
Abstract
Since the approval of fingolimod, several selective sphingosine-1-phosphate receptor modulators have entered clinical development for multiple sclerosis. However, side effects can occur with sphingosine-1-phosphate receptor modulators. By considering short-term data across the drug class and longer term fingolimod data, we aim to highlight the potential of sphingosine-1-phosphate receptor modulators in multiple sclerosis, while offering reassurance that their benefit–risk profiles are suitable for long-term therapy. Short-term fingolimod studies demonstrated the efficacy of this drug class, showed that cardiac events upon first-dose administration are transient and manageable, and showed that serious adverse events are rare. Early-phase studies of selective sphingosine-1-phosphate receptor modulators also show efficacy with a similar or improved safety profile, and treatment initiation effects were reduced with dose titration. Longer term fingolimod studies demonstrated sustained efficacy and raised no new safety concerns, with no increases in macular edema, infection, or malignancy rates. Switch studies identified no safety concerns and greater patient satisfaction and persistence with fingolimod when switching from injectable therapies with no washout period. Better outcomes were seen with short than with long washouts when switching from natalizumab. The specific immunomodulatory effects of sphingosine-1-phosphate receptor modulators are consistent with the low observed rates of long-term, drug-related adverse effects with fingolimod. Short-term data for selective sphingosine-1-phosphate receptor modulators support their potential effectiveness in multiple sclerosis, and improved side-effect profiles may widen patient access to this drug class. The long-term safety, tolerability, and persistence profiles of fingolimod should reassure clinicians that sphingosine-1-phosphate receptor modulators are likely to be suitable for the long-term treatment of multiple sclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Sphingosine-1-phosphate receptor modulators are a new class of oral disease-modifying therapy for multiple sclerosis (MS); this review summarizes both short-term efficacy and safety data available for this drug class, and long-term data for fingolimod. |
Short-term data demonstrate the clinical effectiveness on relapses, magnetic resonance imaging lesions, and disability progression of this drug class in MS, while serious adverse events are rare, and side effects at first dose are transient and manageable. |
Fingolimod is an effective MS therapy in the long term, and neither its long-term use nor switching to fingolimod from other treatments increases the risk of adverse events. |
The positive benefit–risk profile of sphingosine-1-phosphate receptor modulators should reassure clinicians that this drug class is suitable for the long-term treatment of MS. |
1 Introduction
Several selective sphingosine-1-phosphate (S1P) receptor modulators are in clinical development for the treatment of patients with relapsing forms of multiple sclerosis (RMS) or secondary progressive multiple sclerosis (SPMS) [1,2,3]. These compounds were developed following the success of fingolimod [1,2,3,4,5], a functional antagonist of S1P receptor subtypes S1P1,3–5, and the first oral disease-modifying therapy (DMT) approved for the treatment of RMS in USA and of relapsing-remitting multiple sclerosis in Europe [6,7,8]. Sphingosine-1-phosphate receptor modulators under development include ponesimod, siponimod, ozanimod, ceralifimod, GSK2018682, and amiselimod [9,10,11].
Sphingosine-1-phosphate receptor modulators have not been shown to be effective in primary progressive multiple sclerosis. A phase III study of fingolimod in patients with primary progressive multiple sclerosis showed a small but non-significant improvement in confirmed disability progression (CDP) with fingolimod over placebo and similar percentage brain volume loss (BVL) in the two treatment groups [12]. This review therefore focuses on the role of S1P receptor modulators in RMS and SPMS.
Other articles have reviewed the mechanisms of action of S1P receptor modulators [2, 11, 13, 14]; thus, here we provide an overview before summarizing short-term efficacy and safety data (≤2 years) across the drug class, and longer term data (>2 years) available from studies of fingolimod. Real-world and observational studies as well as phase II and III clinical trials are considered to understand outcomes among patients potentially under-represented in controlled trials. Here, we aim to illustrate the potential of S1P receptor modulators in the treatment of patients with RMS and SPMS, and provide reassurance that their benefit–risk profiles are suitable for long-term therapy. Studies are summarized in Online Resource 1 of the Electronic Supplementary Material (ESM) [e1–e40].
2 Mechanism of Action of Sphingosine-1-Phosphate (S1P) Receptor Modulators
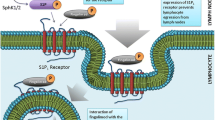
Sphingosine-1-phosphate is a bioactive phospholipid regulating a range of cellular processes, including immunity, inflammation, angiogenesis, heart rate, smooth muscle tone, cell differentiation, cell migration and survival, calcium homeostasis, and endothelium integrity. Its effects are mediated by the G-protein-coupled receptor subtypes S1P1–5 [3, 11]. The pattern of expression and roles of different S1P receptor subtypes are summarized in Fig. 1 [11, 15]. S1P1, S1P2, and S1P3 are ubiquitous [11] but most abundant in immune, cardiovascular, and central nervous system cells [3]; S1P4 is mostly expressed in lymphoid tissues and the lungs [3, 11]; and S1P5 in the central nervous system (on oligodendrocytes), skin, and spleen [3, 11]. S1P1 signaling mediates the trafficking of lymphocytes, particularly their egress from lymph nodes and migration into the blood and target tissues, apparently without inhibiting their activation, proliferation, or effector functions [3, 8, 11, 15,16,17].

Parts a and b reproduced with permission from Jeffery et al. Expert Rev Neurother. 2016;16:31–44 [15]. Part c reproduced/adapted with permission from Subei and Cohen, CNS Drugs. 2015;29:565–75 [11]
Mechanism of action of sphingosine-1-phosphate (S1P) receptor modulators. a The binding of S1P receptor modulators to S1P1 on central memory T-cells (TCM) causes these cells to engulf their own S1P1, resulting in TCM that are unresponsive to S1P signals. Any new S1P receptors being produced inside the cell remain in a state of arrest until S1P receptor modulation is removed. Therefore, TCM do not leave the lymph node in response to S1P signals, and, by inhibiting the movement of TCM into the circulation, S1P receptor modulators prevent these autoreactive cells from migrating into the central nervous system. In contrast, the levels of peripheral effector memory T-cells (TEM) are largely unaffected by S1P receptor modulators, thus preserving immunosurveillance and the capacity to respond to and contain locally invading pathogens. b Interaction of S1P receptor modulators with S1P receptor subtypes. c Interaction of fingolimod and selective S1P receptor drugs with S1P receptor subtypes. aAmiselimod is selective, but its selectivity is unknown. CCR7 C-C chemokine receptor type 7, CV cardiovascular, S1P 1–5 sphingosine-1-phosphate receptor subtypes 1–5, T H 17 T helper cell 17, T N naïve T-cell.
