00; p < 0.001) in the likelihood of achieving an increase in Hb of ≥ 1 g/dL in the tocilizumab cohort than in the tofacitinib (n = 3126), other bDMARD (n = 55,694) and non-biologic DMARD (n = 91,236) cohorts, with no clinically relevant changes in Hb levels in these latter three cohorts. Initiating tocilizumab within 1 year of RA diagnosis was associated with a 95% increase (OR 1.95; 95% CI 1.19–3.21) in the likelihood of achieving an increase in Hb level at 6 months compared with initiating treatment 1 year post RA diagnosis. Conversely, early initiation of treatment in the other cohorts had no impact on the likelihood of achieving better Hb levels (ORs 0.98–1.19). In the tocilizumab, tofacitinib, other bDMARD and non-biologic DMARD group, 26, 29, 21 and 24% of patients, respectively, had anaemia at index date, with corresponding mean times to initiation of therapy from RA diagnosis of 39, 39, 12 and 4 months [37].
2.2 Subcutaneous Tocilizumab
2.2.1 In Clinical Trials
The efficacy of SC tocilizumab monotherapy (MUSASHI [38]) or combination therapy (BREVACTA [39] and SUMMACTA [40]) was investigated in multicentre, phase 3 trials in adults with moderate to severe active RA who had an inadequate response to csDMARD(s) [38,39,40] and/or bDMARD(s) [38]. Each trial comprised a 24-week double-blind phase and a 72- [39, 40] or 84-week [38, 41], open-label phase (with a 1-week dose-interruption period between these two phases in SUMMACTA [40]). All participants in BREVACTA and SUMMACTA received concomitant csDMARDs [39, 40]. In BREVACTA, escape therapy with tocilizumab 162 mg once weekly was permitted from week 12 in patients with an inadequate response (i.e. < 20% improvement from baseline in SJC and TJC) to tocilizumab 162 mg once every 2 weeks or placebo (16.5 vs. 41.1% of patients received escape therapy) [39]. The primary endpoint in all trials was the percentage of patients achieving an ACR20 response at week 24 [38,39,40], with safety a coprimary endpoint in SUMMACTA [40].
The impact of discontinuing methotrexate (i.e. tocilizumab monotherapy) versus continuing methotrexate (i.e. tocilizumab plus methotrexate) in patients who had achieved LDA (i.e. DAS28 ≤ 3.2) after 24 weeks of methotrexate plus tocilizumab 162 mg weekly (patients weighing ≥ 100 kg) or every other week (patients weighing < 100 kg) was evaluated in the 52-week, double-blind, multicentre, phase 3 COMP-ACT trial (abstracts) [42,43,44]. Patients weighing < 100 kg who had not achieved LDA at week 12 could escalate their tocilizumab dosage from 162 mg every other week to 162 mg once weekly [44]. Patients achieving LDA at week 24 were randomized to tocilizumab monotherapy (n = 147 evaluable) or tocilizumab plus methotrexate (n = 147 evaluable) until week 52. The primary outcome was the mean change in DAS28 score from week 24 to 40. At 24 weeks, DAS28 scores were similar in both groups [44]. At baseline patients had a mean disease duration of 6.8 years and mean DAS28 score of 6.3 [43].
2.2.1.1 Versus Placebo
At 24 weeks, the ACR20 response rate was significantly higher with add-on tocilizumab than add-on placebo, as were secondary clinical outcomes of ACR50 and 70 response rates and DAS28 remission rates (Table 3) [39]. In patients who switched to once-weekly tocilizumab escape therapy, the ACR20 response rate 12 weeks after escape in those initially randomized to tocilizumab once every 2 weeks was 58% and in those initially randomized to placebo was 72%. In exploratory subgroup analyses, ACR20, 50 and 70 response rates in the tocilizumab and placebo groups in patients receiving concomitant methotrexate or another DMARD at baseline were generally consistent with those in the overall population, as were these response rates in patients with an inadequate response to a DMARD or TNFi. Radiographic outcomes also favoured tocilizumab combination therapy, with significantly lower mean changes in mTSS (0.62 vs. 1.23; p = 0.0149) and erosion score (0.26 vs. 0.65; p = 0.0078) at 24 weeks in tocilizumab than placebo recipients [39]. Least square mean (LSM) changes in patient-reported outcomes (PROs) also significantly (p < 0.001) favoured tocilizumab combination therapy over add-on placebo at 12 weeks, including SF-36 MCS and PCS scores and HAQ-DI scores (abstract) [45]. In addition, significantly (p < 0.05) more tocilizumab than placebo recipients reported scores that were at least the minimum clinically important difference for all PROs and numerically more tocilizumab recipients reported scores of at least the normative value at week 12 [45].
2.2.1.2 IV Versus SC Tocilizumab
In the Japanese MUSASHI study, SC tocilizumab monotherapy was noninferior to IV tocilizumab monotherapy at 24 weeks in terms of ACR20 response rate in the per-protocol population (Table 3), with sensitivity analyses in the modified intent-to-treat (ITT) population consistent with this result [38]. There were no significant BGDs in terms of secondary outcomes, including ACR50 and 70 response rates (Table 3), and DAS28 (Table 3), CDAI (16 vs. 23%) and Boolean (16 vs. 16%) remission rates [38].
At 24 weeks, there were no significant differences in efficacy between the add-on SC and IV tocilizumab groups for primary and secondary outcomes in SUMMACTA (Table 3) [40]. Improvements in mean HAQ-DI scores from baseline to week 24 were similar with SC and IV tocilizumab combination therapy, as were CDAI remission rates (13.5 vs. 15%) [40]. LSM improvements in PROs were also similar in the SC and IV tocilizumab combination therapy groups at 24 weeks, including scores for HAQ-DI, SF-36 PCS and MCS, with add-on tocilizumab treatment resulting in clinically meaningful improvements in all PROs [45].
2.2.1.3 Impact of Discontinuation of Methotrexate in Patients with Low Disease Activity
In the 52-week COMP-ACT trial, discontinuation of methotrexate at week 24 (tocilizumab monotherapy) was noninferior to continuation of methotrexate (+ tocilizumab) for changes in DAS28 score from week 24 to 40 [mean change in DAS28 score 0.46 vs. 0.14; BGD 0.318 (95% CI 0.045–0.592)], indicating that patients who achieve LDA can effectively discontinue methotrexate therapy [44]. There were also no BGDs for changes in PROs from week 24 to 40, including mean changes in patient global assessment, pain, FACIT-F and HAQ-DI scores [43]. In addition, a similar proportion of patients in the tocilizumab monotherapy and combination therapy groups had an HAQ-DI score of < 0.5 at weeks 24 (randomization), 40 and 52 [43]. There were also no significant BGDs for mean changes from week 24 to week 40 in bone erosion, synovitis, osteitis and cartilage loss scores (assessed in the hands and wrists) or in the proportion of patients with no progression in each of these outcomes [42].
