
Abstract
Psoriatic arthritis (PsA) is a chronic, systemic inflammatory disease. Up to 40 % of patients with psoriasis will go on to develop PsA, usually within 5–10 years of cutaneous disease onset. Both conditions share common pathogenic mechanisms involving genetic and environmental factors. Because psoriasis is typically present for years before PsA-related joint symptoms emerge, dermatologists are in a unique position to detect PsA earlier in the disease process through regular, routine screening of psoriasis patients. Distinguishing clinical features of PsA include co-occurrence of psoriatic skin lesions and nail dystrophy, as well as dactylitis and enthesitis. Patients with PsA are usually seronegative for rheumatoid factor, and radiographs may reveal unique features such as juxta-articular new bone formation and pencil-in-cup deformity. Early treatment of PsA with disease-modifying anti-rheumatic drugs has the potential to slow disease progression and maintain patient quality of life. Optimally, a single therapeutic agent will control both the skin and joint psoriatic symptoms. A number of traditional treatments used to manage psoriasis, such as methotrexate and cyclosporine, are also effective for PsA, but these agents are often inadequately effective, temporary in benefit and associated with significant safety concerns. Biologic anti-tumour necrosis factor agents, such as etanercept, infliximab and adalimumab, are effective for treating patients who have both psoriasis and PsA. However, a substantial number of patients may lose efficacy, have adverse effects or find intravenous or subcutaneous administration inconvenient. Emerging oral treatments, including phosphodiesterase 4 inhibitors, such as apremilast, and new biologics targeting interleukin-17, such as secukinumab, brodalumab and ixekizumab, have shown encouraging clinical results in the treatment of psoriasis and/or PsA. Active and regular collaboration of dermatologists with rheumatologists in managing patients who have psoriasis and PsA is likely to yield more optimal control of psoriatic dermal and joint symptoms, and improve long-term patient outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Psoriasis is a chronic, inflammatory systemic disease typically characterized by erythematous, scaly patches, or plaques on the skin resulting from hyperproliferation of epidermal keratinocytes [1, 2]. The disorder affects approximately 1–3 % of the world population [3–6]. Psoriatic arthritis (PsA) is a spondyloarthritic condition, which is present in up to 42 % of individuals with psoriasis and has an estimated prevalence of 0.1–1.0 % in the general population [7]. PsA is characterized by synovitis, enthesitis, dactylitis and spondylitis [8, 9]. Psoriasis and PsA share common pathophysiologic mechanisms (Fig. 1) [10–14]; nearly all individuals with PsA also have psoriasis [10].

Shared attributes of cutaneous psoriasis and psoriatic arthritis [10–14]. AP activator protein, CPII C-propeptide of type II collagen, C2C collagen fragment neoepitopes Col2-3/4Clong mono, HLA human leukocyte antigen, hsCRP highly sensitive C-reactive protein, IFN interferon, IL interleukin, IL-12B interleukin 12 beta, IL-23r interleukin 23 receptor, MAPK mitogen-activated protein kinase, MMP matrix metalloproteinase, NF nuclear factor, OPG osteoprotegerin, pDC precursor dendritic cell, RANK receptor activator of NF-κB, TNF tumour necrosis factor
Between 6 and 42 % of patients with psoriasis will develop PsA [8], although lower estimates may reflect a missed diagnosis or misdiagnosis [7, 15, 16]. In a study that assessed the prevalence and clinical pattern of PsA in 1,511 patients with psoriasis, 312 (21 %) had existing PsA and 85 % of these cases had been diagnosed for the first time during the study [17]. In a more recent study, 949 psoriasis patients seen serially in dermatology clinics were subsequently evaluated by rheumatologists; 30 % were diagnosed with PsA and 41 % of these had not been aware of the diagnosis [18]. PsA is often undiagnosed or misdiagnosed. Skin disease generally precedes joint disease in the majority (≈75–80 %) of patients, with a typical lag time of about 7–12 years from the onset of psoriasis to diagnosis of PsA [3, 7, 15, 19]. Thus, routinely screening psoriasis patients for PsA in the dermatology clinic is important, as most patients with PsA will present to a dermatologist first, long before joint symptoms emerge. However, in 10–15 % of cases, PsA may precede psoriasis [10].
Patients with psoriasis, PsA or both generally have reduced health-related quality of life (QOL), productivity and functionality similar to those of patients with other serious diseases, such as cancer, heart disease and diabetes [20–30]. In addition, PsA has been associated with long-term work disability, loss of productivity and work absenteeism [24–27]. The burden of illness associated with PsA in patients with psoriasis has been reviewed [31, 32]. Individuals with both psoriasis and PsA may experience emotional distress and discomfort caused by skin involvement and pain, and physical limitations caused by joint involvement, which could synergistically affect their QOL [17, 33, 34]. Compared with psoriasis patients who do not have PsA, psoriasis patients with PsA have greater QOL impairment, including physical and mental components of the 36-Item Short Form Health Survey [32, 34]. In addition, patients may have difficulty determining the appropriate provider for diagnosis and management of their PsA, and patients may lack understanding regarding their treatment options. Because most patients will be treated first for the skin lesions associated with psoriasis, dermatologists are in a unique position to screen for and diagnose early PsA [3, 35, 36].
2 Pathophysiology
2.1 Genetic Variations in Psoriasis and PsA
About 40 years ago, Moll and Wright reported that first-degree relatives of individuals with PsA had a 19-fold increase in psoriasis prevalence compared with the general population [37]. Investigations into the genetic basis of psoriasis and PsA have revealed commonalities as well as distinctions between the two disease processes. Considerable overlap exists in human leukocyte antigens (HLAs) associated with both diseases (e.g. B13, B17, B57, Cw6 and DR7) [10, 38]. The tumour necrosis factor (TNF)-α locus, TNF-238, and the NOD2/PSORAS1 locus on chromosome 16q, which corresponds to a variant in the CARD15 domain previously shown to increase susceptibility to Crohn’s disease, have been identified as regions that may predispose patients to both psoriasis and PsA [10, 39–41], although findings have been inconclusive [42]. Interleukin (IL)-12 beta and IL-23 receptor are also associated with both psoriasis and PsA [38, 43–45]. Although the evidence is preliminary and research is ongoing, certain single-nucleotide polymorphisms have been related to PsA and clinical subphenotypes [46, 47]. In one such study, Jadon and colleagues [46] detected a strong predictive relationship between an IL-12 beta variant and the presence of PsA; they also detected a trend towards a relationship between an IL-23 receptor variant and erosive peripheral joint disease.
Genetic differences between psoriasis and PsA are also being identified. A recent study showed that psoriasis is more closely associated with HLA-C alleles and PsA is more closely associated with HLA-B alleles, including HLA B*27 and B*39 [11, 48]. PSORS1 on chromosome 6p and PSORS2 on chromosome 17q have been confirmed as loci for genetic susceptibility to psoriasis [10]. However, these loci have not shown a reproducible association with PsA [10, 49]. Mapping of the major histocompatibility complex has identified several novel loci for PsA that are independent of known HLA susceptibility alleles [50]. A recent study found that the frequency of C*06:02 was lower in patients with PsA (28.7 %) than in those with psoriasis (57.5 %) [11]. Two separate clinical patterns of major histocompatibility complex effect have been identified; C*06 was associated with more penetrant skin disease and less prevalent musculoskeletal disease, while an HLA-B (B*27) phenotype was associated with more prevalent musculoskeletal disease [11]. Together, the available evidence shows that the genetic aetiology of psoriasis and PsA is complex and appears to be based on multiple shared and unshared genetic factors as well as environmental stimuli [10, 51].
2.2 Inflammatory Processes and Mediators in Psoriasis and PsA
Psoriasis and PsA show significant similarities in inflammatory processes and mediators. The immune system is normally homeostatic, with mechanisms in place to turn off an immune response and avoid tissue damage associated with chronic inflammation [52]. Cyclic adenosine monophosphate (cAMP), a naturally occurring secondary messenger, helps maintain homeostasis by modulating the network of pro-inflammatory and anti-inflammatory mediators [53]. cAMP levels are regulated by phosphodiesterases (PDEs), intracellular enzymes that convert cAMP to AMP [53]. In immune cells, PDE4 is the predominantly active PDE [54]. The conversion of cAMP to AMP leads to increased pro-inflammatory mediator production and decreased anti-inflammatory mediator production [53]. Increased levels of pro-inflammatory mediators are found in psoriatic lesions and the synovium of patients with PsA [55–61].