2.1 Fingolimod
Fingolimod is a structural analog of sphingosine [8] and, after phosphorylation, is a functional antagonist of S1P1,3–5 [11], which acts reversibly to retain circulating central memory T cells and naïve T cells in the lymph nodes, thereby reducing the migration of autoreactive lymphocytes into the central nervous system [8]. Effects of fingolimod on B cells and natural killer cells have also been reported [8], and some experimental evidence exists for a direct neuroprotective effect of fingolimod on brain cells (reviewed in Hunter et al. [8] and Jeffery et al. [15]). The therapeutic effects of fingolimod are primarily mediated via S1P1 on lymphocytes; however, certain adverse events (AEs) involving heart rate and atrioventricular (AV) conduction are also caused by the initial agonistic interaction with S1P1, while those involving macular edema or pulmonary function may be associated with its nonselective binding to other S1P receptors, such as S1P3 [11, 15].
2.2 Other S1P Receptor Modulators
Discovery of the receptor-binding characteristics of fingolimod has triggered a search for S1P receptor modulators with a greater selectivity for S1P1. Agents currently under development include ponesimod, siponimod, amiselimod, and ozanimod [11]; GSK2018682 has completed phase I trials [18], although further development seems unlikely [11], and a phase III study of ceralifimod was terminated prematurely, thus its future development is also uncertain [19, 20]. Ponesimod is an orally active, reversible, and selective S1P1 modulator [21]; siponimod, ozanimod, and ceralifimod are orally active selective modulators of S1P1 and S1P5 [22,23,24,25,26]; GSK2018682 is a selective S1P1 modulator [18] that has some activity at S1P5; and amiselimod is known to be a selective S1P receptor modulator, but the subtype selectivity is unknown [11]. Like fingolimod, the therapeutic effects of these compounds are primarily mediated by rapid internalization, degradation, and functional antagonism of S1P1, leading to lymphocyte sequestration in the lymph nodes; the lack of activity on S1P3–4 may alleviate some side effects, including those at first dose. All of these compounds (apart from amiselimod) also have a shorter half-life and show a reduced time to lymphocyte recovery after treatment discontinuation compared with fingolimod, providing an improved safety profile for patients who need to interrupt medication for any reason. The downstream effects of the interactions between these compounds and different S1P receptor subtypes are shown in Fig. 1 [11, 15], and a summary of the S1P receptor modulator pharmacokinetics and pharmacodynamics is provided in Table 1 [6, 7, 18, 21, 22, 27,28,29,30].
3 Short-Term Efficacy (≤2 Years)
Randomized controlled studies [31,32,33,34] of fingolimod typically range from 6 months to 2 years in duration, whereas data published for ponesimod, siponimod, amiselimod, and ozanimod are from dose-ranging clinical studies of no more than 6 months’ duration [21, 28, 35, 36]; short-term efficacy data from these studies are summarized in Online Resource 2 of the ESM [e1, e2, e4, e7, e20, e25, e28, e32].
3.1 Fingolimod
As of November 2016, approximately 184,000 patients with multiple sclerosis (MS) have been treated with fingolimod, with total patient exposure exceeding 397,000 patient-years [37]. In phase II and III studies [31,32,33,34], significant reductions in annualized relapse rate (ARR) and magnetic resonance imaging (MRI) markers of disease activity (gadolinium-enhancing [Gd+] and T2 lesion counts) were consistently observed with fingolimod compared with placebo or intramuscular (IM) interferon (IFN) β-1a. Significantly less BVL was observed with fingolimod than with placebo or IFNβ-1a in studies with a duration longer than 6 months, and significant reductions in the volume of T1-hypointense ‘black holes’ were seen with fingolimod compared with placebo. In the placebo-controlled studies, there were trends towards proportionally more patients remaining free from 3- and 6-month CDP with fingolimod than with placebo; this difference was statistically significant in the FREEDOMS trial [32].
3.2 Other S1P Receptor Modulators
Trials of ponesimod, amiselimod, siponimod, and ozanimod have shown treatment effects on MRI disease activity and ARR in patients with RMS [21, 28, 35, 36]; however, CDP was not assessed in these studies. In patients receiving ponesimod, significant reductions in the numbers of Gd + lesions and combined unique active lesions were reported at all doses compared with placebo. Lower ARRs (significant at 40 mg) and smaller cumulative numbers of T2 lesions (significant at 20 mg) were also reported with ponesimod than with placebo [21]. A dose-dependent numerical reduction in BVL was observed with ponesimod, although p values were not reported.
Amiselimod provided significant dose-dependent reductions in the mean total numbers of T1 Gd + lesions compared with placebo from weeks 8 to 24 in patients with RMS [36]. The mean total number of new or enlarged T2 lesions from weeks 8 to 24 was also significantly reduced with amiselimod 0.2 and 0.4 mg compared with placebo. Annualized relapse rates were lower with amiselimod 0.2 and 0.4 mg than with placebo, although the difference was significant only in the 0.4-mg group. Brain volume loss after 24 weeks of treatment was similar in the amiselimod and placebo groups, although reductions in gray matter volume were significantly smaller with all amiselimod doses than with placebo.
After 3 months of siponimod treatment, lesion activity was reduced compared with placebo; significant relative reductions were observed in the numbers of combined unique active lesions (doses ≥0.5 mg), new Gd + lesions (doses ≥0.5 mg), and T2 lesions (doses ≥1.25 mg). At 6 months, there were significant relative reductions in the number of new Gd+ and T2 lesions with siponimod compared with placebo at doses of ≥0.5 and ≥2 mg, respectively. Over 6 months, ARRs were generally lower with siponimod than with placebo, but differences were significant only in the 2-mg group [35]. In the extension study, reductions in Gd + lesions and ARR were sustained over 2 years with ≥1.25 mg; at the 2-mg dose, 57.7% of patients remained free from combined unique active lesions at 2 years [10]. Positive outcomes with ozanimod have also been reported, with significantly lower cumulative numbers of Gd+ and T2 lesions during weeks 12–24 at both the 0.5- and 1-mg doses compared with placebo. Over 24 weeks, numerically lower ARRs were also reported at both doses compared with placebo [28].
Siponimod has been evaluated in patients with SPMS. In the phase III trial of siponimod 2 mg, the risk of 3- and 6-month CDP was significantly reduced by 21 and 26%, respectively, compared with placebo [38]. Full results from this study have not been published to date.
4 Short-Term Safety (≤2 Years)
Although the therapeutic effects of S1P receptor modulators are primarily mediated via binding to S1P1 on lymphocytes, interactions with this and other S1P receptor subtypes may lead to AEs, for example decreases in heart rate and AV conduction delays after the first dose(s), mild increases in blood pressure, macular edema, and bronchoconstriction. Short-term safety data are summarized in Online Resource 3 of the ESM [e1, e2, e4, e7, e16, e17, e20, e21, e24, e25, e28, e32].