2.2.1.4 Longer-Term Treatment
The efficacy of tocilizumab was maintained during the 72- and 84-week extension phases in SUMMACTA [40] and MUSASHI [41] (no data reported for BREVACTA [39]), and during a further 84-week, open-label, single-arm, phase 3b extension study of BREVACTA and SUMMACTA [46]. In the phase 3b study, mean DAS28, CDAI and SDAI scores remained stable with add-on tocilizumab once weekly (n = 173) or once every 2 weeks (n = 44) [46]. Overall, at week 36 (i.e. after ≈ 132 weeks’ treatment), 62% of patients achieved an ACR20 response, 49 and 36% achieved LDA and disease remission by DAS28 criteria, 41 and 11% achieved LDA and remission by CDAI criteria, and 41 and 14% achieved LDA and remission by SDAI criteria; after week 36, patient numbers were insufficient for analyses to be conducted [46].
2.2.2 In the Real-World Setting
In TOZURA, a multinational (total of 22 countries), umbrella project involving 11 single-arm, multicentre studies, 24 weeks’ tocilizumab provided similar efficacy in patients with moderate to severe RA, irrespective of whether it was used as monotherapy (n = 353) or in combination with a csDMARD (n = 1451) (abstract) [47]. Retention rates at 24 weeks were similar in the monotherapy and combination therapy groups (79.3 and 85.6%), with no significant BGDs for mean changes from baseline in DAS28 and CDAI scores, the proportion of patients achieving DAS28 (Table 3) or CDAI remission, and ACR20 (Table 3), ACR50 (Table 3), ACR70 (Table 3) and ACR90 response rates [47].
3 Safety Profile
SC and IV tocilizumab as monotherapy or in combination with csDMARDs were generally well tolerated in the clinical trial and clinical practice settings after ≤ 9 years’ treatment, based on studies discussed in Sect. 2. Very common adverse reactions (incidence ≥ 10%) occurring during tocilizumab monotherapy or combination therapy were upper respiratory tract infection (URTI) and hypercholesterolaemia [2]. In a pooled analysis of 24-week, phase 3 RCTs of IV tocilizumab (8 mg/kg once every 4 weeks), the most common adverse reactions (i.e. incidence ≥ 5 and ≥ 1% higher than in the methotrexate group; n = 288 and 284) with tocilizumab monotherapy were URTI (7 vs. 5%), nasopharyngitis (7 vs. 6%), headache (7 vs. 2%), hypertension (6 vs. 2%) and increased ALT level (6 vs. 4%), and those with tocilizumab combination therapy were URTI (8 vs. 6% with placebo + DMARDs; n = 1582 and 1170), nasopharyngitis (6 vs. 4%) and headache (5 vs. 3%) [48].
The safety profile of SC tocilizumab was consistent with that of IV tocilizumab, with the exception of injection-site reactions (ISRs), and remained stable over time in the 97-week SUMMACTA trial [40]. ISR rates per 100 patient-years (PYs) of exposure with SC and IV tocilizumab were 57.97 and 32.59 at 24 weeks and 26.05 and 33.63 at 97 weeks. The number of PYs’ exposure in the SC and IV groups at 24 weeks was 289.82 and 288.39, with respective values at 97 weeks of 1013.26 and 816.53. The cumulative serious adverse event (SAE) rates at 24 and 97 weeks with SC tocilizumab were 11.73 and 14.61 events/100 PYs’ exposure, with these rates similar to those for IV tocilizumab (14.91 and 15.43 events/100 PYs’ exposure at 24 and 97 weeks) [40].
The tolerability profile of IV tocilizumab monotherapy was generally similar to that of SC adalimumab monotherapy in ADACTA, with treatment-emergent adverse events (TEAEs) occurring in 82 and 83% of patients, and 6% of patients in both groups discontinuing treatment because of these events [6]. The most commonly reported TEAEs were URTI (11% with tocilizumab vs. 11% with adalimumab), nasopharyngitis (11 vs. 8%) and worsening of RA symptoms (7 vs. 10%). SAEs occurred in 12% of tocilizumab recipients and 10% of adalimumab recipients, with serious infections the most common of these SAEs (4% in both groups) [6].
In ACT-iON (Sect. 2.1.2), the safety profile of tocilizumab (n = 423) was generally similar to that of TNF-inhibitors (n = 793), although the cumulative probability of drug discontinuation was significantly lower with tocilizumab than with TNFi therapy (15 vs. 27%; p < 0.001) [27]. Adverse events (AEs) occurred in 49% of tocilizumab recipients (124.1 events/100 PYs’ exposure) and 57% of TNFi recipients (130.3 events/100 PYs’ exposure) and resulted in treatment withdrawal in 2.1 and 1.6% of patients. Unadjusted SAE rates were similar in the tocilizumab and TNFi group (6.44 and 11.99 events/100 PYs’ exposure), as was the incidence of death (0.7 and 0.8%; 0.74 and 0.77 events/100 PYs’ exposure). The number of PYs of exposure in the tocilizumab and TNFi safety populations was 403.7 and 775.8 [27].
Tocilizumab should not be used during pregnancy unless clearly necessary, with no adequate data available on its use in this population [2]. A slight increase in the risk of spontaneous abortion/embryo-fetal death at a high dose (> 100 times human exposure) was observed in an animal study, with the potential risk to humans unknown [2]. In a retrospective analysis of a global safety database (clinical trial and post-marketing data), there appeared to be no substantial increase in the risk of malformations in women exposed to tocilizumab shortly before conception or early in the first trimester based on 180 prospectively reported pregnancies and 108 retrospective cases [49]. Of the prospective and retrospective cases, 60.6 and 50.9%, respectively, resulted in live births, 21.7 and 28.7% in spontaneous abortions and 17.2 and 20.4% of pregnancies were electively terminated. Amongst the prospective cases, there was also one stillbirth. In the retrospective group, there were three cases (infants/fetuses) of congenital abnormalities. The malformation rate was 4.5% and there was an increased rate of preterm births (accounting for 31.2% of births) relative to the general population. The risk of adverse pregnancy outcomes was not increased after paternal exposure to tocilizumab in 13 pregnancies with known outcomes [49].