Pro-inflammatory mediators that drive psoriasis and PsA are released by a variety of cell types, including innate immune cells, adaptive immune cells and resident immune cells (Fig. 2) [1, 57]. Plasmacytoid dendritic cells act as regulators of innate and active immune responses and play a pivotal role in T cell-mediated immune responses [1, 62, 63]. Large numbers of plasmacytoid dendritic cells are found in psoriatic skin and psoriatic synovium [10]. Activated dendritic cells present antigens and produce interferon (IFN)-α and pro-inflammatory mediators, such as IL-12 and IL-23 [1]. T cells respond to antigen presented by myeloid dendritic cells by proliferating and differentiating into type 1 and type 17 T helper cells, which increase the secretion of inflammatory cytokines [1]. In one study, messenger RNA (mRNA) levels of the T cell regulatory cytokine IL-7, which stimulates production of pro-inflammatory cytokines, were increased in the synovial fluid of patients with spondyloarthritis [64]. CD11+ dendritic cells express nitric oxide synthase and TNF-α [65], and CD8+ and CD4+ T cells produce IFN-γ, TNF-α and IL-2 [66].

Mechanisms of systemic, chronic inflammation in psoriasis and psoriatic arthritis. From Nestle et al. [1]. Copyright © 2009, Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. CCL chemokine (C-C motif) ligand, CXCL chemokine (C-X-C motif) ligand, Th T helper, TNF tumour necrosis factor
Activated dendritic cells and associated inflammatory signalling molecules also exert effects on other cell types, including keratinocytes, leukocytes, neutrophils, endothelial cells and vascular smooth muscle cells, marked by chemotaxis, proliferation and production of additional inflammatory mediators [63]. Chronic disruptions in inflammatory signalling are believed to lead to lasting changes in resident cells of the skin and joints, which underlie the clinical hallmarks of psoriatic disease [1, 67]. Angiogenesis is another common but less often recognized feature of both psoriasis and PsA [8, 68]. Angiogenic markers, such as vascular endothelial growth factor (VEGF), placental growth factor, VEGF receptor 2 and neuropilin-1, are increased in psoriatic disease [67]. Chronic inflammatory signalling also plays a role in psoriatic joint disease. Circulating levels of Dikkopf-1 and macrophage colony stimulating factor, both soluble mediators of bone remodelling, have been found to be higher in PsA patients than in psoriasis patients and healthy controls; levels of these mediators have been correlated with radiographic progression [69].
3 Clinical Features of PsA and Screening
Dermatologists play an important role in screening and diagnosing patients with early PsA. Active monitoring of psoriasis patients for signs of joint or arthritic involvement and familiarity with PsA screening, diagnosis and treatment options can help dermatologists positively impact the clinical course of psoriatic disease [3, 8, 35, 36]. Irreversible joint damage and PsA progression begin within the first 2 years after disease onset, with an increase in the number of joints affected over time; thus, routine screening in the dermatology clinic can be key to early detection [8, 36, 70–72]. Early intervention in the PsA disease process can inhibit or delay structural joint damage, and patients are more likely to experience improvements in pain, fatigue, depression and QOL [8, 35, 72].
Because the symptoms of PsA may overlap with those of many other inflammatory and rheumatologic conditions, including osteoarthritis, rheumatoid arthritis (RA) and other spondyloarthritic conditions [36], PsA is often misdiagnosed (Table 1) [8, 73–81]. Alternatively, a patient may not consider it relevant or appropriate to mention musculoskeletal symptoms to a dermatologist and may assume that the symptoms will resolve or that there is nothing to be done for them, thus leading to inattention. PsA diagnosis is further complicated by the broad range of potential musculoskeletal symptoms and an often waxing-and-waning clinical course [8]. To identify patients who may have PsA, dermatologists can routinely ask psoriasis patients about key signs/symptoms of PsA, including whether they have experienced morning stiffness in joints lasting longer than 30 min, swelling in a finger or toe, or nail changes. A number of screening tools have been developed for diagnosis of PsA (Table 2) [82–88], several of which show good sensitivity or specificity in the setting of dermatology clinics or general medical clinics. If PsA is suspected, referral to a rheumatologist should be considered, depending on the dermatologist’s comfort level with managing PsA [8, 15, 89].
4 Patient Assessment, Staging and Treatment Plan
4.1 Assessment and Staging
Patients in whom there is a strong suspicion of PsA, based on clinical presentation of arthritis, enthesitis, dactylitis and/or spondylitis (pain, swelling, tenderness, stiffness), should undergo a more thorough examination. The Classification Criteria for Psoriatic Arthritis (CASPAR) [Table 3] were developed as classification criteria for the purpose of identifying standardized groups of PsA subjects for research where specificity is most important—not as individual diagnostic criteria, wherein sensitivity is paramount [90]. Nonetheless, clinicians may find utility in applying the elements of CASPAR when considering a diagnosis of PsA. CASPAR bases diagnosis on clinical presentation, history and radiographic and laboratory evidence, and exhibits 99 % specificity for classification of PsA [75, 90]. The stem of the criteria requires clinician judgment that the patient has inflammatory arthritis, enthesitis and/or spondylitis; if this stem is fulfilled, then the other elements of the criteria can be applied. Inflammatory features of PsA include stiffness, pain, swelling and tenderness of the joints, ligament and tendon insertions into bone (enthesitis), and spine, and dactylitis (swelling of an entire digit). The severity and pattern (e.g. symmetrical versus asymmetrical, monoarticular versus polyarticular) of peripheral and axial joint disease vary widely among patients [8]. Enthesitis may be one of the earliest signs of PsA [91]. Dactylitis occurs in up to 50 % of patients with PsA and is a marker for disease progression [92]. Spondylitis, characterized by inflammation in joints, entheses and bones of the spine and sacroiliac joints, may occur in a significant proportion of PsA patients [91]. PsA is characterized by joint erosions, joint space narrowing, bony proliferation, osteolysis (e.g. pencil-in-cup deformity), acro-osteolysis, ankylosis, spur formation and spondylitis on radiographs [8, 90, 93]. PsA patients are typically seronegative for rheumatoid factor [10, 74, 90, 94].
Controversy exists as to whether psoriatic nail involvement is predictive of PsA. Approximately 45 % of psoriasis patients have characteristic psoriatic nail pitting, onycholysis and hyperkeratosis but do not have concurrent PsA [7]. Nevertheless, nail dystrophy appears to be a clinically valuable marker that should raise suspicion of psoriatic joint disease. In a cohort study of 1,593 patients with psoriasis, nail dystrophy was a significant predictor of co-existing PsA (hazard ratio 2.93, 95 % confidence interval 1.68–5.12) [95]. Nail involvement can also help distinguish between PsA and RA, a condition where nail dystrophy does not occur [7, 96]. Currently, CASPAR includes psoriatic nail dystrophy among the criteria for PsA [90].
Even when PsA appears likely, differential diagnosis of PsA versus other arthritic and rheumatologic conditions should be considered [8, 74, 76, 97]. This can be challenging, given the many overlapping clinical features among these conditions (Table 1) [8, 73–81]. Hallmark clinical features differentiating PsA from other, similar arthropathies include the presence of psoriatic skin involvement in nearly all cases, nail dystrophy and dactylitis. Diagnostic laboratory markers for PsA are lacking, and those used to date have typically helped differentiate PsA from other conditions rather than specifically diagnosing PsA [98]. Evaluation of the erythrocyte sedimentation rate and C-reactive protein level has limited utility in diagnosis of PsA, as these markers have been shown to be elevated in only about half of the patients with PsA; however, in PsA patients who do have an elevated erythrocyte sedimentation rate and elevated C-reactive protein levels, they are useful to assess disease activity and thus worthwhile to assess in patients who are suspected to have PsA.
Antibodies to cyclic citrullinated peptides, which are present in up to 13 % of PsA patients, have also been evaluated; although their utility in diagnosis is uncertain, evidence suggests that this marker may be useful in identifying patients with erosive features and multiple joint involvement [99]. When evaluating patients with suspected PsA, imaging studies are important tools. In addition to radiography, which is the primary method used, ultrasound has been recommended as an imaging tool to evaluate joint inflammation in patients with suspected PsA [100].
Assessment tools used by rheumatologists to stage and treat patients with PsA have been adapted from measures used in RA and include global assessment questionnaires and examination of small and large joints, entheses and digits, including a swollen and tender joint count. An overall assessment of the skin and nails is also performed [8, 9, 101, 102]. Adequate assessment of disease severity and risk of progression is key to making sound treatment decisions. In clinical trials and registries, baseline PsA severity and response to treatment are currently gauged by American College of Rheumatology (ACR) and Disease Activity Score (DAS) criteria defined for RA on the basis of tender and swollen joint counts, as well as individual measures of enthesitis and dactylitis [102]. New PsA-specific composite indices, which assess varying combinations of joint involvement, skin involvement, disease activity, pain, health-related QOL and functional impact—such as the Composite Disease Activity Index (CPDAI), PsA Disease Activity Score (PSADAS) and Arithmetic Mean of Desirability Functions (AMDF)—have recently been developed, and their psychometric properties are being evaluated [102, 103]. Risk of disease progression is judged on the basis of history and initial clinical presentation. Patients presenting with established PsA for more than 2 years are at relatively greater risk of disease progression (i.e. clinical damage) than those with a shorter history of joint involvement [91]. Other recognized predictors of disease progression are baseline joint damage, baseline acute-phase reactants, polyarticular presentation and the number of inflamed joints at each visit [104].