4.1 Fingolimod
In the phase III FREEDOMS, FREEDOMS II, and TRANSFORMS trials [32,33,34], AEs occurred in similar proportions of patients receiving fingolimod 0.5 mg (86–98%), IFNβ-1a (91.6%), and placebo (92–97%); similarly, serious AEs (SAEs) and discontinuations owing to AEs occurred in 5.8–15% and 3.7–18% of patients, respectively, and proportions were similar across treatment groups in each study [32,33,34]. First-dose effects of fingolimod treatment were consistently mild, with transient decreases in heart rate of 8.0–8.5 bpm that started to attenuate after 6 h. Bradycardia was reported in 0.7–2.1% of patients receiving fingolimod 0.5 mg; all cases resolved within 24 h. Second-degree AV block was rarely reported and was absent in one study [33]; where reported, it occurred in 0.2–0.7% of patients. Elevated alanine aminotransferase levels were found in 8.0–8.5% of patients, but all were asymptomatic. Overall incidences of infection were similar in patients receiving fingolimod 0.5 mg, placebo, or IFNβ-1a (42–74%); however, herpes virus, upper respiratory tract, and urinary tract infections were consistently more common with fingolimod than with controls. Macular edema was reported in 0.5–1.0% of patients receiving fingolimod 0.5 mg, but all affected patients were asymptomatic or had stable visual acuity; only one case of macular edema was classified as an SAE. Malignancies were reported at low frequencies across the phase III studies, with basal cell carcinoma being most common, affecting 0.7–3.0% of patients receiving fingolimod 0.5 mg. Incidences of AEs, SAEs, and discontinuations owing to AEs in real-world studies of fingolimod were generally lower than, or at the lower range of, those observed in the phase III trials [39,40,41,42,43]. In a phase II study, overall safety profiles of fingolimod 1.25 and 5.0 mg were similar to that seen with fingolimod 0.5 mg [31].
4.2 Other S1P Receptor Modulators
Safety data from short-term studies of ponesimod, siponimod, amiselimod, and ozanimod are broadly similar to those from fingolimod studies [10, 21, 28, 35, 36]; AEs occurred at similar rates for active treatment and placebo in each of the trials, the most common AEs, such as headache, nasopharyngitis, dizziness, and influenza, occurring at rates similar to those reported with fingolimod. Serious AEs in patients receiving ponesimod and siponimod were somewhat more frequent than with placebo, as were AEs causing discontinuation (ponesimod, 5.3–13.4%, placebo, 2.5%; siponimod, 12–20% placebo, 4.0%) [21, 35]. With amiselimod, incidences of AEs causing discontinuation and of SAEs were similar in the amiselimod and placebo groups [36]. With ponesimod and siponimod, first-dose effects (decreased heart rate, bradycardia, and second-degree AV block) and elevated alanine aminotransferase levels occurred at rates similar to those seen with fingolimod; however, cardiac events were reported as reasons for discontinuation in 2.6% of patients receiving ponesimod and 3.4% of those receiving siponimod [21, 35], and second-degree AV block was the only SAE seen with siponimod that was reported in at least two patients in the same treatment group [21, 35].
In the siponimod extension study, dose titration effectively reduced the first-dose effects on heart rate seen with higher doses, with no cardiac SAEs reported [10]. Proportionally more cases of macular edema were classified as SAEs with ponesimod than with siponimod, and these cases resulted in treatment discontinuation. Incidences of infections were similar among patients treated with placebo, ponesimod, or siponimod, with urinary and upper respiratory tract infections being most common; unlike with fingolimod treatment, no cases of herpes virus infection were reported following treatment with ponesimod or siponimod [21, 35]. Malignancies occurred at low rates with ponesimod and siponimod [10, 21, 35].
The safety profiles of amiselimod and ozanimod differed somewhat from those reported for the other compounds. First-dose effects were not observed with amiselimod [36]. Mean heart rate remained stable during the 6 h after initial dosing and throughout the study, and there was no clinically relevant bradycardia, sinus pause, or AV block in any amiselimod group. One episode each of second-degree AV block and non-sustained ventricular tachycardia were reported as SAEs with amiselimod; however, both patients were asymptomatic, and these events were not considered clinically significant by the cardiologist on the safety monitoring board [36]. Moreover, no cases of macula edema were reported with amiselimod. Incidences of infections were similar among patients treated with placebo and amiselimod (26–32%), with nasopharyngitis and upper respiratory tract infections being most common. Headache was reported more frequently in each of the amiselimod groups (10%) than with placebo (4%); frequencies of cardiac disorders were low and similar across treatment groups (4–8%) [36]. Liver enzyme abnormalities led to discontinuations in three patients (one each in the placebo [1%], amiselimod 0.2-mg [1%], and amiselimod 0.4-mg [1%] groups), and slight dose-dependent increases in mean values for alanine aminotransferase, aspartate aminotransferase, and γ-glutamyltransferase were seen with amiselimod; however, no symptoms were associated with these abnormal values, thus they were not considered to be clinically significant [36]. With ozanimod, there were no discontinuations owing to an AE, and no cases of macular edema, notable infections, notable cardiovascular AEs, or malignancies; and with dose titration, heart rate was reduced at first dose by ≤2 bpm [28].
5 Long-Term Efficacy (>2 Years)
The efficacy benefits of fingolimod reported during the 2-year FREEDOMS trial were sustained in the 2-year FREEDOMS extension study [44]. In the continuous-fingolimod patient groups, the ARR was lower (p < 0.0001), BVL was reduced (p < 0.05), and a greater proportion of patients were free from 3-month CDP (p < 0.05) compared with the placebo–fingolimod switch-patient groups [44]. In the placebo–fingolimod switch groups, ARR was lower (p < 0.001 for both groups), and BVL was reduced (p < 0.01 for the placebo–fingolimod 0.5 mg group) after switching to fingolimod [44]. Similarly, the low levels of clinical and MRI markers of disease activity reported with fingolimod in the FREEDOMS II trial were maintained in the extension among patients who continued on treatment [45]. From baseline to the end of the study, ARR was significantly lower in the continuous-fingolimod groups than in the placebo–fingolimod switch groups (p < 0.001); ARR, MRI disease activity, and BVL rates remained low with continuous fingolimod treatment in the trial extension [45].
In the TRANSFORMS extension study, effects of fingolimod were maintained in the long term (for up to 4.5 years), with low rates of disease activity and improvements in efficacy observed after patients switched from IFNβ-1a IM to fingolimod [46]. From baseline to the end of study, ARR was significantly lower in patients receiving continuous fingolimod than in patients in the IFNβ-1a–fingolimod switch group (0.17 vs. 0.27, respectively; 35% reduction in relapse risk; p < 0.001) [46]. In a post hoc analysis, the proportion of patients with no evidence of disease activity (relapses, 3-month CDP, MRI activity; no evidence of disease activity-3) increased by approximately 50% in the 1st year after switching to fingolimod (from 44.3 to 66.0%) [46].
After 5 years in the ongoing LONGTERMS study, a single-arm open-label extension to various phase II, III, and IIIb trials of fingolimod (Fig. 2) [47], clinical disease activity remained low in patients treated with fingolimod [48]. Annualized relapse rate was low during the 1st year and remained low during each subsequent year up to interim analysis [48]. Mean Expanded Disability Status Scale scores remained stable, with most patients not reaching an Expanded Disability Status Scale score higher than 3 after receiving fingolimod [48]. Overall, most patients remained free from 6-month CDP in the FREEDOMS (78%) and TRANSFORMS (75%) cohorts [48].