3.1 Adverse Events of Special Interest
In a pooled long-term safety analysis of RCTs and LTE studies (clinical trial group n = 7647; 22,394 PYs’ exposure) and of global postmarketing reports (n = 606,937; 440,000 PYs’ exposure), the safety profile of tocilizumab during 7 years’ postmarketing experience was consistent with the safety profile in clinical trials, with no evidence of an increased safety risk with increasing exposure to tocilizumab (abstract) [50]. In the RCT all-exposed population, SAEs of special interest occurred with an overall rate of 14.16 events/100 PYs’ exposure, with the rate consistent for each 6-month period over a 5-year period. The overall spontaneous reporting rate of AEs of special interest in the global postmarketing safety population was 9.37 cases/100 patients, with the rate consistent for each 6-month period over a 7-year period [50]. These data confirm evidence from earlier integrated safety analyses of RCTs [51] and post-marketing data [24, 26, 28, 52]. During long-term tocilizumab treatment in the clinical practice setting, the type and incidence of malignancies were consistent with those expected in RA patients and remained stable over time [53].
In the clinical practice setting, the long-term safety profile of tocilizumab was similar to those of abatacept and rituximab in RA patients, based on a multicentre study utilizing three French registries [namely REGATE (tocilizumab; n = 1498); AIR (rituximab; n = 1984) and ORA (abatacept; n = 1016)] (abstracts) [54, 55]. The primary outcome of the study was the drug retention rate without failure at month 24, with failure defined as all-cause death, study drug discontinuation, initiation of a new biologic or a combination of csDMARDs, or an increase from baseline in corticosteroid dose of > 10 mg/day on two consecutive visits [54]. At 24 months’ follow-up, total exposure rates in the tocilizumab, rituximab and abatacept cohorts were 3,441 PYs, 10,545 PYs and 4912 PYs, respectively [54, 55]. Corresponding drug retention without failure rates at 24 months in the tocilizumab, rituximab and abatacept cohorts were 61, 65 and 40%, with the risk of this occurring significantly (p < 0.001) more likely in abatacept than in tocilizumab [hazard ratio (HR) 1.82] or rituximab (HR 2.00) recipients [54]. There were no statistically significant differences between the tocilizumab, rituximab and abatacept cohorts in the incidence rate ratios for AEs of special interest at 24 months, including serious infections, major adverse cardiovascular (CV) events (MACE), cancers or all-cause deaths [55].
3.1.1 Infections and Infestations
Serious and sometimes fatal infections have been reported during immunosuppressive therapy, including with tocilizumab [2]. In 6-month RCTs, the rate of all infections with IV tocilizumab (8 mg/kg) plus DMARD therapy was 127 events/100 PYs’ exposure compared with 112 events/100 PYs’ exposure with placebo (+ DMARD) therapy. Corresponding rates of serious infections in these two groups were 5.3 and 3.9 events/100 PYs’ exposure. In the long-term exposure population, the overall rate of infections with tocilizumab was 108 events/100 PYs’ exposure and that for serious infections was 4.7 events/100 PYs’ exposure [2]. There appeared to be no association between the risk of serious infections and reductions in absolute neutrophil count (ANC) observed in some patients receiving tocilizumab, based on a pooled analysis of RCTs and LTEs, with ANC reductions manageable with dosage interruptions [56].
In ACT-UP trials, the most common AEs and SAEs occurring during IV tocilizumab monotherapy or combination therapy were infections and infestations [23]. Serious infections and infestations occurred in 8.5% of tocilizumab monotherapy recipients and 7.5% of tocilizumab plus csDMARD recipients, corresponding to event rates of 4 and 5 events/100 PYs’ exposure [23]. Similar rates were seen in the German ROUTINE postmarketing study [26]. In the German ICHIBAN postmarketing study, during IV tocilizumab therapy in RA patients aged < 50 (n = 261), 50–65 (n = 438) or > 65 (n = 203) years, there was no increase in rate of infections of any grade (27.2, 19.8 and 19.2 events/100 PYs’ exposure, respectively) or serious infections (2.9, 2.9 and 3.1 events/100 PYs’ exposure) with increasing age [28].
In the REGATE registry (n = 1491 patients; mean follow-up 27.6 months), the incident rate for first serious infections was 4.7/100 PYs’ tocilizumab exposure, with a mean time between initiation of tocilizumab and first serious infection of 12.8 months [57]. The most frequent sites of infection were the lung and respiratory tract (28% of cases) and skin and soft tissue (26%), with most bacterial infections responding to anti-bacterial drugs. In multivariate analyses, positivity for anti-citrullinated protein antibodies at baseline was associated with a significantly lower risk of serious infections during tocilizumab treatment (HR 0.56; p = 0.012), whereas predictive factors associated with a higher risk of serious infection were an initial ANC of > 5.0 × 109/L (HR 1.94; p < 0.001) and concomitant leflunomide treatment (HR 2.18; p = 0.009; leflunomide alone vs. no treatment) [57].
3.1.2 Gastrointestinal Perforations
Events of diverticular perforations as complications of diverticulitis have been reported uncommonly with tocilizumab therapy in RA patients [2]. In 6-month RCTs, the overall rate of gastrointestinal perforations (GIP) was 0.26 events/100 PYs’ tocilizumab exposure, with a similar rate observed in the long-term tocilizumab exposure population (0.28 events/100 PYs’ exposure) [2]. Safety analyses of data from clinical trial (providing 17,906 PYs’ exposure), global postmarketing (382,621 PYs’ exposure) and US healthcare claims (3268 PYs’ exposure) populations indicated that adjusted GIP incident rates during IV tocilizumab treatment were similar in these three settings (1.9, 1.2 and 1.8 events/1000 PYs’ exposure, respectively) and were consistent over time [58]. The majority of these events occurred in the lower GI tract [58]. The incidence rate of GIP in this healthcare claims population were consistent with those reported previously for a US healthcare claims population [59].
The incidence rate of lower GIP was significantly higher with tocilizumab therapy than with TNFi, csDMARDs, abatacept or rituximab, based on data from real-world registries in Germany (RABBIT [60]) and the USA [58, 59]. For example, the crude incidence rate of lower GIPs was significantly higher in the tocilizumab cohort (2.7 events/1000 PYs’ exposure; 95% CI 1.4–4.8) than in the csDMARD (0.6 events/1000 PYs’ exposure; 95% CI 0.3–1.1), TNFi (0.5 events/1000 PYs’ exposure; 95% CI 0.3–0.9), rituximab (0.2 events/1000 PYs’ exposure; 95% CI 0.01–1.1) and abatacept (0.5 events/1000 PYs’ exposure; 95% CI 0.01–2.8) cohorts in the RABBIT registry [60]. These crude incidence rates were consistent with those observed in RCTs and corresponded to a number needed to harm with tocilizumab, csDMARD and TNFi treatment of 371, 1647 and 1911, respectively. Univariate analyses indicated that the risk of lower GIP was 4.5 times higher with tocilizumab treatment than with csDMARDs (HR 4.48; 95% CI 2.0–10.0), with no increase in the risk of these events relative to csDMARDs with TNFi (HR 1.04; 95% CI 0.5–2.3) or other bDMARDs (HR 0.33; 95% CI 0.1–1.4). Lower GIPs resulted in death within 30 days of the event in five tocilizumab recipients (n = 11 cases), two csDMARD recipients (n = 11 cases) and two TNFi recipients (n = 13 cases). The analysis included 877 tocilizumab recipients (4082 PYs’ exposure), 4423 csDMARD recipients (18,113 PYs’ exposure), 6711 TNFi recipients (24,851 PYs’ exposure), 928 rituximab recipients (4950 PYs’ exposure) and 371 abatacept recipients (1976 PYs’ exposure) [60].