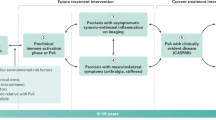
Ideally, dermatologists initiate treatment with a medication that is effective for both psoriasis and PsA, while considering proven efficacy in axial disease and clinical features such as enthesitis and dactylitis. Alternatively, referral to a rheumatologist for treatment should occur, especially if the PsA manifestations are not responding adequately. Recently, the European League Against Rheumatism (EULAR) established a PsA treatment algorithm (Fig. 3) [89]. This algorithm may serve as a guide for dermatologists in defining appropriate treatment options for psoriatic disease to optimally manage these patients, whether they are treating them alone or in conjunction with a rheumatologist. The long-term goals of the EULAR algorithm are to maintain health-related QOL, limit skin and joint signs and symptoms, and prevent or slow structural damage [89]. For patients with active PsA, initial treatment with a traditional disease-modifying anti-rheumatic drug (DMARD) should be considered; if the response is inadequate, an anti-TNF-α biologic agent should be considered [89] (Table 4 [2, 105–128]). According to EULAR recommendations, initial treatment with an anti-TNF-α biologic agent may be prudent for patients with extensive involvement of skin and joints [89]. The Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) has also published international treatment recommendations for PsA [129], which are currently in the process of being updated. These guidelines take into account each of the key clinical domains of PsA (arthritis, enthesitis, dactylitis, spondylitis and skin disease) and encourage the clinician to evaluate the patient comprehensively in the treatment algorithm, in terms of both the clinical severity of each domain and the impact on function and QOL.

European League Against Rheumatism (EULAR) recommended treatment algorithm for management of psoriatic arthritis. The recommendations have been divided into four phases. Small fonts within the ellipses in phases II and III refer to dose modifications or an alternative therapy, as detailed within the body of the recommendations. aBecause of the variable nature of the disease, not all situations can be covered by this figure; therefore, it is important to consult the full text to which the numbers or letters in parentheses refer; dotted lines refer to situations where deleting a phase is recommended. bActive disease: ≥1 tender and inflamed joint and/or tender enthesis point, and/or dactylitic digit, and/or inflammatory back pain; adverse prognostic factors: ≥5 active joints; or high functional impairment due to activity; or damage; or past glucocorticoid use. cThe treatment target is clinical remission or, if remission is unlikely to be achievable, at least low disease activity; clinical remission is the absence of signs and symptoms. Reproduced from Gossec et al. [89]. Copyright © 2012 with permission from BMJ Publishing Group Ltd. DMARD disease-modifying anti-rheumatic drug, MTX methotrexate, TNF tumour necrosis factor
4.2 Traditional Treatments
Phototherapy, fumaric acid esters and systemic retinoids can be effective first-line strategies for psoriasis but do not improve signs and symptoms of PsA [129]. Non-steroidal anti-inflammatory drugs (NSAIDs) are first-line treatment options for mild PsA and may improve joint symptoms [89, 129]. However, NSAIDs are not recommended for treatment of psoriasis, because they may worsen skin lesions [130–134]. Similarly, traditional regimens of systemic corticosteroids are not recommended for treatment of psoriasis, because clinical trial data are not available and because of their association with psoriasis flares during or after tapering [129].
4.2.1 Methotrexate
Methotrexate is approved in the USA by the Food and Drug Administration (FDA) for severe, recalcitrant, disabling psoriasis and in the UK, Germany, France, Sweden and Spain for severe psoriasis. European approvals of methotrexate for treatment of concomitant psoriasis and PsA are limited. Although formal clinical evidence is lacking on the efficacy of methotrexate in PsA, it is commonly used as first-line therapy and in combination with biologics. Two randomized, placebo-controlled studies assessed the efficacy and safety of methotrexate in PsA patients; each study demonstrated significant improvements in global assessment ratings with active treatment [105, 106]. However, neither study showed a positive effect of methotrexate on the basis of objective measures such as tender and swollen joint counts [105, 106]. Methotrexate has not been conclusively found to have a positive impact on radiographic progression seen over 24 months [107]. An observational cohort study, however, found a mild positive impact of methotrexate on PsA progression [108].
Comparative studies between methotrexate and other treatments in psoriasis patients have generally shown greater efficacy with other treatment options, such as cyclosporine A [135], adalimumab [136] and briakinumab [137]. Treatment with methotrexate has been associated with hepatic, pulmonary and bone marrow toxicity, as well as teratogenicity [138]. Furthermore, in psoriasis patients with risk factors for liver disease, such as obesity (fatty liver) and alcoholism, the potential for transaminase elevation and liver pathology limits long-term use of methotrexate.
4.2.2 Sulfasalazine
Sulfasalazine is not approved by the FDA for the treatment of patients with psoriasis or PsA [138]. Although the precise mechanism of action of sulfasalazine is unknown, it is thought to have anti-inflammatory effects mediated through inhibition of the 5-lipoxygenase pathway [139]. Modest improvements in psoriasis have been reported in randomized, double-blind, controlled studies of sulfasalazine [138, 140]. Other studies have demonstrated the effectiveness of sulfasalazine in PsA [109–111, 141–143], with short-term data in one small study also showing dermatologic improvements [110]. The impact of sulfasalazine on radiographic progression, however, has not been reported [109–111, 141–143]. Sulfasalazine has been associated with gastrointestinal intolerance, arthralgia, reversible oligospermia, leukopenia and agranulocytosis [138].
4.2.3 Leflunomide
Leflunomide is a pyrimidine synthesis inhibitor and has been shown to inhibit T cell activation and proliferation [144]. It is not approved by the FDA for psoriasis or PsA, but it is approved by the European Medicines Agency (EMA) for PsA. In randomized, double-blind, placebo-controlled studies of patients with both psoriasis and PsA, leflunomide improved skin and joint symptoms [112–114]. Leflunomide has been associated with gastrointestinal toxicity (e.g. diarrhoea and nausea), elevated liver enzymes, increased risk of infections and leukopenia [138].
4.2.4 Cyclosporine A
Cyclosporine A is a potent immunosuppressant and is thought to act on the immune system at multiple pathways [145]. It is approved by the FDA for severe, recalcitrant psoriasis on the basis of positive findings from a number of controlled clinical trials [138, 146–152]. Three studies have also demonstrated the safety and modest efficacy of cyclosporine A in PsA [115–117]. Cyclosporine A has been associated with nephrotoxicity and hypertension [138] and is recommended only for short-term use (up to 12 months).
4.2.5 Biologics
4.2.5.1 Anti-TNF-α Agents
TNF-α plays a pivotal role in the chronic inflammation and aberrant immune responses that underlie psoriasis and PsA. Biologic agents that inhibit TNF-α, including a fusion protein, etanercept, and antibody-based treatments such as infliximab and adalimumab, are indicated for and have shown efficacy in psoriasis and PsA, including inhibition of radiographic progression [2, 118–121, 126, 136, 153–155]. Golimumab, a newer human monoclonal antibody against TNF-α, is also indicated for and has shown efficacy in the treatment of PsA on the basis of positive findings from a 24-week, randomized, placebo-controlled study [122] and from open-label extension studies [123]. Golimumab is not approved by the FDA for treatment of psoriasis; however, clinical trial evidence suggests that this agent can have a positive effect on psoriatic skin lesions in patients with PsA [122]. Certolizumab pegol (Cimzia®; UCB Pharma, Brussels, Belgium) is a human anti-TNF-α antibody conjugated with a 40 kDa polyethylene glycol molecule, which is administered subcutaneously. It is FDA approved for treatment of PsA and has received a recommendation for marketing authorization in Europe by the EMA Committee for Medicinal Products for Human Use [156, 157]. Because it contains only a single Fab′ of anti-TNF antibody and lacks an Fc′ portion, certolizumab pegol hypothetically may be less likely to induce antibody-related complement activation, apoptosis or cellular toxicity [158]. Results from a randomized, double-blind phase II study of certolizumab pegol showed efficacy in patients with moderate to severe psoriasis [158]. A phase III, multicentre, double-blind, parallel-group study in patients with PsA showed improvement in all PsA clinical domains, including skin manifestations and physical functioning at week 24 [127, 128]. Twenty percent of the study patients had previously experienced inadequate responses to an anti-TNF agent and, despite this, they demonstrated responses similar to those of the overall study population [127]. Adverse events were consistent with those seen in other studies of certolizumab pegol.