Adapted with permission from Khatri, Ther Adv Neurol Disord. 2016;9:130–47 [47]
Study design of the pivotal and extension studies of fingolimod, and of the other sphingosine-1-phosphate (S1P) receptor modulators. Areas shaded gray indicate that the S1P receptor subtypes are unaffected by the drugs in each section. aAmiselimod is selective, but its selectivity is unknown. BOLD BAF312 on MRI lesion given once daily, DREAMS a study of the safety and efficacy of ONO-4641 in patients with relapsing–remitting multiple sclerosis, EPOC evaluate patient outcomes, EU European Union, EXPAND exploring the efficacy and safety of siponimod in patients with secondary progressive multiple sclerosis, FIRST fingolimod initiation and cardiac safety trial, FREEDOMS FTY720 research evaluating effects of daily oral therapy in multiple sclerosis, GA glatiramer acetate, IFN interferon, IM intramuscular, LONGTERMS long-term study of fingolimod in multiple sclerosis patients from the FTY clinical program, MOMENTUM a randomized, double-blind, placebo-controlled, phase II, dose-finding study of MT-1303 in subjects with relapsing–remitting multiple sclerosis, OPTIMUM oral ponesimod versus teriflunomide in relapsing multiple sclerosis, PANGAEA post authorization noninterventional German safety of Gilenya in relapsing–remitting multiple sclerosis patients, PREFERMS evaluation of patient retention of fingolimod versus currently approved disease-modifying therapy in patients with relapsing–remitting multiple sclerosis, RADIANCE safety and efficacy of the selective sphingosine-1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis, S1P 1–5 sphingosine-1-phosphate receptor subtypes 1–5, SC subcutaneous, START study to validate telemetric ECG systems for first-dose administration of fingolimod, SUNBEAM phase III study of RPC1063 in relapsing multiple sclerosis, TOFINGO disease control and safety in patients with relapsing–remitting multiple sclerosis switching from natalizumab to fingolimod, TRANSFORMS trial assessing injectable interferon versus FTY720 oral in relapsing–remitting multiple sclerosis, TRANSITION a two-year observational study to evaluate the safety profile of fingolimod in patients with multiple sclerosis who switch from natalizumab to fingolimod, VERIFY investigating the effect of recent immunization in patients receiving fingolimod therapy.
Results of the 36-month interim analysis [49] of the PANGAEA study, a 5-year prospective study designed to collect real-world efficacy, safety, and pharmacoeconomic data from patients receiving fingolimod in Germany [50], supported the positive efficacy profile demonstrated by fingolimod in the phase III clinical trials and extensions [49]. A mean baseline ARR of 1.5 was reduced to 0.43, 0.41, and 0.37 at 12, 24, and 36 months, respectively. In the first, second, and third years of the study, 67.9, 68.3, and 70.1% of patients, respectively, were relapse-free [49]. The mean Expanded Disability Status Scale score (3.0) was stable over 36 months, and 70.5% of patients had no clinical disease activity in year 3 [49]. Treatment satisfaction improved by 3 points over 24 months [49].
6 Long-Term Safety Data (>2 Years)
In the extension [44] to the 2-year FREEDOMS trial [32], long-term fingolimod treatment was well tolerated, and no new safety indications were observed [44]. Adverse events associated with treatment initiation were seen in the switch group, but the overall incidences and patterns of AEs (91.7–95.5%) and SAEs (7.1–11.7%) were similar across groups [44]. Cardiac effects associated with initiation of fingolimod were transient; blood pressure remained stable in the continuous-treatment groups [44].
In the 2-year extension to FREEDOMS II [45, 51], the safety profile of fingolimod was consistent with that observed in the core study. The incidences of AEs were similar across the continuous-fingolimod and treatment-switch groups (85.3% and 87.9%) [51]; macular edema was confirmed in one patient in each switch group [51], and there was one reported occurrence of squamous cell carcinoma and one of basal cell carcinoma in the continuous-fingolimod 0.5-mg group [51].
In the TRANSFORMS extension, the safety profile was consistent with that observed in the core study [46]. The incidences of AEs (94.7% and 92.2%) and SAEs (15.4 and 12.6%) were similar in the continuous-fingolimod and IFNβ-1a–fingolimod switch groups, respectively [46]. Transient effects on heart rate at treatment initiation and effects on liver function were observed, but there were no cases of macular edema [46]. The incidence of herpes viral infections was similar in both treatment groups (continuous-fingolimod group, n = 36 [10.1%]; treatment-switch group, n = 25 [15%]) [46]. There was an imbalance in the incidence of cancer between the continuous-fingolimod and treatment-switch groups (n = 14 and n = 1, respectively) [46], but the incidences of basal cell carcinoma and other cancer types were similar to those reported in a pooled analysis of 3553 patients treated with fingolimod in the core and extension trials [46].
In the LONGTERMS study, the safety profile of fingolimod was confirmed at interim analysis, and no long-term safety or tolerability concerns were raised [52, 53]. Incidence rate ratios [95% confidence interval (CI)] for AEs and SAEs were 0.74 (0.68–0.79) and 0.73 (0.60–0.91), respectively, relative to events reported in a cohort of patients pooled from the phase III trials, suggesting that incidence does not increase with prolonged fingolimod exposure [52, 53]. Incidence rate ratios for AEs of special interest (including bradyarrhythmia post first-dose, hypertension, infections, liver transaminase elevation, lymphopenia, macular edema, malignant neoplasms, reproductive toxicity, respiratory conditions, skin cancer, and thromboembolic events) were <1.0, with the exception of lymphopenia [52, 53]. Physicians had access to lymphocyte counts in LONGTERMS, but these data were blinded in the phase III trials unless they reached a threshold of 0.2 × 109/L; therefore, lymphopenia may have been reported more frequently in LONGTERMS [52, 53]. The incidence of lymphopenia was not associated with an increased risk of infection (incidence rate ratio, 0.7), and no opportunistic infections occurred in patients with sustained low actual lymphocyte counts [52]. Results of a 36-month interim analysis [49] of the PANGAEA study [50] also supported the positive safety and tolerability profiles seen in the phase III trials [49]. In the first and second years, 10.8 and 9.1% of patients discontinued therapy, 4.3 and 2.3% owing to AEs, respectively; 45.4% of patients experienced no AEs [49].
7 S1P Receptor Modulator Switch Studies
Studies examining switching treatment to an S1P receptor modulator are currently confined to studies of fingolimod. In the randomized 12-month PREFERMS study of treatment retention with fingolimod and injectable DMTs (iDMTs), retention was significantly greater with fingolimod (81.3% [n = 352] vs. 29.2% [n = 125]; p < 0.0001, respectively). Among patients who switched treatment, fewer discontinued fingolimod [1.1% (n = 5)] than iDMTs [9.8% (n = 43)], and patients receiving fingolimod also experienced greater treatment satisfaction at all assessments than those given iDMT (p < 0.001). The rate of AEs per patient-year was lower with fingolimod (4.01) than with iDMTs (7.01) [39].