3.1.3 Liver Enzymes and Hepatic Events
Tocilizumab treatment, particularly in combination with methotrexate, may be associated with elevations in hepatic transaminases [2]. In a pooled safety analysis of phase 3 RCTs, LTE studies, a pharmacology study and a phase 4 study, mean ALT and AST levels increased above the upper limit of normal (ULN) at least once in 71 and 59% of patients treated with IV tocilizumab (4, 8 or 10 mg/kg doses; ± DMARDs), with most elevations of transaminase enzymes occurring within 12 months of initiating tocilizumab therapy [61]. In the all-exposed tocilizumab population (n = 4171), the mean duration of treatment was 3.9 years, representing a total exposure of 16,205 PY. The risk of ALT and AST elevations did not increase with increased exposure to tocilizumab. In the first 12 months, increased ALT levels of > 1–3, > 3–5 and > 5 × ULN occurred in 50, 6 and 2% of patients, respectively, with corresponding frequencies for AST elevations of 42, 2 and 4%. During months 73 to 84, increased ALT levels of > 1–3, > 3–5 and > 5 × ULN occurred in 24, 0.7 and 0.2% of patients, respectively, with corresponding frequencies for AST elevations of 13, 0.4 and 0.3%. In most patients (80%), elevations in transaminase levels of > 3 × ULN (typically single occurrences) returned to normal levels, with a median time to normalization of 5.6 weeks. Relatively few patients (2.5%) discontinued tocilizumab treatment because of elevated transaminase levels, most of whom discontinued treatment during the initial 12-month period. The frequency and severity of transaminase elevations with tocilizumab monotherapy were similar to those with methotrexate monotherapy, with tocilizumab (+ methotrexate/other DMARD) combination therapy associated with numerically higher rates than methotrexate or other DMARD alone [61].
In the all-exposed population, hepatic adverse events (HAEs) occurred with an overall rate of 0.78 events/100 PYs’ exposure and remained stable with increasing exposure to tocilizumab [61]. There were no serious HAEs reported during the placebo-controlled period of RCTs, with all seven serious HAEs reported during the LTE studies (overall rate 0.04 events/100 PYs’ exposure). Investigators determined the relationship of serious HAEs to tocilizumab treatment was variable [61].
3.1.4 Cardiovascular Safety
Compared with the general population, RA is associated with an increased risk of CV disease (CVD), with this risk increasing with the duration of RA and evident from the early stages of the disease [62]. In a pooled, retrospective post hoc analysis of RCTs and LTE studies (n = 3986; 14,683 PYs’ follow-up; mean duration of treatment 3.7 years), IV tocilizumab treatment was associated with a MACE event rate of 3.4 events/1000 PYs’ exposure (total of 50 independently adjudicated cases of MACE), with a median time after initiating tocilizumab to the first MACE event of 680 days [63]. The MACE event rate did not appear to increase over the duration of these RCTs and LTE studies. In multivariate analyses, baseline factors independently predictive of a future MACE event were older age (HR 1.07; p < 0001), a history of cardiac disorders (HR 2.32; p = 0.0161), a higher DAS28 score (HR 1.36; p = 0.0158) and higher total cholesterol to HDL cholesterol ratio (HR 1.33; p = 0.0109). MACE was defined as definite or probable myocardial infarction (MI), nonfatal stroke or death caused by CVD. During tocilizumab treatment in RCTs, a higher DAS28 score and higher SJC and TJC scores, but not lipid parameter changes, at 24 weeks were predictive of a future MACE. This study had several limitations that should be considered carefully when interpreting results, including its retrospective, post hoc design, the results were not adjudicated for multiplicity and the study was not powered to evaluate CV safety [63].
The phase 4, multicentre, noninferiority ENTRACTE trial evaluated the CV safety of IV tocilizumab (n = 1538) compared with etanercept (n = 1542) in RA patients (aged ≥ 50 years) with an inadequate response to ≥ 1 non-biologic DMARD and who had ≥ 1 CVD risk factor, extra-articular RA manifestations, or a history of a CVD event (abstract) [64]. The average follow-up time was 3.2 years. The primary outcome was the time to first occurrence of an adjudicated MACE, defined as CVD death, non-fatal MI or non-fatal stroke. In the primary ITT analysis, 83 MACE events occurred in the tocilizumab group over 4900 PYs’ exposure and 78 occurred in the etanercept group over 4891 PYs’ exposure (HR 1.05; 95% CI 0.77–1.43), excluding a > 43% relative increase in the risk of MACE in tocilizumab recipients. Results in the on-treatment analysis were consistent with these findings. Compared with etanercept, tocilizumab treatment was associated with significant increases in total cholesterol, LDL-C, HDL-C and triglyceride levels by week 4, after which time average lipid levels remained constant throughout the study [64].
In a US multi-database, population-based, propensity-score matched cohort study in RA patients who had previously received ≥ 1 bDMARD, there was no significant difference in the risk of the composite CV outcome of hospitalization for MI or stroke in patients initiating tocilizumab versus those initiating TNFi treatment (primary outcome) (36 vs. 89 composite CV outcome events; incidence rate 0.52 vs. 0.59 events/100 PYs’ exposure) [65]. The mean follow-up duration was 0.9 years, with a maximal observation period of 4.5 years for the primary as-treated analysis. The propensity-score matched tocilizumab and TNFi cohorts consisted of 9218 and 18,810 patients, with these cohorts similar with regard to CV comorbidities, comorbidity index, medication use and healthcare utilization patterns [65].