The evidence surrounding anti-TNF agents and risk reduction of adverse cardiac events is not definitive. Data on the cardioprotectiveness of these agents in PsA are not currently available. A large retrospective Kaiser Permanente Southern California (KPSC) health plan study concluded that TNF inhibitor use in psoriasis was associated with a significantly reduced myocardial infarction risk, compared with topical treatment. Although the KPSC data show promising results, variables in the KPSC study, such as the use of age as a dichotomous variable in multivariate analyses and the lack of a longer TNF treatment duration, raise questions as to whether the use of age as a continuous variable or a longer study period would have impacted the findings [159]. Additional studies are needed to further evaluate the cardioprotective effects of anti-TNF agents in both psoriasis and PsA.
4.2.5.2 Ustekinumab
The anti-IL-12/23 monoclonal antibody ustekinumab is indicated for and has shown efficacy in psoriasis [160, 161]. In a phase II study of PsA, ustekinumab showed improvement in ACR response rates and significant improvement in skin disease, enthesitis, dactylitis and physical functioning [124]. The study doses were higher than those used for managing psoriasis [124, 161]. Similar positive results were recently reported with ustekinumab in phase III studies in PsA patients who had previously received anti-TNF and DMARD treatment [162, 163]. Results from the Active Comparator (CNTO1275/Enbrel) Psoriasis Trial (ACCEPT) in psoriasis demonstrated that, compared with etanercept treatment, ustekinumab treatment resulted in greater improvement in efficacy parameters at week 12, including achievement of a 75 % improvement in the Psoriasis Area and Severity Index score (PASI 75; the primary endpoint), a Physician’s Global Assessment (PGA) score of 0 or 1, and achievement of a 90 % improvement in the PASI score (PASI 90) [164]. Of note, however, although greater efficacy was seen with ustekinumab, more than half of the patients in the etanercept group met the primary endpoint, and almost half obtained a PGA score of 0 or 1 [164].
Direct comparison studies have not yet been conducted for many of the biologic therapies, which may make it difficult to determine whether one agent is more beneficial in treating specific patient symptoms. Overall, TNF agents may be a better choice than topical treatments when patients have extensive skin involvement, and the overuse of some topical agents and associated adverse effects may be of concern [89]. Guidelines note that efficacy with etanercept may be lesser or delayed when it comes to PsA skin involvement. Although there have been no head-to-head comparison studies with etanercept in PsA, a comparison study in psoriasis demonstrated that at 12 weeks, treatment with ustekinumab resulted in superior efficacy in skin outcomes, compared with etanercept. With respect to joint involvement, no evident differences in efficacy have been observed among the various TNF inhibitors but, again, head-to-head comparison studies are not available [89]. Although comparator studies can provide insight into the efficacy and safety of an agent in clinical trials, it is always important to remember the impact that individual patient characteristics can have on drug performance, thus highlighting the importance of an individualized approach to drug selection.
Biologics have a number of limitations for patients with psoriasis and PsA. A substantial proportion of patients with psoriasis or PsA do not have a sustained response to TNF-α inhibitors [165, 166]. In addition, some biologics, such as infliximab, are dosed intravenously, which can be inconvenient for patients. Loss of response over time, cost and safety issues affect decisions to use nonbiologic and biologic DMARDs over the long term [167, 168]. An unmet need exists for an efficacious, well-tolerated, safe and easy-to-use treatment option for patients with psoriasis and PsA that improves both skin and joint disease.
4.2.6 Combination Therapy
The available treatment algorithms put forth by the American Academy of Dermatology (AAD) [101], GRAPPA [129] and EULAR [89] position combination therapy with a DMARD/TNF inhibitor or DMARD/other biologic as treatment for moderate or severe PsA in patients who still have symptoms after conventional DMARD use. Minimal literature on the effectiveness of biologic combination therapy is available, warranting the need for studies evaluating the combination of two biologics for PsA [169–173]. The EULAR guidelines further note that clinical trials to date have not shown superiority with combination use of TNF inhibitors and synthetic DMARDs versus TNF inhibitor monotherapy, and additional trials are needed [89].
5 New and Future Treatment Options
5.1 PDE4 Inhibition
The large PDE enzyme family is the sole route for enzymatic degradation of cAMP, an intracellular secondary messenger, which controls a wide array of cellular functions [174]. Apremilast is an orally available small molecule, which specifically inhibits the activity of PDE4, an isoform found predominantly in immune cells such as monocytes, T cells and neutrophils [174]. By inhibiting PDE4-mediated breakdown of cAMP, apremilast works intracellularly to regulate inflammatory mediators that are considered to play an important role in the pathogenesis of psoriasis and PsA [175, 176].
5.1.1 Apremilast
Phase II studies of apremilast have demonstrated efficacy in patients with psoriasis and PsA. In one study, apremilast 20 or 30 mg twice daily reduced the severity of moderate to severe plaque psoriasis over 24 weeks and improved pruritus [177]. In another study, apremilast 20 mg twice daily or 40 mg once daily improved the signs and symptoms of PsA over 24 weeks [178]. In both studies, apremilast was generally well tolerated; the majority (>90 %) of treatment-related adverse events in the clinical studies were mild to moderate and did not lead to study discontinuation [177, 178]. The most common treatment-related adverse events were headache, nausea, diarrhoea, nasopharyngitis, fatigue and upper respiratory tract infection [177, 178]. No opportunistic infections were reported [177, 178].
Apremilast is currently being studied in phase III clinical trial programmes for psoriasis and PsA; additional studies are assessing its efficacy in ankylosing spondylitis, Behçet’s disease and RA. The Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis (ESTEEM) programme is assessing the use of apremilast in patients with moderate to severe plaque psoriasis in two 52-week, randomized, placebo-controlled trials with long-term, open-label extensions. Preliminary results from ESTEEM 1 have demonstrated the clinical efficacy of apremilast in significantly reducing the severity of moderate to severe psoriasis and its effectiveness in difficult-to-treat areas such as the nails and scalp. In addition, apremilast was generally well tolerated, with no new safety or laboratory findings [179, 180]. The Psoriatic Arthritis Long-term Assessment of Clinical Efficacy (PALACE) programme is assessing the effects of apremilast on the signs and symptoms of PsA in four randomized, placebo-controlled trials with long-term, open-label extensions. PALACE 3 is evaluating the efficacy and safety of apremilast in patients with PsA and a qualifying psoriasis lesion. Preliminary results from PALACE 1 have confirmed the clinical efficacy of apremilast in patients with PsA who have prior experience with traditional and biologic DMARDs, with no new safety signals and improved tolerability, compared with the results of phase II studies [181, 182].
5.2 JAK Inhibitors
Janus kinase (JAK) inhibitors serve as intracellular signal transduction molecules linked to surface receptors for multiple pro-inflammatory cytokines, including IL-21, which are crucial for T cell activation and functioning. Tofacitinib is a small-molecule inhibitor of JAK1, JAK3 and, to a lesser extent, JAK2, and is administered orally [183]. Tofacitinib 5 mg twice daily is approved in the USA for patients with RA who are unresponsive to DMARDs. With tofacitinib, JAK inhibition may interrupt key components of the immune and inflammatory responses that underlie both psoriasis and PsA. Tofacitinib has shown efficacy across various parameters in RA [183–185]. The potential role of tofacitinib in treatment of psoriasis patients is not yet known; however, in a phase I, randomized, controlled, dose-ranging study, tofacitinib was effective in patients with psoriasis [186]. Similarly, in a 12-week, phase IIb, double-blind, placebo-controlled study, tofacitinib (2, 5 and 15 mg twice daily) was effective in treatment of patients with moderate to severe plaque psoriasis [187]. Decreases in mean neutrophil counts and haemoglobin values and increases in lipoprotein levels were observed [187]. Phase III studies with tofacitinib 5 and 10 mg twice daily are ongoing in patients with moderate to severe plaque psoriasis. No clinical efficacy and safety data for PsA are available but, on the basis of the results in RA and psoriasis, evaluation of tofacitinib in PsA is warranted.
5.3 Anti-IL-17 Agents
IL-17 is a pro-inflammatory cytokine involved in the pathogenesis of psoriasis and PsA, as well as in other immune-mediated chronic inflammatory disorders. As such, it has been the object of intense drug development efforts, and a number of biologic agents targeting this molecule are being investigated in clinical studies of psoriasis and PsA.