TRANSITION is a 2-year, prospective, observational, single-cohort, real-world study examining the safety profile of fingolimod in patients with RMS who switched from natalizumab (target enrollment: N = 1500) [54]. In the 1-year interim analysis (n = 639) [55], AEs and SAEs were experienced by 48 and 8% of patients, respectively [54]. Among AEs of interest, exposure-adjusted incidences (95% CI) per 100 patient-years were: hypertension, 2.80 (1.66–4.42); lymphopenia, 1.85 (0.96–3.23); leukopenia, 0.92 (0.34–1.99); liver transaminase elevation, 0.46 (0.09–1.34); second-degree AV block, 0.30 (0.04–1.10); macular edema, 0.30 (0.04–1.10); basal cell carcinoma, 0.30 (0.04–1.10); and infections, 15.2 (12.2–18.7). The incidence of infections reported as SAEs was 1.38 (0.63–2.62) [55].
Evaluate patient outcomes (EPOC) was a randomized, open-label, 6-month, multicenter study of treatment satisfaction in patients who switched from an iDMT to fingolimod 0.5 mg without a washout period, compared with patients who remained on iDMT or switched to an alternative iDMT [56]. Patient-reported satisfaction measures (including effectiveness, side effects, and convenience), and measures of fatigue and depression, were significantly improved in patients who switched to fingolimod compared with those receiving iDMT (all p < 0.001) [56]. Adverse events and SAEs were experienced by 78.8 and 4% of patients taking fingolimod, and by 62 and 2% of patients on iDMTs, respectively. The three most frequent AEs in the fingolimod group were headache (12.4%), fatigue (11.5%), and upper respiratory tract infection (6.5%). Multiple sclerosis relapse (0.6%), lymphopenia (0.3%), and non-cardiac chest pain (0.3%) were the only SAEs with more than one case in the fingolimod group, while only MS relapse was reported in the iDMT group (0.8%). Adverse events leading to study-drug discontinuation occurred in 5.2% of patients taking fingolimod (most commonly macular edema [0.5%] and fatigue [0.5%]) compared with 1.6% of patients taking iDMT. Infections were reported in similar proportions of patients in the fingolimod group (30.1%) and the iDMT group (27.8%). Similarly to LONGTERMS, low lymphocyte counts were not associated with increased infection rate in fingolimod-treated patients, and no significant differences were found in lymphocyte counts between fingolimod-treated patients who experienced an infection and those who did not [56].
The randomized, double-blind, placebo-controlled, 32-week TOFINGO study examined the effect of washout duration in patients with relapsing-remitting multiple sclerosis switching from natalizumab to fingolimod (n = 142) [57]. Adverse events were experienced by 68% of patients and were mostly mild or moderate with a similar incidence across groups. No unusually severe relapses or opportunistic infections occurred after treatment switch, and shorter washout periods were associated with less MRI disease activity than longer periods [57].
8 Safety Precautions for Fingolimod Therapy
As the only approved S1P receptor modulator, fingolimod has the greatest amount of safety data available, together with a large amount of clinical experience, with total patient exposure exceeding 397,000 patient-years [37]. Factors that should be considered before initiation of fingolimod or that require monitoring on treatment, including first-dose monitoring, pregnancy, diabetes mellitus, posterior reversible encephalopathy syndrome, basal cell carcinoma, infections such as varicella, opportunistic cryptococcal infections, and progressive multifocal leukoencephalopathy, are summarized in Table 2 [6, 7, 58,59,60,61].
First-dose monitoring for cardiovascular effects is required with fingolimod treatment and is likely to be required for the second generation of S1P receptor modulators; and as such is unlikely to be discontinued in the near future. However, clinical data with fingolimod indicate that the first-dose effects are not as widespread or as severe as previously thought, allowing the first dose of fingolimod treatment to be initiated outside of the clinic setting. Interim results from a study of fingolimod initiation (n = 3951) reported that 0.7% of patients experienced a bradycardia of <45 bpm, and 1.6% had an AV block of grade ≥2 following the first dose [58]. Furthermore, limited first-dose effects have been observed with ozanimod and amiselimod [28, 36]; therefore, the need for first-dose monitoring may be reduced in future.
Fingolimod showed developmental toxicity in animal studies, and the S1P receptor is known to be involved in vascular formation during embryogenesis; women should therefore avoid becoming pregnant while taking fingolimod, and treatment discontinuation is recommended if pregnancy occurs [6, 7]. Minor dose-dependent reductions in values for forced expiratory volume and diffusion capacity for carbon monoxide were observed with fingolimod treatment, starting as early as 1 month after treatment initiation and remaining stable thereafter [6, 7]. Fingolimod should therefore be used with caution in patients with severe respiratory disease, pulmonary fibrosis, or chronic obstructive pulmonary disease [6]. Basal cell carcinoma has been associated with fingolimod use, with increased incidence reported in clinical trials compared with placebo; healthcare professionals and patients are therefore advised to monitor for suspicious skin lesions [6, 7]. Tumefactive lesions, which can be part of a spectrum of the MS course itself, have been noted with fingolimod during therapy as well as after withdrawal of fingolimod [62,63,64,65,66]. As with natalizumab, a high-efficacy DMT, disease reactivation, often labeled as ‘rebound’, has been reported after withdrawal of fingolimod therapy [67,68,69,70,71].
A dose-dependent increase in the risk of macular edema was observed in the fingolimod clinical development program [6, 7]. Macular edema occurred predominantly during the first 3–4 months of therapy and generally resolved after fingolimod discontinuation; macular edema has also been reported in the post-marketing setting, usually within the first 6 months of therapy. Patients are required to undergo examination of the fundus including the macula prior to treatment and at 3–4 months after starting treatment, and should also be examined at any time when they report visual disturbances while receiving therapy [6, 7]. Patients with a history of uveitis and patients with diabetes mellitus are at increased risk of macular edema with fingolimod; thus regular follow-up examinations are recommended for these patients during treatment [6, 7].
The dose-dependent reduction in peripheral lymphocyte count with fingolimod treatment can increase the risk of infections [6, 7, 60]. In clinical trials, some infections (such as herpes virus and respiratory tract infections) were more common with fingolimod than with placebo, and there was a higher incidence of serious infections with fingolimod than with placebo. Serious infections with opportunistic pathogens including viruses, fungi, and bacteria have been reported in the post-marketing setting [6, 7]. These include serious life-threatening events of disseminated varicella zoster and herpes simplex infections (including one fatal event), and cases of Kaposi’s sarcoma. Cryptococcal infections have also been reported, albeit rarely, with fingolimod in the post-marketing setting, typically occurring after approximately 2–3 years of treatment [6, 7, 60]. Patients with signs or symptoms consistent with any of these infections should undergo prompt diagnostic evaluation and appropriate management.