In another propensity-score matched cohort study utilizing the same three US healthcare claims databases as the study discussed above, there was no increase in the risk of the primary composite CV outcome of hospitalization of any duration for MI and stroke in RA patients initiating tocilizumab treatment compared with those initiating abatacept (abstract) [66]. For each individual database, the risk of the primary composite CV outcome was similar in the tocilizumab and abatacept cohorts (incidence rate range 0.37–1.64 vs. 0.59–1.69 events/100 PYs’ exposure), with an overall HR of 0.82 (95% CI 0.55–1.22) [66].
3.1.5 Infusion and Injection-Site Reactions
In 6-month RCTs, infusion reactions occurring during and within 24 h post-infusion were reported in 7% of patients in the tocilizumab 8 mg/kg (+ DMARD) group and 5% of patients in the placebo (+ DMARD) group [2]. Those occurring during the infusion were primarily episodes of hypertension and those occurring during the subsequent 24-h period were headache and skin rash, with none of these events treatment limiting. During RCTs and LTE studies, the rate of anaphylactic reactions was several fold higher with the 4 mg/kg dose than with 8 mg/kg dose, with an overall frequency of 0.2% (8/4009 patients). Fatal cases of anaphylaxis have been reported during postmarketing use of tocilizumab. Tocilizumab-related clinically significant hypersensitivity reactions occurred in 1.4% (56/4009 patients) of patients, with most occurring during the second to fifth infusion of tocilizumab [2].
During the 6-month controlled period of RCTs, 10.1% (64/631 patients) and 2.4% (15/631) of patients in the once-weekly SC tocilizumab and placebo groups (both + csDMARDs) experienced ISRs, including erythema, pruritus, pain and haematoma [2]. ISRs were of mild to moderate severity, resolved without treatment and did not result in drug discontinuation [2].
3.1.6 Immunogenicity
The immunogenicity risk is low with tocilizumab treatment [67]. In 6-month RCTs of IV tocilizumab, 1.6% (46/2876 patients) of tocilizumab-treated patients developed anti-drug antibodies (ADAs) [2]. Of the 46 patients who developed ADAs, six had an associated medically significant hypersensitivity reaction, five of which resulted in permanent discontinuation of tocilizumab. Thirty patients (1.1%) developed neutralizing antibodies [2]. The rate of development of ADAs was low with both SC (1.5% of patients) and IV (1.2%) tocilizumab, based on a pooled analysis of five RCTs of SC tocilizumab (n = 3099; ≤ 3.5 years’ treatment) and eight RCTs and a clinical pharmacology study of IV tocilizumab (n = 5875; ≤ 5 years’ treatment) [67]. The majority of patients who developed ADAs were also positive for neutralizing antibodies (85.1 and 78.3% with SC and IV tocilizumab). Rates of ADA development were low (< 2% of patients) irrespective of whether patients received SC or IV tocilizumab monotherapy or combination therapy (+ csDMARDs). The development of ADAs appeared to have no impact on the PK, efficacy or safety (including anaphylaxis, hypersensitivity or ISRs) of tocilizumab [67]. The low immunogenicity risk in tocilizumab-treated RA patients may reflect the effects of tocilizumab-mediated IL-6 blockade of B-cell responses and the function of follicular helper CD4 T cells [68].
4 Dosage and Administration
In the EU, IV and SC tocilizumab (+ methotrexate) are indicated for the treatment of severe, active and progressive RA in adults not previously treated with methotrexate, and in the treatment of moderate to severe active RA in adults who have either responded inadequately to or who were intolerant of previous therapy with ≥ 1 DMARD or TNFi [2]. In the latter patient population, tocilizumab may be given as monotherapy. The recommended IV dosage of tocilizumab is 8 mg/kg once every 4 weeks (doses > 800 mg are not recommended). The recommended SC dosage of tocilizumab is 162 mg once weekly (fixed-dose pre-filled syringe); for patients switching from IV to SC tocilizumab, the once weekly dosing interval should be followed [2]. Local prescribing information should be consulted for detailed information, including contraindications, warnings, dosage adjustments and monitoring requirements.
5 Place of Tocilizumab in the Management of Rheumatoid Arthritis
RA places a considerable burden on society and healthcare systems worldwide and significantly impacts on a patient’s HRQOL and risks for comorbidities and death [62, 69, 70]. Disease management primarily targets achieving remission, with no active joint inflammation and no erosion or functional deterioration or, where appropriate, achieving low/minimal disease activity [70, 71]. Other key aims include maximizing long-term HRQOL through symptom control, prevention of structural damage and normalization of social and work participation. Pharmacotherapy is essential to abrogate inflammation and prevent adverse clinical outcomes in RA patients, and should be initiated as early as possible in the course of the disease utilizing a treat-to-target strategy [70, 71]. The management of RA was revolutionized by the introduction of bDMARDs over 20 years ago and it appears likely that the advent of biosimilars will play an increasing role in future disease management, especially given their substantial cost savings and bioequivalent efficacy to the reference product; however, long-term pharmacovigilance is required to fully define their safety [72].
The mainstays of pharmacotherapy are csDMARDs (e.g. methotrexate, leflunomide, sulfasalazine), bDMARDs [TNFi (adalimumab, certolizumab pegol, etanercept, golimumab, infliximab), other bDMARDs such as IL-6R antagonists (tocilizumab), T cell co-stimulation inhibitor (abatacept) or the anti- B cell agent (rituximab)] and targeted synthetic DMARDs (tsDMARDs; namely the JAK inhibitors tofacitinib and baricitinib) [70, 71]. Methotrexate remains the anchor drug for initiating treatment and as a backbone of combination regimens, reflecting its low cost and well established efficacy and safety profile. Current treatment guidelines recommend a sequential, step-wise, individual approach based on response, with treatment typically initiated with a csDMARD (unless contraindicated), followed by addition of another DMARD [70, 71]. More specifically, as second-line therapy, 2016 EULAR guidelines recommend a bDMARD (e.g. tocilizumab, abatacept, rituximab or a TNFi, or the respective EMA/FDA approved biosimilar) or tsDMARD if prognostically unfavourable factors are present, or switching to/adding a second csDMARD if unfavourable prognostic factors are absent, with subsequent therapy including a change in any first bDMARD to any other bDMARD (but not another biosimilar of the same reference agent) [70].
Extensive experience in the clinical trial and real-world settings over the last decade has firmly established the short- and long-term efficacy of IV and SC tocilizumab as monotherapy or combination therapy in adults with moderate to severe RA (Sect. 2), including in both early-stage (Sect. 2.1.1.2) and longer-duration established (Sect. 2.1.1.1) disease, with both formulations exhibiting similar efficacy in a RCT (Sect. 2.2.1.2). Tocilizumab monotherapy or combination (+ csDMARD) therapy provided rapid, marked improvements in clinical and radiographic outcomes and improved HRQOL, with these benefits maintained during long-term treatment (Sect. 2).