5.3.1 Secukinumab
Secukinumab (AIN-457; Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA) is a fully human, targeted monoclonal antibody against IL-17a, and is administered subcutaneously or intravenously [188, 189]. Preliminary results from phase II studies of secukinumab have shown efficacy in psoriasis with 150 mg subcutaneous regimens, 3 × 75 mg and 3 × 150 mg subcutaneous doses and 3 mg/kg, 10 mg/kg and 3 × 10 mg/kg intravenous doses [188, 189]. With subcutaneous administration of secukinumab, the rates of serious adverse events and infections were comparable to those observed with placebo [188, 189]. Phase III trials of secukinumab for psoriasis (ClinicalTrials.gov study identifier NCT01365455) and PsA (ClinicalTrials.gov study identifier NCT01392326) are under way. Data are limited on the clinical efficacy and safety of secukinumab in PsA. A short-term phase II proof-of-concept study supported a significant phase III programme, which is currently under way [190].
5.3.2 Brodalumab
Brodalumab (AMG 827; Amgen, Thousand Oaks, CA, USA) is a fully human monoclonal antibody, which binds to the IL-17 receptor and blocks IL-17 signalling [191]. Brodalumab binds with high affinity to human IL-17RA and blocks the biologic activity of IL-17A, IL-17F, IL-17A/F heterodimer and IL-17E (IL-25). A phase II study of subcutaneous brodalumab demonstrated efficacy in patients with moderate to severe psoriasis [191]. Brodalumab has also demonstrated efficacy in PsA in a phase II study, with similar efficacy noted in anti-TNF-experienced and -inexperienced patients [192]. Given these positive results, phase III studies of brodalumab in patients with psoriasis are under way (ClinicalTrials.gov study identifiers NCT01708603 and NCT01708629).
5.3.3 Ixekizumab
Ixekizumab (LY2439821; Eli Lilly and Company, Indianapolis, IN, USA) is a humanized anti-IL-17A monoclonal antibody, which is administered subcutaneously. In a phase II study, ixekizumab improved clinical symptoms in patients with moderate to severe psoriasis [193]. Phase III studies of ixekizumab in psoriasis (ClinicalTrials.gov study identifier NCT01474512) and PsA (ClinicalTrials.gov study identifier NCT01695239) are ongoing; no data are yet available with this agent in patients with PsA.
5.4 Protein Kinase C Inhibitor
Protein kinase C isoforms are important for activation and proliferation of T cells, as well as for antigen receptor function [194]. Activated T cells are considered central to the psoriatic disease process [194]. Sotrastaurin (AEB071; Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA) is a protein kinase C inhibitor, which is administered orally. Sotrastaurin has strong and specific activity on PKCθ, PKCα and PKCβ, and lesser activity on PKCδ, PKCε and PKCη [194]. Preliminary phase II proof-of-concept data suggest its efficacy in psoriasis [194]; additional phase II study results are expected. No clinical efficacy and safety data in PsA are available.
5.5 Other Mechanisms
5.5.1 CF101
Adenosine A3 receptors are over-expressed in peripheral blood mononuclear cells in patients with psoriasis, RA and Crohn’s disease [195]. CF101 (Can-Fite BioPharma, Petah Tikva, Israel) is an oral adenosine A3 receptor inhibitor, which has been shown in preclinical investigations to decrease production of pro-inflammatory cytokines, including TNF-α and nuclear factor kappa B (NF-κB), and to induce inflammatory cell apoptosis, leading to a broad anti-inflammatory effect [195]. In a phase II, randomized, double-blind, dose-ranging, placebo-controlled study, CF101 showed progressive improvement over 12 weeks in the mean change from baseline in the PASI score, compared with placebo, in patients with plaque psoriasis [195].
Additional investigational treatments for psoriasis and PsA in phase II studies are summarized in Table 5 [158, 177, 178, 181, 186, 187, 191, 193, 195–200].
6 Conclusions
Psoriatic arthritis is a chronic and progressive inflammatory arthritis closely associated with psoriasis and can lead to significant morbidity. Dermatologists who treat psoriasis patients are able to regularly and routinely screen for early PsA by asking about possible joint pain, stiffness or tenderness. Rapid screening questionnaires may also help in identifying early PsA. In patients with suspected PsA, dermatologists who are familiar with the distinguishing clinical features of PsA and the current diagnostic CASPAR can conduct a more thorough evaluation. Depending on their comfort level in dealing with joint symptoms, dermatologists may gauge PsA severity and the risk of progression and initiate treatment. Optimal interventions for active PsA should aim to control skin and joint signs and symptoms. For patients with more severe or complicated symptoms, dermatologists and rheumatologists must collaborate to adequately manage both skin and joint psoriatic involvement over the long term.
References
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509.
Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58(5):826–50.
Chang CA, Gottlieb AB, Lizzul PF. Management of psoriatic arthritis from the view of the dermatologist. Nat Rev Rheumatol. 2011;7(10):588–98.
Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003–2004. J Am Acad Dermatol. 2009;60(2):218–24.
Augustin M, Reich K, Glaeske G, et al. Co-morbidity and age-related prevalence of psoriasis: analysis of health insurance data in Germany. Acta Derm Venereol. 2010;90(2):147–51.
Stern RS, Nijsten T, Feldman SR, et al. Psoriasis is common, carries a substantial burden even when not extensive, and is associated with widespread treatment dissatisfaction. J Investig Dermatol Symp Proc. 2004;9(2):136–9.
Gladman DD, Antoni C, Mease P, et al. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64(Suppl 2):ii14–7.
Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol. 2008;58(5):851–64.
Mease PJ. Psoriatic arthritis: update on pathophysiology, assessment and management. Ann Rheum Dis. 2011;70(Suppl 1):i77–84.
Ciocon DH, Kimball AB. Psoriasis and psoriatic arthritis: separate or one and the same? Br J Dermatol. 2007;157(5):850–60.
Winchester R, Minevich G, Steshenko V, et al. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2012;64(4):1134–44.
Chandran V, Cook RJ, Edwin J, et al. Soluble biomarkers differentiate patients with psoriatic arthritis from those with psoriasis without arthritis. Rheumatology (Oxford). 2010;49(7):1399–405.
Ritchlin CT, Haas-Smith SA, Li P, et al. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111(6):821–31.
Batliwalla FM, Li W, Ritchlin CT, et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol Med. 2005;11(1–12):21–9.
Gottlieb AB, Mease PJ, Jackson JM, et al. Clinical characteristics of psoriatic arthritis and psoriasis in dermatologists’ offices. J Dermatolog Treat. 2006;17(5):279–87.
Radtke MA, Reich K, Blome C, et al. Prevalence and clinical features of psoriatic arthritis and joint complaints in 2009 patients with psoriasis: results of a German national survey. J Eur Acad Dermatol Venereol. 2009;23(6):683–91.
Reich K, Kruger K, Mossner R, et al. Epidemiology and clinical pattern of psoriatic arthritis in Germany: a prospective interdisciplinary epidemiological study of 1511 patients with plaque-type psoriasis. Br J Dermatol. 2009;160(5):1040–7.
Mease PJ, Papp KA, Gladman D, et al. The prevalence of rheumatologist-diagnosed psoriatic arthritis in psoriasis patients in European/North American dermatology clinics: results of the PREPARE study [abstract P007]. Dermatol Ther. 2012;2(10):S3–4.
Qureshi AA, Husni ME, Mody E. Psoriatic arthritis and psoriasis: need for a multidisciplinary approach. Semin Cutan Med Surg. 2005;24(1):46–51.
Rapp SR, Feldman SR, Exum ML, et al. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3 Pt 1):401–7.
Meyer N, Paul C, Feneron D, et al. Psoriasis: an epidemiological evaluation of disease burden in 590 patients. J Eur Acad Dermatol Venereol. 2010;24(9):1075–82.
Wu Y, Mills D, Bala M. Impact of psoriasis on patients’ work and productivity: a retrospective, matched case-control analysis. Am J Clin Dermatol. 2009;10(6):407–10.
Chan B, Hales B, Shear N, et al. Work-related lost productivity and its economic impact on Canadian patients with moderate to severe psoriasis. J Cutan Med Surg. 2009;13(4):192–7.
Verstappen SM, Watson KD, Lunt M, et al. Working status in patients with rheumatoid arthritis, ankylosing spondylitis and psoriatic arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology (Oxford). 2010;49(8):1570–7.
Wallenius M, Skomsvoll JF, Koldingsnes W, et al. Work disability and health-related quality of life in males and females with psoriatic arthritis. Ann Rheum Dis. 2009;68(5):685–9.
Salaffi F, Carotti M, Gasparini S, et al. The health-related quality of life in rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis: a comparison with a selected sample of healthy people. Health Qual Life Outcomes. 2009;7:25.
Brodszky V, Balint P, Geher P, et al. Disease burden of psoriatic arthritis compared to rheumatoid arthritis. Hungarian experiment. Rheumatol Int. 2009;30(2):199–205.
Singh JA, Strand V. Spondyloarthritis is associated with poor function and physical health-related quality of life. J Rheumatol. 2009;36(5):1012–20.
Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and metabolic syndrome: a systematic review and meta-analysis of observational studies. J Am Acad Dermatol. 2013;68(4):654–62.
Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 2013;149(1):84–91.
Boehncke WH, Menter A. Burden of disease: psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2013;14(5):377–88.
Mease PJ, Menter MA. Quality-of-life issues in psoriasis and psoriatic arthritis: outcome measures and therapies from a dermatological perspective. J Am Acad Dermatol. 2006;54(4):685–704.
Kimball AB, Jacobson C, Weiss S, et al. The psychosocial burden of psoriasis. Am J Clin Dermatol. 2005;6(6):383–92.
Lundberg L, Johannesson M, Silverdahl M, et al. Health-related quality of life in patients with psoriasis and atopic dermatitis measured with SF-36, DLQI and a subjective measure of disease activity. Acta Derm Venereol. 2000;80(6):430–4.
Mease PJ. Psoriatic arthritis—update on pathophysiology, assessment, and management. Bull NYU Hosp Jt Dis. 2010;68(3):191–8.
Landells I, MacCallum C, Khraishi M. The role of the dermatologist in identification and treatment of the early stages of psoriatic arthritis. Skin Ther Lett. 2008;13(4):4–7.
Moll JM, Wright V. Familial occurrence of psoriatic arthritis. Ann Rheum Dis. 1973;32(3):181–201.
Duffin KC, Chandran V, Gladman DD, et al. Genetics of psoriasis and psoriatic arthritis: update and future direction. J Rheumatol. 2008;35(7):1449–53.
Hohler T, Kruger A, Schneider PM, et al. A TNF-alpha promoter polymorphism is associated with juvenile onset psoriasis and psoriatic arthritis. J Invest Dermatol. 1997;109(4):562–5.
Rahman P, Siannis F, Butt C, et al. TNFalpha polymorphisms and risk of psoriatic arthritis. Ann Rheum Dis. 2006;65(7):919–23.
Rahman P, Bartlett S, Siannis F, et al. CARD15: a pleiotropic autoimmune gene that confers susceptibility to psoriatic arthritis. Am J Hum Genet. 2003;73(3):677–81.
Zhu K, Yin X, Tang X, et al. Meta-analysis of NOD2/CARD15 polymorphisms with psoriasis and psoriatic arthritis. Rheumatol Int. 2012;32(7):1893–900.
Huffmeier U, Lascorz J, Bohm B, et al. Genetic variants of the IL-23R pathway: association with psoriatic arthritis and psoriasis vulgaris, but no specific risk factor for arthritis. J Invest Dermatol. 2009;129(2):355–8.
Castelino M, Barton A. Genetic susceptibility factors for psoriatic arthritis. Curr Opin Rheumatol. 2010;22(2):152–6.
Filer C, Ho P, Smith RL, et al. Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis Rheum. 2008;58(12):3705–9.
Jadon D, Tillett W, Wallis D, et al. Association of ERAP1, IL12B and IL23R gene polymorphisms with subphenotypes of psoriatic arthritis [abstract 1366]. Arthritis Rheum. 2011;63(10 Suppl):S534.
Ellinghaus E, Stuart PE, Ellinghaus D, et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J Invest Dermatol. 2012;132(4):1133–40.
Eder L, Pellett F, Chandran V, et al. HLA-B*27 and Cw*06 are risk alleles for psoriatic arthritis among psoriasis patients [abstract]. Arthritis Rheum. 2010;62(Suppl 10):1974.
Giardina E, Predazzi I, Sinibaldi C, et al. PSORS2 markers are not associated with psoriatic arthritis in the Italian population. Hum Hered. 2006;61(2):120–2.
Rahman P, Roslin NM, Pellett FJ, et al. High resolution mapping in the major histocompatibility complex region identifies multiple independent novel loci for psoriatic arthritis. Ann Rheum Dis. 2011;70(4):690–4.
Alenius GM, Friberg C, Nilsson S, et al. Analysis of 6 genetic loci for disease susceptibility in psoriatic arthritis. J Rheumatol. 2004;31(11):2230–5.
Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280(5361):243–8.
Serezani CH, Ballinger MN, Aronoff DM, et al. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39(2):127–32.
Baumer W, Hoppmann J, Rundfeldt C, et al. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets. 2007;6(1):17–26.
Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101(5):701–5.
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol. 2009;129(1):79–88.
Yawalkar N, Tscharner GG, Hunger RE, et al. Increased expression of IL-12p70 and IL-23 by multiple dendritic cell and macrophage subsets in plaque psoriasis. J Dermatol Sci. 2009;54(2):99–105.
Johansen C, Usher PA, Kjellerup RB, et al. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. 2009;160(2):319–24.
Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359(1–2):419–29.
Piskin G, Sylva-Steenland RMR, Bos JD, et al. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006;176(3):1908–15.
Lowes MA, Chamian F, Abello MV, et al. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci USA. 2005;102(52):19057–62.
Williams IR, Kupper TS. Immunity at the surface: homeostatic mechanisms of the skin immune system. Life Sci. 1996;58(18):1485–507.
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, et al. Identification of TNF-related apoptosis-inducing ligand and other molecules that distinguish inflammatory from resident dendritic cells in patients with psoriasis. J Allergy Clin Immunol. 2010;125(6):1261–8.
Rihl M, Kellner H, Kellner W, et al. Identification of interleukin-7 as a candidate disease mediator in spondylarthritis. Arthritis Rheum. 2008;58(11):3430–5.
Chu CC, Di Meglio P, Nestle FO. Harnessing dendritic cells in inflammatory skin diseases. Semin Immunol. 2011;23(1):28–41.
Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113(5):752–9.
Henno A, Blacher S, Lambert CA, et al. Histological and transcriptional study of angiogenesis and lymphangiogenesis in uninvolved skin, acute pinpoint lesions and established psoriasis plaques: an approach of vascular development chronology in psoriasis. J Dermatol Sci. 2010;57(3):162–9.
Coates LC, Anderson RR, Fitzgerald O, et al. Clues to the pathogenesis of psoriasis and psoriatic arthritis from imaging: a literature review. J Rheumatol. 2008;35(7):1438–42.
Dalbeth N, Pool B, Smith T, et al. Circulating mediators of bone remodeling in psoriatic arthritis: implications for disordered osteoclastogenesis and bone erosion. Arthritis Res Ther. 2010;12(4):R164.
Gladman DD, Stafford-Brady F, Chang CH, et al. Longitudinal study of clinical and radiological progression in psoriatic arthritis. J Rheumatol. 1990;17(6):809–12.
McHugh NJ, Balachrishnan C, Jones SM. Progression of peripheral joint disease in psoriatic arthritis: a 5-yr prospective study. Rheumatology (Oxford). 2003;42(6):778–83.
Kane D, Stafford L, Bresnihan B, et al. A prospective, clinical and radiological study of early psoriatic arthritis: an early synovitis clinic experience. Rheumatology (Oxford). 2003;42(12):1460–8.
Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum. 1973;3(1):55–78.
Brockbank J, Gladman D. Diagnosis and management of psoriatic arthritis. Drugs. 2002;62(17):2447–57.
Mease PJ, Gladman DD, Keystone EC. Alefacept in combination with methotrexate for the treatment of psoriatic arthritis: results of a randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2006;54(5):1638–45.
Tikly M, Makda MA. A diagnostic approach to the common arthritic conditions. SA Fam Pract. 2009;51(3):188–93.
Marchesoni A, Atzeni F, Spadaro A, et al. Identification of the clinical features distinguishing psoriatic arthritis and fibromyalgia. J Rheumatol. 2012;39(4):849–55.
Hauser W, Hayo S, Biewer W, et al. Diagnosis of fibromyalgia syndrome-a comparison of Association of the Medical Scientific Societies in Germany, survey, and American College of Rheumatology criteria. Clin J Pain. 2010;26(6):505–11.
VanItallie TB. Gout: epitome of painful arthritis. Metabolism. 2010;59(Suppl 1):S32–6.
Vazquez-Mellado J, Hernandez-Cuevas CB, Alvarez-Hernandez E, et al. The diagnostic value of the proposal for clinical gout diagnosis (CGD). Clin Rheumatol. 2012;31(3):429–34.
Mease PJ. Inflammatory musculoskeletal disease: identification and assessment. J Rheumatol. 2011;38(3):557–61.
Khraishi M, Landells I, Mugford G. The self-administered Psoriasis and Arthritis Screening Questionnaire (PASQ): a sensitive and specific tool for the diagnosis of early and established psoriatic arthritis. Psoriasis Forum. 2010;16(2):9–16.
Alenius GM, Stenberg B, Stenlund H, et al. Inflammatory joint manifestations are prevalent in psoriasis: prevalence study of joint and axial involvement in psoriatic patients, and evaluation of a psoriatic and arthritic questionnaire. J Rheumatol. 2002;29(12):2577–82.