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the John Cunningham virus; it typically occurs only in patients who are immunocompromised, and may be fatal or result in severe disability. Cases of PML have been reported in patients treated with fingolimod [6, 7, 61]. In many instances, patients had received prior treatment with natalizumab, which is associated with an increased risk of PML [61]. However, as of February 2017, 13 cases of probable or definite PML have been reported in patients treated with fingolimod without prior natalizumab therapy [61, 72]. These cases occurred in patients who had been receiving fingolimod therapy for between 18 and 65 months, and who were aged 34–63 years [61, 72]. These cases of PML were not associated with sustained grade 4 lymphopenia [61]; more research is needed to determine the relationship between lymphopenia and the risk of developing PML [73]. The low frequency of PML with fingolimod therapy means that there is currently limited information available to identify factors associated with an increased risk of this infection. Fingolimod should be withheld at the first sign or symptom suggestive of PML and an appropriate diagnostic evaluation performed; vigilance for PML-suspicious lesions during routine MRI is also important [6, 7]. In a recent article contextualizing the risk of PML with DMTs in MS, natalizumab was classified as having a high potential risk of PML, while fingolimod was grouped together with dimethyl fumarate as having a low risk [61, 72].
9 Conclusions
The ability of S1P receptor modulators to block immune cell trafficking specifically [3, 8, 11, 15,16,17] makes them particularly interesting for the treatment of a number of autoimmune diseases including MS [1, 3]. Fingolimod is the only approved agent with extensively characterized efficacy and safety profiles, but several S1P receptor modulators are currently in development. The specificity of the immunomodulatory effects of S1P receptor modulators suggests that the likelihood of off-target AEs in the long term is small, unlike with other drugs licensed for the treatment of MS associated with broad-range immune suppression. The short-term efficacy data available for the S1P receptor modulators under development support the potential effectiveness of this drug class in MS, and the available safety data are consistent across the class and with the known mechanism of action. The long-term safety, tolerability, and persistence profiles of fingolimod, including upon switching from other treatments, should reassure clinicians regarding the suitability of S1P receptor modulators as treatment options for patients with MS.
References
Gonzalez-Cabrera PJ, Brown S, Studer SM, Rosen H. S1P signaling: new therapies and opportunities. F1000Prime Rep. 2014;6:109. doi:10.12703/P6-109.
Radick L, Mehr SR. The latest innovations in the drug pipeline for multiple sclerosis. Am Health Drug Benefits. 2015;8(8):448–53.
Urbano M, Guerrero M, Rosen H, Roberts E. Modulators of the sphingosine 1-phosphate receptor 1. Bioorg Med Chem Lett. 2013;23(23):6377–89. doi:10.1016/j.bmcl.2013.09.058.
Aktas O, Kury P, Kieseier B, Hartung HP. Fingolimod is a potential novel therapy for multiple sclerosis. Nat Rev Neurol. 2010;6(7):373–82. doi:10.1038/nrneurol.2010.76.
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. doi:10.1097/WNF.0b013e3181cbf825.
European Medicines Agency. Gilenya (fingolimod) summary of product characteristics. 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf. Accessed 25 May 2016.
US Food and Drug Administration. Gilenya (fingolimod) prescribing information. 2016. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/gilenya.pdf. Accessed 25 May 2016.
Hunter SF, Bowen JD, Reder AT. The direct effects of fingolimod in the central nervous system: implications for relapsing multiple sclerosis. CNS Drugs. 2015;30(2):135–47. doi:10.1007/s40263-015-0297-0.
Hammond ER. Perspectives on safety and efficacy: the BOLD phase 2 extension study of siponimod in relapsing-remitting multiple sclerosis. JAMA Neurol. 2016;73(9):1052–4. doi:10.1001/jamaneurol.2016.2284.
Kappos L, Li DK, Stuve O, Hartung HP, Freedman MS, Hemmer B, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD study. JAMA Neurol. 2016;73(9):1089–98. doi:10.1001/jamaneurol.2016.1451.
Subei AM, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs. 2015;29(7):565–75. doi:10.1007/s40263-015-0261-z.
Lublin F, Miller DH, Freedman MS, Cree BAC, Wolinsky JS, Weiner H, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–84. doi:10.1016/S0140-6736(15)01314-8.
Candido K, Soufi H, Bandyopadhyay M, Dasgupta S. Therapeutic impact of sphingosine 1-phosphate receptor signaling in multiple clerosis. Mini Rev Med Chem. 2016;16(7):547–54.
Guerrero M, Urbano M, Roberts E. Sphingosine 1-phosphate receptor 1 agonists: a patent review (2013–2015). Expert Opin Ther Pat. 2016;26(4):455–70. doi:10.1517/13543776.2016.1157165.
Jeffery DR, Rammohan KW, Hawker K, Fox E. Fingolimod: a review of its mode of action in the context of its efficacy and safety profile in relapsing forms of multiple sclerosis. Expert Rev Neurother. 2016;16(1):31–44. doi:10.1586/14737175.2016.1123094.
Francis G, Kappos L, O’Connor P, Collins W, Tang D, Mercier F, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult Scler. 2014;20(4):471–80. doi:10.1177/1352458513500551.
Kappos L, Cohen J, Collins W, de Vera A, Zhang-Auberson L, Ritter S, et al. Fingolimod in relapsing multiple sclerosis: an integrated analysis of safety findings. Mult Scler Relat Disord. 2014;3(4):494–504. doi:10.1016/j.msard.2014.03.002.
Xu J, Gray F, Henderson A, Hicks K, Yang J, Thompson P, et al. Safety, pharmacokinetics, pharmacodynamics, and bioavailability of GSK2018682, a sphingosine-1-phosphate receptor modulator, in healthy volunteers. Clin Pharmacol Drug Dev. 2014;3(3):170–8. doi:10.1002/cpdd.98.
EU Clinical Trials Register. EudraCT number: 2013-003126-83. A phase III, randomized, double-blind, double dummy, multicenter trial comparing the efficacy and safety of 2 doses of daily oral ONO 4641 (0.05 mg and 0.1 mg) versus interferon-β-1a 30 µg IM weekly in subjects with relapsing multiple sclerosis: efficacy and safety of ONO-4641 versus interferon-ß-1a in patients with multiple sclerosis. Available from: http://www.clinicaltrialsregister.eu/ctr-search/search?query=ceralifimod. Accessed 9 Aug 2017.
EU Clinical Trials Register. EudraCT number: 2013-002351-15. A phase III, randomized, double-blind, double dummy, multicenter trial comparing the efficacy and safety of 2 doses of daily oral ONO 4641 (0.05 mg and 0.1 mg) versus interferon-beta-1a 30 mcg IM weekly in subjects with relapsing-remitting multiple sclerosis. Available from: http://www.clinicaltrialsregister.eu/ctr-search/search?query=ceralifimod. Accessed 9 Aug 2017.
Olsson T, Boster A, Fernandez O, Freedman MS, Pozzilli C, Bach D, et al. Oral ponesimod in relapsing–remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry. 2014;85(11):1198–208. doi:10.1136/jnnp-2013-307282.