Albeit the advent of bDMARDs resulted in a paradigm shift in the treatment of RA, their immunomodulatory mechanism of action (like all DMARDs) has also raised concerns regarding potential safety issues, including serious infections and infusion or injection-site reactions [73, 74]. By inhibiting the activity of overexpressed signaling proteins, such as IL-6-mediated signaling, DMARDs also block important signaling pathways of the normal immune response, resulting in an increased risk of infections [73, 74]. In RCTs and in the real-world setting, the most common AEs and SAEs occurring during tocilizumab treatment were infections and infestations, with similar rates of infections irrespective of age or whether patients received monotherapy or combination therapy (Sect. 3.1.1). Real-world data also indicated that infection rates were similar during tocilizumab treatment to those with abatacept and rituximab therapy (Sect. 3.1). Other adverse events of special interest with individual DMARDs include an increased risk of GIP with tocilizumab (Sect. 3.1.2), cytopenias and hepatotoxicity with csDMARDs, progressive focal leucoencephalopathy with rituximab, and congestive heart failure and demyelinating disease with TNFi [73, 74].
Biologics, including some TNFi such as adalimumab and infliximab, have been associated with immunogenicity (i.e. development of ADAs) that may, in turn, cause hypersensitivity reactions and lead to subtherapeutic serum drug concentrations and reduced efficacy [73, 75]. This risk may be mitigated by combination therapy with methotrexate [73, 75]. Conversely, tocilizumab exhibited low immunogenicity in clinical trials, most likely reflecting its mechanism of action, with the development of ADAs having no impact on the PKs, efficacy and safety of tocilizumab (Sect. 3.1.6).
In conclusion, extensive clinical experience has firmly established the short- and long-term efficacy and safety of tocilizumab (monotherapy or in combination with csDMARDs) in adults with early-stage and longer-duration established RA. In the clinical trial and real-world settings, tocilizumab monotherapy or combination therapy provided rapid and sustained improvements in clinical and radiographic outcomes and HRQOL. The safety profile of tocilizumab is consistent over time in both of these settings and, in general, is consistent with that of other immunomodulatory agents. Given its low risk of immunogenicity, the flexibility of IV and SC administration and the convenience of the once-weekly, self-administered, SC regimen, tocilizumab provides an effective treatment for severe, active and progressive RA in adults not previously treated with methotrexate, and an effective biologic first- or subsequent-line treatment for moderate to severe active RA in adults who have either responded inadequately to or were intolerant of previous therapy with ≥ 1 csDMARD or TNFi.
Data Selection Tocilizumab: 512 records identified
Duplicates removed | 100 |
Excluded at initial screening (e.g. press releases; news reports; not relevant drug/indication) | 88 |
Excluded during initial selection (e.g. preclinical study; reviews; case reports; not randomized trial) | 133 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 116 |
Cited efficacy/tolerability articles | 65 |
Cited articles not efficacy/tolerability | 10 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were tocilizumab, RoActemra, Actemra, TCZ, rheumatoid arthritis. Records limited to those in English language. Searches last updated 4 October 2017 | |
Change history
19 December 2017
The article Tocilizumab: A Review in Rheumatoid Arthritis, written by Lesley J. Scott, was originally published Online First without open access. After publication in volume 77, issue 17, pages 1865–1879 F. Hoffmann-La Roche Ltd requested that the article be Open Choice to make the article an open access publication. Post-publication open access was funded by F. Hoffmann-La Roche Ltd. Further details may be found at http://www.medengine.com/Redeem/68FBF06068F81EA7 . The article is forthwith distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License ( http://creativecommons.org/licenses/by-nc/4.0/ ), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
Dhillon S. Intravenous tocilizumab: a review of its use in adults with rheumatoid arthritis. BioDrugs. 2014;28(1):75–106.
European Medicines Agency. Tocilizumab (RoActemra) 20 mg/mL concentrate solution for infusion/162 mg solution for injection in pre-filled syringe: summary of product characteristics. 2016. http://www.ema.europa.eu. Accessed 2 Oct 2017.
European Medicines Agency. EPAR summary for the public: RoActemra (tocilizumab). 2017. http://www.ema.europa.eu. Accessed 4 Oct 2017.
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(1):88–96.
Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371(9617):987–97.
Gabay C, Emery P, van Vollenhoven R, et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet. 2013;381(9877):1541–50.
Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X-ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. 2007;66(9):1162–7.
Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75(6):1081–91.
Kremer JM, Blanco R, Brzosko M, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthr Rheumatol. 2011;63(3):609–21.
Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67(11):1516–23.
Yazici Y, Curtis JR, Ince A, et al. Efficacy of tocilizumab in patients with moderate to severe active rheumatoid arthritis and a previous inadequate response to disease-modifying antirheumatic drugs: the ROSE study. Ann Rheum Dis. 2012;71(2):198–205.
Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthr Rheumatol. 2008;58(10):2968–80.
Bijlsma JW, Welsing PM, Woodworth TG, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): a multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet. 2016;388(10042):343–55.
Dougados M, Kissel K, Sheeran T, et al. Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24-week symptomatic and structural results of a 2-year randomised controlled strategy trial in rheumatoid arthritis (ACT-RAY). Ann Rheum Dis. 2013;72(1):43–50.
Fleischmann RM, Halland AM, Brzosko M, et al. Tocilizumab inhibits structural joint damage and improves physical function in patients with rheumatoid arthritis and inadequate responses to methotrexate: LITHE study 2-year results. J Rheumatol. 2013;40(2):113–26.
Kremer JM, Blanco R, Halland A-M, et al. Clinical efficacy and safety maintained up to 5 years in patients with rheumatoid arthritis treated with tocilizumab in a randomised trial. Clin Exp Rheumatol. 2016;34(4):625–33.
Jones G, Wallace T, McIntosh MJ, et al. Five-year efficacy and safety of tocilizumab monotherapy in patients with rheumatoid arthritis who were methotrexate- and biologic-naive or free of methotrexate for 6 months: the AMBITION study. J Rheumatol. 2017;44(2):142–6.
Genovese MC, Rubbert-Roth A, Smolen JS, et al. Long-term safety and efficacy of tocilizumab in patients with rheumatoid arthritis: a cumulative analysis of up to 4.6 years of exposure. J Rheumatol. 2013;40(6):768–80.
Nishimoto N, Ito K, Takagi N. Safety and efficacy profiles of tocilizumab monotherapy in Japanese patients with rheumatoid arthritis: meta-analysis of six initial trials and five long-term extensions. Mod Rheumatol. 2010;20(3):222–32.
Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab combination therapy or monotherapy or methotrexate monotherapy in methotrexate-naive patients with early rheumatoid arthritis: 2-year clinical and radiographic results from the randomised, placebo-controlled FUNCTION trial. Ann Rheum Dis. 2017. doi:10.1136/annrheumdis-2016-210561.
Dougados M, Kissel K, Conaghan PG, et al. Clinical, radiographic and immunogenic effects after 1 year of tocilizumab-based treatment strategies in rheumatoid arthritis: the ACT-RAY study. Ann Rheum Dis. 2014;73(5):803–9.
Huizinga TWJ, Conaghan PG, Martin-Mola E, et al. Clinical and radiographic outcomes at 2 years and the effect of tocilizumab discontinuation following sustained remission in the second and third year of the ACT-RAY study. Ann Rheum Dis. 2015;74(1):35–43.
Haraoui B, Casado G, Czirjak L, et al. Patterns of tocilizumab use, effectiveness and safety in patients with rheumatoid arthritis: core data results from a set of multinational observational studies. Clin Exp Rheumatol. 2017. (epub ahead of print).
Bykerk VP, Ostor AJK, Alvaro-Gracia J, et al. Tocilizumab in patients with active rheumatoid arthritis and inadequate responses to DMARDs and/or TNF inhibitors: a large, open-label study close to clinical practice. Ann Rheum Dis. 2012;71(12):1950–4.
Flipo R-M, Maillefert J-F, Chazerain P, et al. Factors influencing the use of tocilizumab as monotherapy in patients with rheumatoid arthritis in a real-life setting: results at 1 year of the ACT-SOLO study. Rheum Musculoskelet Dis. 2016. doi:10.1136/rmdopen-2016-000340.
Iking-Konert C, von Hinuber U, Richter C, et al. ROUTINE: a prospective, multicentre, non-interventional, observational study to evaluate the safety and effectiveness of intravenous tocilizumab for the treatment of active rheumatoid arthritis in daily practice in Germany. Rheumatology. 2016;55(4):624–35.
Choy EH, Bernasconi C, Aassi M, et al. Treatment of rheumatoid arthritis with anti-tumor necrosis factor or tocilizumab therapy as first biologic in a global comparative observational study. Arthr Care Res. 2017. doi:10.1002/acr.23303.
Specker C, Kaufmann J, Kellner H, et al. Safe and effective tocilizumab therapy in elderly patients with rheumatoid arthritis [abstract no. FRI0202]. Ann Rheum Dis. 2016;75(Suppl. 2):504.
Weinblatt ME, Kremer J, Cush J, et al. Tocilizumab as monotherapy or in combination with nonbiologic disease-modifying antirheumatic drugs: twenty-four-week results of an open-label, clinical practice study. Arthr Care Res. 2013;65(3):362–71.
Terreaux W, Masson C, Eschard JP, et al. Incidence of paradoxical reactions in patients treated with tocilizumab for rheumatoid arthritis: data from the French registry REGATE. Jt Bone Spine. 2017. doi:10.1016/j.jbspin.2017.01.002.
Gabay C, Riek M, Hetland ML, et al. Effectiveness of tocilizumab with and without synthetic disease-modifying antirheumatic drugs in rheumatoid arthritis: results from a European collaborative study. Ann Rheum Dis. 2016;75(7):1336–42.
Lauper K, Nordström DC, Pavelka K, et al. Retention of tocilizumab as monotherapy versus TNF inhibitors with conventional synthetic DMARDs in rheumatoid arthritis patients with inadequate response to TNF inhibitors: a study from the TOCERRA collaboration [abstract no. Sat0206]. In: 18th Annual EULAR. 2017.
Koike T, Harigai M, Inokuma S, et al. Effectiveness and safety of tocilizumab: postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J Rheumatol. 2014;41(1):15–23.
Kihara M, Davies R, et al. Use and effectiveness of tocilizumab among patients with rheumatoid arthritis: an observational study from the British Society for Rheumatology Biologics Register for rheumatoid arthritis. Clin Rheumatol. 2017;36:241–50.
Backhaus M, Kaufmann J, Richter C, et al. Comparison of tocilizumab and tumour necrosis factor inhibitors in rheumatoid arthritis: a retrospective analysis of 1603 patients managed in routine clinical practice. Clin Rheumatol. 2015;34(4):673–81.
Harrold LR, Reed GW, Best J, et al. Comparative effectiveness of tocilizumab (TCZ) monotherapy with tumor necrosis factor inhibitors (TNFi) in combination with varying doses of methotrexate (MTX) in patients with rheumatoid arthritis [abstract no. 2449]. Arthr Rheumatol. 2017;69(Suppl 10).
Paul SK, Montvida O, Best JH, et al. Effectiveness of biologic and non-biologic antirheumatic drugs on anaemia markers in 153,788 patients with rheumatoid arthritis: new evidence from real-world data. Semin Arthr Rheum. 2017. doi:10.1016/j.semarthrit.2017.08.001.
Ogata A, Tanimura K, Sugimoto T, et al. Phase III study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthr Care Res. 2014;66(3):344–54.
Kivitz A, Olech E, Borofsky M, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthr Care Res. 2014;66(11):1653–61.
Burmester GR, Rubbert-Roth A, Cantagrel A, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75(1):68–74.
Ogata A, Amano K, Dobashi H, et al. Longterm safety and efficacy of subcutaneous tocilizumab monotherapy: results from the 2-year open-label extension of the MUSASHI study. J Rheumatol. 2015;42(5):799–809.
Peterfy C, Kremer J, Rigby W, et al. MRI results following discontinuation of methotrexate in patients with rheumatoid arthritis treated with subcutaneous tocilizumab: results from a randomized controlled trial [abstract no. 2962]. Arthr Rheumatol. 2017;69(Suppl 10).
Kremer JM, Rigby WFC, Singer N, et al. Patient-reported outcomes following discontinuation of methotrexate in patients with rheumatoid arthritis treated with subcutaneous tocilizumab: results from a randomized controlled trial [abstract no. 2460]. Arthr Rheumatol. 2017;69(Suppl 10).
Kremer J, Rigby W, Singer N, et al. Sustained response following discontinuation of methotrexate in patients with rheumatoid arthritis treated with subcutaneous tocilizumab: results from a randomized controlled trial [abstract no. 1905]. Arthr Rheumatol. 2017;69(Suppl 10).
Strand V, Lampl K, Birchwood C, et al. Patient-reported outcomes in patients with rheumatoid arthritis treated with subcutaneous tocilizumab compared with placebo or intravenous tocilizumab in combination with csDMARDs [abstract no. Fri0253]. In: 18th Annual EULAR. 2017.