Husni ME, Meyer KH, Cohen DS, et al. The PASE questionnaire: pilot-testing a psoriatic arthritis screening and evaluation tool. J Am Acad Dermatol. 2007;57(4):581–7.
Ibrahim GH, Buch MH, Lawson C, et al. Evaluation of an existing screening tool for psoriatic arthritis in people with psoriasis and the development of a new instrument: the Psoriasis Epidemiology Screening Tool (PEST) questionnaire. Clin Exp Rheumatol. 2009;27(3):469–74.
Chandran V, Gladman DD. Toronto Psoriatic Arthritis Screening (ToPAS) questionnaire: a report from the GRAPPA 2009 Annual Meeting. J Rheumatol. 2011;38(3):546–7.
Khraishi M, Mong J, Mugford G, et al. The electronic Psoriasis and Arthritis Screening Questionnaire (ePASQ): a sensitive and specific tool to diagnose psoriatic arthritis patients. J Cutan Med Surg. 2011;15(3):143–9.
Tinazzi I, Adami S, Zanolin EM, et al. The early psoriatic arthritis screening questionnaire: a simple and fast method for the identification of arthritis in patients with psoriasis. Rheumatology (Oxford). 2012;51(11):2058–63.
Gossec L, Smolen JS, Gaujoux-Viala C, et al. European League Against Rheumatism recommendations for the management of psoriatic arthritis with pharmacological therapies. Ann Rheum Dis. 2012;71(1):4–12.
Taylor W, Gladman D, Helliwell P, et al. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665–73.
Gladman DD, Chandran V. Review of clinical registries of psoriatic arthritis: lessons learned? Value for the future? Curr Rheumatol Rep. 2011;13(4):346–52.
Brockbank JE, Stein M, Schentag CT, et al. Dactylitis in psoriatic arthritis: a marker for disease severity? Ann Rheum Dis. 2005;64(2):188–90.
Mease PJ, van der Heijde D. Joint damage in psoriatic arthritis: how is it assessed and can it be prevented? Int J Adv Rheumatol. 2006;4(2):38–48.
Wright V. Psoriasis and arthritis. Ann Rheum Dis. 1956;15(4):348–56.
Wilson FC, Icen M, Crowson CS, et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population-based study. Arthritis Rheum. 2009;61(2):233–9.
McGonagle D, Palmou Fontana N, Tan AL, et al. Nailing down the genetic and immunological basis for psoriatic disease. Dermatology. 2010;221(Suppl 1):15–22.
Mease P. Management of psoriatic arthritis: the therapeutic interface between rheumatology and dermatology. Curr Rheumatol Rep. 2006;8(5):348–54.
Punzi L, Podswiadek M, Oliviero F, et al. Laboratory findings in psoriatic arthritis. Reumatismo. 2007;59(Suppl 1):52–5.
Bogliolo L, Alpini C, Caporali R, et al. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol. 2005;32(3):511–5.
Bonifati C, Elia F, Francesconi F, et al. The diagnosis of early psoriatic arthritis in an outpatient dermatological centre for psoriasis. J Eur Acad Dermatol Venereol. 2012;26(5):627–33.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol. 2011;65(1):137–74.
Mease PJ. Measures of psoriatic arthritis: Tender and Swollen Joint Assessment, Psoriasis Area and Severity Index (PASI), Nail Psoriasis Severity Index (NAPSI), Modified Nail Psoriasis Severity Index (mNAPSI), Mander/Newcastle Enthesitis Index (MEI), Leeds Enthesitis Index (LEI), Spondyloarthritis Research Consortium of Canada (SPARCC), Maastricht Ankylosing Spondylitis Enthesis Score (MASES), Leeds Dactylitis Index (LDI), Patient Global for Psoriatic Arthritis, Dermatology Life Quality Index (DLQI), Psoriatic Arthritis Quality of Life (PsAQOL), Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), Psoriatic Arthritis Response Criteria (PsARC), Psoriatic Arthritis Joint Activity Index (PsAJAI), Disease Activity in Psoriatic Arthritis (DAPSA), and Composite Psoriatic Disease Activity Index (CPDAI). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S64–85.
Helliwell PS, Fitzgerald O, Fransen J, et al. The development of candidate composite disease activity and responder indices for psoriatic arthritis (GRACE project). Ann Rheum Dis. 2013;72(6):986–91.
Bond SJ, Farewell VT, Schentag CT, et al. Predictors for radiological damage in psoriatic arthritis: results from a single centre. Ann Rheum Dis. 2007;66(3):370–6.
Willkens RF, Williams HJ, Ward JR, et al. Randomized, double-blind, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. Arthritis Rheum. 1984;27(4):376–81.
Kingsley GH, Kowalczyk A, Taylor H, et al. A randomized placebo-controlled trial of methotrexate in psoriatic arthritis. Rheumatology (Oxford). 2012;51(8):1368–77.
Abu-Shakra M, Gladman DD, Thorne JC, et al. Longterm methotrexate therapy in psoriatic arthritis: clinical and radiological outcome. J Rheumatol. 1995;22(2):241–5.
Chandran V, Schentag CT, Gladman DD. Reappraisal of the effectiveness of methotrexate in psoriatic arthritis: results from a longitudinal observational cohort. J Rheumatol. 2008;35(3):469–71.
Clegg DO, Reda DJ, Mejias E, et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs Cooperative Study. Arthritis Rheum. 1996;39(12):2013–20.
Gupta AK, Grober JS, Hamilton TA, et al. Sulfasalazine therapy for psoriatic arthritis: a double blind, placebo controlled trial. J Rheumatol. 1995;22(5):894–8.
Dougados M, vam der Linden S, Leirisalo-Repo M, et al. Sulfasalazine in the treatment of spondyloarthropathy. A randomized, multicenter, double-blind, placebo-controlled study. Arthritis Rheum. 1995;38(5):618–27.
Kaltwasser JP, Nash P, Gladman D, et al. Efficacy and safety of leflunomide in the treatment of psoriatic arthritis and psoriasis: a multinational, double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2004;50(6):1939–50.
Nash P, Thaci D, Behrens F, et al. Leflunomide improves psoriasis in patients with psoriatic arthritis: an in-depth analysis of data from the TOPAS study. Dermatology. 2006;212(3):238–49.
Ash Z, Gaujoux-Viala C, Gossec L, et al. A systematic literature review of drug therapies for the treatment of psoriatic arthritis: current evidence and meta-analysis informing the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis. 2012;71(3):319–26.
Fraser AD, van Kuijk AW, Westhovens R, et al. A randomised, double blind, placebo controlled, multicentre trial of combination therapy with methotrexate plus ciclosporin in patients with active psoriatic arthritis. Ann Rheum Dis. 2005;64(6):859–64.
Salvarani C, Macchioni P, Olivieri I, et al. A comparison of cyclosporine, sulfasalazine, and symptomatic therapy in the treatment of psoriatic arthritis. J Rheumatol. 2001;28(10):2274–82.
Mahrle G, Schulze HJ, Brautigam M, et al. Anti-inflammatory efficacy of low-dose cyclosporin A in psoriatic arthritis. A prospective multicentre study. Br J Dermatol. 1996;135(5):752–7.
Antoni CE, Kavanaugh A, Kirkham B, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT). Arthritis Rheum. 2005;52(4):1227–36.
Kavanaugh A, Antoni CE, Gladman D, et al. The Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT): results of radiographic analyses after 1 year. Ann Rheum Dis. 2006;65(8):1038–43.
Mease PJ, Gladman DD, Ritchlin CT, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52(10):3279–89.
Gladman DD, Mease PJ, Ritchlin CT, et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial. Arthritis Rheum. 2007;56(2):476–88.
Kavanaugh A, McInnes I, Mease P, et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum. 2009;60(4):976–86.
Kavanaugh A, van der Heijde D, McInnes IB, et al. Golimumab in psoriatic arthritis: one-year clinical efficacy, radiographic, and safety results from a phase III, randomized, placebo-controlled trial. Arthritis Rheum. 2012;64(8):2504–17.
Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009;373(9664):633–40.
Fitzgerald O, Helliwell P, Mease P, et al. Application of composite disease activity scores in psoriatic arthritis to the PRESTA data set. Ann Rheum Dis. 2012;71(3):358–62.
Mease PJ, Kivitz AJ, Burch FX, et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum. 2004;50(7):2264–72.
Mease PJ, Fleischmann R, Deodhar AA, et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a phase 3 double-blind randomised placebo-controlled study (RAPID-PsA). Ann Rheum Dis. 2014;73(1):48–55.
van der Heijde D, Fleischmann R, Wollenhaupt J, et al. Effect of different imputation approaches on the evaluation of radiographic progression in patients with psoriatic arthritis: results of the RAPID-PsA 24-week phase III double-blind randomised placebo-controlled study of certolizumab pegol. Ann Rheum Dis. 2014;73(1):233–7.