Gergely P, Nuesslein-Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167(5):1035–47. doi:10.1111/j.1476-5381.2012.02061.x.
Komiya T, Sato K, Shioya H, Inagaki Y, Hagiya H, Kozaki R, et al. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin Exp Immunol. 2013;171(1):54–62. doi:10.1111/j.1365-2249.2012.04669.x.
O’Sullivan C, Schubart A, Mir AK, Dev KK. The dual S1PR1/S1PR5 drug BAF312 (siponimod) attenuates demyelination in organotypic slice cultures. J Neuroinflammation. 2016;13:31. doi:10.1186/s12974-016-0494-x.
Pan S, Gray NS, Gao W, Mi Y, Fan Y, Wang X, et al. Discovery of BAF312 (siponimod), a potent and selective S1P receptor modulator. ACS Med Chem Lett. 2013;4(3):333–7. doi:10.1021/ml300396r.
Scott FL, Clemons B, Brooks J, Brahmachary E, Powell R, Dedman H, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016;173(11):1778–92. doi:10.1111/bph.13476.
Brossard P, Derendorf H, Xu J, Maatouk H, Halabi A, Dingemanse J. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br J Clin Pharmacol. 2013;76(6):888–96. doi:10.1111/bcp.12129.
Cohen JA, Arnold DL, Comi G, Bar-Or A, Gujrathi S, Hartung JP, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373–81. doi:10.1016/S1474-4422(16)00018-1.
Olson A, Timony G, Hartung JP, Smith H, Peach RJ, Rosen H, et al. Sa1222 results of a safety and tolerability study of Rpc 1063, a novel orally administered sphingosine-1-phosphate receptor 1 (S1p1r) agonist, in healthy adult volunteers. Gastroenterology. 2013;44(5 Suppl 1):S-234. doi:10.1016/S0016-5085(13)60828-8.
Sugahara K, Maeda Y, Shimano K, Mogami A, Kataoka H, Ogawa K, et al. Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br J Pharmacol. 2017;174(1):15–27. doi:10.1111/bph.13641.
Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–40. doi:10.1056/NEJMoa052643.
Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi:10.1056/NEJMoa0909494.
Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–56. doi:10.1016/S1474-4422(14)70049-3.
Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15. doi:10.1056/NEJMoa0907839.
Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756–67. doi:10.1016/S1474-4422(13)70102-9.
Kappos L, Arnold DL, Bar-Or A, Camm J, Derfuss T, Kieseier BC, et al. Safety and efficacy of amiselimod in relapsing multiple sclerosis (MOMENTUM): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016;15(11):1148–59. doi:10.1016/S1474-4422(16)30192-2.
Novartis. Novartis patient exposure. Q3 Novartis Pharmaceuticals annual report. 2016.
Kappos L, Bar-Or A, Cree B, Fox R, Giovannoni G, Gold R, et al. Efficacy and safety of siponimod in secondary progressive multiple sclerosis: results of the placebo controlled, double-blind, phase III EXPAND study. ECTRIMS Online Libr. 2016;147077:250.
Cree BAC, Wynn D, Cascione M, Meng X, Schofield L, Tenenbaum N. Key results from PREFERMS: real-world patient retention and outcomes on fingolimod versus platform injectable disease-modifying therapies in early relapsing–remitting multiple sclerosis. Neurology. 2016;78:P3–115 (Meeting Abstracts).
Gold R, Comi G, Palace J, Siever A, Gottschalk R, Bijarnia M, et al. Assessment of cardiac safety during fingolimod treatment initiation in a real-world relapsing multiple sclerosis population: a phase 3b, open-label study. J Neurol. 2014;261(2):267–76. doi:10.1007/s00415-013-7115-8.
Ontaneda D, Hara-Cleaver C, Rudick RA, Cohen JA, Bermel RA. Early tolerability and safety of fingolimod in clinical practice. J Neurol Sci. 2012;323(1–2):167–72. doi:10.1016/j.jns.2012.09.009.
Ordonez-Boschetti L, Rey R, Cruz A, Sinha A, Reynolds T, Frider N, et al. Safety and tolerability of fingolimod in Latin American patients with relapsing-remitting multiple sclerosis: the open-label FIRST LATAM study. Adv Ther. 2015;32(7):626–35. doi:10.1007/s12325-015-0224-2.
Wynn D, LaGanke C, Schofield L, Meng X, Tenenbaum N. Real-world fingolimod first-dose effects in patients with pre-existing hypertension, pre-existing cardiac conditions and in those receiving selective serotonin reuptake inhibitors. Neurology. 2016;78:P3.065 (Meeting Abstracts).
Kappos L, O’Connor P, Radue EW, Polman C, Hohlfeld R, Selmaj K, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology. 2015;84(15):1582–91. doi:10.1212/WNL.0000000000001462.
Reder AT, Jeffery D, Goodin D, Kappos L, Lublin FD, Radue EW, et al. Long-term efficacy of fingolimod in patients with relapsing remitting multiple sclerosis: results from the phase 3 FREEDOMS II extension study. Mult Scler. 2013;19(Suppl. 11):510–1.
Cohen JA, Khatri B, Barkhof F, Comi G, Hartung HP, Montalban X, et al. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. J Neurol Neurosurg Psychiatry. 2016;87(5):468–75. doi:10.1136/jnnp-2015-310597.
Khatri BO. Fingolimod in the treatment of relapsing-remitting multiple sclerosis: long-term experience and an update on the clinical evidence. Ther Adv Neurol Disord. 2016;9(2):130–47. doi:10.1177/1756285616628766.
Kappos L, Cohen JA, Barkhof F, Cappiello L, Zhang Y, Von Rosenstiel P. Relapse rates and disability remain consistently low with long-term fingolimod therapy: five-year interim results of the LONGTERMS extension study. Mult Scler. 2013;19(Suppl. 11):486–7.
Ziemssen T, Albrecht H, Haas J, Klotz L, Lang M, Lassek C, et al. 36 months PANGAEA: a 5-year non-interventional study of safety, efficacy and pharmacoeconomic data for fingolimod patients in daily clinical practice. Value Health. 2015;18:A749.
Ziemssen T, Kern R, Cornelissen C. The PANGAEA study design: a prospective, multicenter, non-interventional, long-term study on fingolimod for the treatment of multiple sclerosis in daily practice. BMC Neurol. 2015;15:93. doi:10.1186/s12883-015-0342-0.
Rammohan K, Jeffery D, Goodin DS, Kappos L, Lublin F, Radue EW, et al. Long-term safety of fingolimod in patients with relapsing-remitting multiple sclerosis: results from phase 3 FREEDOMS II extension study. Neurology. 2013;80(Suppl. P01.165).
Cohen J, Kappos L, Selmaj K, Gottschalk R, Pradhan A, Chen Y, et al. Long-term safety and effectiveness of fingolimod: 7 year data from the LONGTERMS study. ECTRIMS Online Libr. 2015;115507:P591.
Cohen J, Von Rosenstiel P, Gottschalk R, Pradhan A, Zhang Y, Kappos L. Long-term safety of fingolimod: an interim analysis of the LONGTERMS cohort. Neurology. 2015;18(Suppl. 14):S4.006.