Kivitz A, Wallace T, Olech E, et al. Long-term safety and efficacy of subcutaneously administered tocilizumab for adult rheumatoid arthritis: a multicenter phase 3b long-term extension study. Rheumatol Ther. 2016;3(2):291–304.
Choy E, Caporali R, Xavier R, et al. Subcutaneous tocilizumab monotherapy or combined with a csDMARD in patients with rheumatoid arthritis: TOZURA, a pooled analysis of phase IV studies in 22 countries [abstract no. Sat0199]. Ann Rheum Dis. 2017;76(Suppl 2):847–8.
Genentech Inc. Actemra (tocilizumab): US prescribing information. 2017. https://www.gene.com/download/pdf/actemra_prescribing.pdf. Accessed 2 Oct 2017.
Hoeltzenbein M, Beck E, Rajwanshi R, et al. Tocilizumab use in pregnancy: analysis of a global safety database including data from clinical trials and post-marketing data. Sem Arthr Rheum. 2016;466(2):238–45.
Mohan S, Michalska M, Yourish J, et al. Long-term safety of tocilizumab from large clinical trial and postmarketing populations [abstract no. OP0105]. In: 18th Annual EULAR. 2017.
Schiff MH, Kremer JM, Jahreis A, et al. Integrated safety in tocilizumab clinical trials. Arthr Res Ther. 2011;13(5):R141.
Curtis JR, Perez-Gutthann S, Suissa S, et al. Tocilizumab in rheumatoid arthritis: a case study of safety evaluations of a large postmarketing data set from multiple data sources. Semin Arthr Rheum. 2015;44:381–8.
Rubbert-Roth A, Sebba A, Brockwell L, et al. Malignancy rates in patients with rheumatoid arthritis treated with tocilizumab. RMD Open. 2016;2(1):e000213.
Gottenberg JE, Morel J, Constantin A, et al. Long-term registry data in 4498 patients with rheumatoid arthritis indicate a similar safety but a different drug retention between abatacept, rituximab and tocilizumab [abstract no. 1998]. Arthr Rheumatol. 2016;68(Suppl 10):2550–3.
Gottenberg JE, Morel J, Constantin A, et al. Similar rates of death, serious infections, cancers, major cardiovascular events in patients treated with abatacept, rituximab and tocilizumab: long-term registry data in 4498 patients with rheumatoid arthritis [abstract no. 2614]. Arthr Rheumatol. 2016;68(Suppl 10):3536–7.
Moots RJ, Sebba A, Rigby W, et al. Effect of tocilizumab on neutrophils in adult patients with rheumatoid arthritis: pooled analysis of data from phase 3 and 4 clinical trials. Rheumatology. 2017;56(4):541–9.
Morel J, Constantin A, Baron G, et al. Risk factors of serious infections in patients with rheumatoid arthritis treated with tocilizumab in the French Registry REGATE. Rheumatology. 2017. doi:10.1093/rheumatology/kex238.
Monemi S, Berber E, Sarsour K, et al. Incidence of gastrointestinal perforations in patients with rheumatoid arthritis treated with tocilizumab from clinical trial, postmarketing, and real-world data sources. Rheumatol Ther. 2016;3(2):337–52.
Xie F, Yun H, Bernatsky S, et al. Risk of gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologic treatments. Arthr Rheumatol. 2016;68(11):2612–7.
Strangfeld A, Richter A, Siegmund B, et al. Risk for lower intestinal perforations in patients with rheumatoid arthritis treated with tocilizumab in comparison to treatment with other biologic or conventional synthetic DMARDs. Ann Rheum Dis. 2017;76(3):504–10.
Genovese MC, Kremer JM, Van Vollenhoven RF, et al. Transaminase levels and hepatic events during tocilizumab treatment: pooled analysis of long-term clinical trial safety data in rheumatoid arthritis. Arthr Rheumatol. 2017. doi:10.1002/art.40176.
Symmons PM, Gabriel SE. Epidemiology of CVD in rheumatic disease, with a focus on RA and SLE. Rheumatology. 2011;7:399–408.
Rao VU, Pavlov A, Klearman M, et al. An evaluation of risk factors for major adverse cardiovascular events during tocilizumab therapy. Arthr Rheumatol. 2015;67(2):372–80.
Giles JT, Sattar N, Gabriel SE, et al. Comparative cardiovascular safety of tocilizumab vs etanercept in rheumatoid arthritis: results of a randomized, parallel-group, multicenter, noninferiority, phase 4 clinical trial [abstract no. 3L]. Arthr Rheumatol. 2016;68(Suppl. 10):4357–9.
Kim SC, Solomon DH, Rogers JR, et al. Cardiovascular safety of tocilizumab versus tumor necrosis factor inhibitors in patients with rheumatoid arthritis: a multi-database cohort study. Arthr Rheumatol. 2017;69(6):1154–64.
Kim SC, Solomon DH, Rogers JR, et al. Cardiovascular safety of tocilizumab versus abatacept in patients with rheumatoid arthritis: a multi-database study [abstract no. 527]. Arthr Rheumatol. 2017;69(Suppl 10).
Burmester GR, Choy E, Kivitz A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–85.
Sigaux J, Hamze M, Daien C, et al. Immunogenicity of tocilizumab in patients with rheumatoid arthritis. Jt Bone Spine. 2017;84(1):39–45.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77.
Smolen JS, Breedveld FC, Burmester GR, et al. Treating rheumatoid arthritis to target: 2014 update of recommendations of an international task force. Ann Rheum Dis. 2016;75:3–15.
Dorner T, Strand V, Cornes P, et al. The changing landscape of biosimilars. Ann Rheum Dis. 2016;75:974–82.
Canete JD, Hernandez MV, Sanmarti R. Safety profile of biological therapies for treating rheumatoid arthritis. Expert Opin Biol Ther. 2017;17(9):1089–103.
Ruderman EM. Overview of safety of non-biologic and biologic DMARDs. Rheumatology. 2012;51(Suppl 6):637–43.
van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):164–72.
Acknowledgements
During the peer review process, the manufacturer of tocilizumab was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Lesley Scott is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information about this Adis Drug Review can be found at http://www.medengine.com/Redeem/68FBF06068F81EA7.
Additional information
The manuscript was reviewed by: M. T. Nurmohamed, Amsterdam Rheumatology Immunology Center, VU University Medical Center and Reade Rheumatology, Amsterdam, Netherlands; F. Verhoeven, CHRU Besançon, Rheumatology, Besançon, France.
A correction to this article is available online at https://doi.org/10.1007/s40265-017-0856-4.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 77, 1865–1879 (2017). https://doi.org/10.1007/s40265-017-0829-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0829-7