Cohan S, Trojano M, Sinha A, Patwardhan M, Tomic D. Study design of TRANSITION: an observational study to evaluate the safety profile of fingolimod in patients switched from natalizumab. Int J MS Care. 2014;16(Suppl. 3):63 (abstract DX2).
Butzkueven H, Weller B, Giacomini PS, Cohan S, Ziemssen T, Tomic D, et al. Baseline characteristics and interim analysis results of TRANSITION: a 2-year observational study evaluating the safety profile of patients with multiple sclerosis who switched from natalizumab to fingolimod. Mult Scler. 2015;21(11 Suppl.):P596 (abstract).
Fox E, Edwards K, Burch G, Wynn DR, LaGanke C, Crayton H, et al. Outcomes of switching directly to oral fingolimod from injectable therapies: results of the randomized, open-label, multicenter, Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Mult Scler Relat Disord. 2014;3(5):607–19. doi:10.1016/j.msard.2014.06.005.
Kappos L, Radue EW, Comi G, Montalban X, Butzkueven H, Wiendl H, et al. Switching from natalizumab to fingolimod: a randomized, placebo-controlled study in RRMS. Neurology. 2015;85(1):29–39. doi:10.1212/WNL.0000000000001706.
Ayzenberg I, Hoepner R, Kleiter I. Fingolimod for multiple sclerosis and emerging indications: appropriate patient selection, safety precautions, and special considerations. Ther Clin Risk Manag. 2016;12:261–72. doi:10.2147/TCRM.S65558.
Dubey D, Cano CA, Stuve O. Update on monitoring and adverse effects of approved second-generation disease-modifying therapies in relapsing forms of multiple sclerosis. Curr Opin Neurol. 2016;29(3):278–85. doi:10.1097/WCO.0000000000000321.
Winkelmann A, Loebermann M, Reisinger EC, Hartung HP, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. 2016;12(4):217–33. doi:10.1038/nrneurol.2016.21.
Berger JR. Classifying PML risk with disease modifying therapies. Mult Scler Relat Disord. 2017;12:59–63. doi:10.1016/j.msard.2017.01.006.
Berger B, Baumgartner A, Rauer S, Mader I, Luetzen N, Farenkopf U, et al. Severe disease reactivation in four patients with relapsing-remitting multiple sclerosis after fingolimod cessation. J Neuroimmunol. 2015;282:118–22. doi:10.1016/j.jneuroim.2015.03.022.
Hakiki B, Portaccio E, Giannini M, Razzolini L, Pasto L, Amato MP. Withdrawal of fingolimod treatment for relapsing-remitting multiple sclerosis: report of six cases. Mult Scler. 2012;18(11):1636–9. doi:10.1177/1352458512454773.
Hatcher SE, Waubant E, Nourbakhsh B, Crabtree-Hartman E, Graves JS. Rebound syndrome in patients with multiple sclerosis after cessation of fingolimod treatment. JAMA Neurol. 2016;73(7):790–4. doi:10.1001/jamaneurol.2016.0826.
Piscolla E, Hakiki B, Pasto L, Razzolini L, Portaccio E, Amato MP. Rebound after fingolimod suspension in a pediatric-onset multiple sclerosis patient. J Neurol. 2013;260(6):1675–7. doi:10.1007/s00415-013-6933-z.
Sempere AP, Berenguer-Ruiz L, Feliu-Rey E. Rebound of disease activity during pregnancy after withdrawal of fingolimod. Eur J Neurol. 2013;20(8):e109–10. doi:10.1111/ene.12195.
Boangher S, Goffette S, Van Pesch V, Mespouille P. Early relapse with tumefactive MS lesion upon initiation of fingolimod therapy. Acta Neurol Belg. 2016;116(1):95–7. doi:10.1007/s13760-015-0495-z.
Faissner S, Hoepner R, Lukas C, Chan A, Gold R, Ellrichmann G. Tumefactive multiple sclerosis lesions in two patients after cessation of fingolimod treatment. Ther Adv Neurol Disord. 2015;8(5):233–8. doi:10.1177/1756285615594575.
Harirchian MH, Taalimi A, Siroos B. Emerging tumefactive MS after switching therapy from interferon-beta to fingolimod: a case report. Mult Scler Relat Disord. 2015;4(5):400–2. doi:10.1016/j.msard.2015.05.007.
Hashimoto Y, Shinoda K, Tanaka E, Uehara T, Matsushita T, Yamasaki R, et al. Re-emergence of a tumefactive demyelinating lesion after initiation of fingolimod therapy. J Neurol Sci. 2017;379:167–8. doi:10.1016/j.jns.2017.06.002.
Pröbstel A-K, Radü E, Mueller-Lenke N, Zhang-Auberson L, Bischof D, Merschhemke M, et al. Tumefactive multiple sclerosis lesions under fingolimod: case series from an MS centre and review of phase 2 and 3 clinical trial data. Mult Scler J. 2016;22(Suppl. 3):706–827 (abstract EP1491).
Novartis Pharma AG. Data on file, August 2017.
Anton R, Haas M, Arlett P, Weise M, Balabanov P, Mazzaglia G, et al. Drug-induced progressive multifocal leukoencephalopathy in multiple sclerosis: European regulators’ perspective. Clin Pharmacol Ther. 2017;102(2):283–9. doi:10.1002/cpt.604.
Acknowledgements
Medical writing and editorial support were provided by Oxford PharmaGenesis Ltd (Oxford, UK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access fees and medical writing and editorial support were funded by Novartis Pharma AG.
Conflict of interest
Giancarlo Comi has received consultancy fees and/or research support from Almirall, Bayer Schering Pharma, Biogen Dompé, Celgene, Excemed, Forward Pharma, Genzyme, Merck Serono International, Novartis, Receptos, Roche, Sanofi-Aventis, Serono Symposia International Foundation, and Teva. Hans-Peter Hartung has received speaking or consultancy fees from Bayer Healthcare, Baxter, Biogen Idec, CSL Behring, GeNeuro, Genzyme, MedImmune, Merck Serono, Novartis, Opexa, Receptos, Roche, Sanofi, and Teva. Rajesh Bakshi is an employee of Novartis Pharma AG. Ian M. Williams is an employee of Oxford PharmaGenesis Ltd. Heinz Wiendl has received consultancy fees and/or research support from Bayer, Bayer Healthcare, Bayer Schering AG, Bayer Vital GmBH, Biogen, CSL Behring, Fresenius Medical Care, Genzyme, GlaxoSmithKline, GW Pharmaceuticals, Lundbeck, Merck Serono, Novartis, OmniaMed, Sanofi-Aventis, Sanofi-Genzyme, Sanofi-Genzyme Germany, Sanofi US, and Teva.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Comi, G., Hartung, HP., Bakshi, R. et al. Benefit–Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis. Drugs 77, 1755–1768 (2017). https://doi.org/10.1007/s40265-017-0814-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0814-1