Ritchlin CT, Kavanaugh A, Gladman DD, et al. Treatment recommendations for psoriatic arthritis. Ann Rheum Dis. 2009;68(9):1387–94.
Meyerhoff JO. Exacerbation of psoriasis with meclofenamate. N Engl J Med. 1983;309(8):496.
Clark DW, Coulter DM. Psoriasis associated with rofecoxib. Arch Dermatol. 2003;139(9):1223.
Ben-Chetrit E, Rubinow A. Exacerbation of psoriasis by ibuprofen. Cutis. 1986;38(1):45.
Reshad H, Hargreaves GK, Vickers CF. Generalized pustular psoriasis precipitated by phenylbutazone and oxyphenbutazone. Br J Dermatol. 1983;109(1):111–3.
Powles AV, Griffiths CE, Seifert MH, et al. Exacerbation of psoriasis by indomethacin. Br J Dermatol. 1987;117(6):799–800.
Flytstrom I, Stenberg B, Svensson A, et al. Methotrexate vs ciclosporin in psoriasis: effectiveness, quality of life and safety. A randomized controlled trial. Br J Dermatol. 2008;158(1):116–21.
Saurat JH, Stingl G, Dubertret L, et al. Efficacy and safety results from the randomized controlled Comparative Study of Adalimumab vs Methotrexate vs Placebo in Patients with Psoriasis (CHAMPION). Br J Dermatol. 2008;158(3):558–66.
Reich K, Langley RG, Papp KA, et al. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N Engl J Med. 2011;365(17):1586–96.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451–85.
Nielsen OH, Bukhave K, Elmgreen J, et al. Inhibition of 5-lipoxygenase pathway of arachidonic acid metabolism in human neutrophils by sulfasalazine and 5-aminosalicylic acid. Dig Dis Sci. 1987;32(6):577–82.
Gupta AK, Ellis CN, Siegel MT, et al. Sulfasalazine improves psoriasis. A double-blind analysis. Arch Dermatol. 1990;126(4):487–93.
Farr M, Kitas GD, Waterhouse L, et al. Treatment of psoriatic arthritis with sulphasalazine: a one year open study. Clin Rheumatol. 1988;7(3):372–7.
Farr M, Kitas GD, Waterhouse L, et al. Sulphasalazine in psoriatic arthritis: a double-blind placebo-controlled study. Br J Rheumatol. 1990;29(1):46–9.
Combe B, Goupille P, Kuntz JL, et al. Sulphasalazine in psoriatic arthritis: a randomized, multicentre, placebo-controlled study. Br J Rheumatol. 1996;35(7):664–8.
Breedveld FC, Dayer JM. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2000;59(11):841–9.
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47(2–3):119–25.
Berth-Jones J, Henderson CA, Munro CS, et al. Treatment of psoriasis with intermittent short course cyclosporin (Neoral). A multicentre study. Br J Dermatol. 1997;136(4):527–30.
Ellis CN, Fradin MS, Messana JM, et al. Cyclosporine for plaque-type psoriasis. Results of a multidose, double-blind trial. N Engl J Med. 1991;324(5):277–84.
Faerber L, Braeutigam M, Weidinger G, et al. Cyclosporine in severe psoriasis. Results of a meta-analysis in 579 patients. Am J Clin Dermatol. 2001;2(1):41–7.
Ho VC, Griffiths CE, Albrecht G, et al. Intermittent short courses of cyclosporin (Neoral(R)) for psoriasis unresponsive to topical therapy: a 1-year multicentre, randomized study. The PISCES Study Group. Br J Dermatol. 1999;141(2):283–91.
Ho VC, Griffiths CE, Berth-Jones J, et al. Intermittent short courses of cyclosporine microemulsion for the long-term management of psoriasis: a 2-year cohort study. J Am Acad Dermatol. 2001;44(4):643–51.
Mrowietz U, Farber L, Henneicke-vonZepelin HH, et al. Long-term maintenance therapy with cyclosporine and posttreatment survey in severe psoriasis: results of a multicenter study. German Multicenter Study. J Am Acad Dermatol. 1995;33(3):470–5.
Shupack J, Abel E, Bauer E, et al. Cyclosporine as maintenance therapy in patients with severe psoriasis. J Am Acad Dermatol. 1997;36(3 Pt 1):423–32.
Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349(21):2014–22.
Tyring S, Gottlieb A, Papp K, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367(9504):29–35.
Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366(9494):1367–74.
Cimzia [package insert]. Smyrna: UCB, Inc.; 2013.
UCB receives CHMP positive opinion for Cimzia® (certolizumab pegol) in active psoriatic arthritis [press release]. UCB, 2013. http://www.ucb.presscentre.com/News/UCB-receives-CHMP-positive-opinion-for-Cimzia-certolizumab-pegol-in-active-psoriatic-arthritis-469.aspx (Accessed 17 Jan 2014).
Reich K, Ortonne JP, Gottlieb AB, et al. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab’ certolizumab pegol: results of a phase II randomized, placebo-controlled trial with a re-treatment extension. Br J Dermatol. 2012;167(1):180–90.
Armstrong AW. Do TNF inhibitors reduce the risk of myocardial infarction in psoriasis patients? JAMA. 2013;309(19):2043–4.
Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008;371(9625):1665–74.
Griffiths CE, Strober BE, van de Kerkhof P, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362(2):118–28.
McInnes IB, Kavanaugh A, Gottlieb AB, et al. Ustekinumab in patients with active psoriatic arthritis: results of the phase 3, multicenter, double-blind, placebo-controlled PSUMMIT I study [abstract OP0158]. Ann Rheum Dis. 2012;71(Suppl 3):107.
Ritchlin CT, Gottlieb AB, McInnes IB, et al. Ustekinumab in active psoriatic arthritis including patients previously treated with anti-TNF agents: results of a phase 3, multicenter, double-blind, placebo-controlled study [abstract 2557]. Arthritis Rheum. 2012;64(10 Suppl):S1080–1.
Young MS, Horn EJ, Cather JC. The ACCEPT study: ustekinumab versus etanercept in moderate-to-severe psoriasis patients. Expert Rev Clin Immunol. 2011;7(1):9–13.
Alwawi EA, Krulig E, Gordon KB. Long-term efficacy of biologics in the treatment of psoriasis: what do we really know? Dermatol Ther. 2009;22(5):431–40.
Gniadecki R, Kragballe K, Dam TN, et al. Comparison of drug survival rates for adalimumab, etanercept and infliximab in patients with psoriasis vulgaris. Br J Dermatol. 2011;164(5):1091–6.
Saurat JH, Guerin A, Yu AP, et al. High prevalence of potential drug-drug interactions for psoriasis patients prescribed methotrexate or cyclosporine for psoriasis: associated clinical and economic outcomes in real-world practice. Dermatology. 2010;220(2):128–37.
Ravindran V, Scott DL, Choy EH. A systematic review and meta-analysis of efficacy and toxicity of disease modifying anti-rheumatic drugs and biological agents for psoriatic arthritis. Ann Rheum Dis. 2008;67(6):855–9.
Kitamura G, Mehr N, Anderson N, et al. A case of tuberculosis in a patient on efalizumab and etanercept for treatment of refractory palmopustular psoriasis and psoriatic arthritis. Dermatol Online J. 2009;15(2):11.
Hamilton TK. Treatment of psoriatic arthritis and recalcitrant skin disease with combination therapy. J Drugs Dermatol. 2008;7(11):1089–93.
Adisen E, Karaca F, Gurer MA. When there is no single best biological agent: psoriasis and psoriatic arthritis in the same patient responding to two different biological agents. Clin Exp Dermatol. 2008;33(2):164–6.
Heinecke GM, Luber AJ, Levitt JO, et al. Combination use of ustekinumab with other systemic therapies: a retrospective study in a tertiary referral center. J Drugs Dermatol. 2013;12(10):1098–102.
Cuchacovich R, Garcia-Valladares I, Espinoza LR. Combination biologic treatment of refractory psoriasis and psoriatic arthritis. J Rheumatol. 2012;39(1):187–93.
Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10(22):1503–19.
Schafer PH, Parton A, Gandhi AK, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–55.
Schafer PH. Effects of apremilast on innate and adaptive immune responses [poster]. 41st Annual Meeting of the European Society for Dermatological Research, Barcelona; 2011.
Papp K, Cather J, Rosoph L, et al. The efficacy of apremilast, a phosphodiesterase-4 inhibitor, in the treatment of moderate to severe psoriasis: results of a phase 2 randomised study. Lancet. 2012;380:738–46.
Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2012;64(10):3156–67.
Reich K, Papp K, Leonardi C, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe psoriasis: 16-week results of a phase 3, randomized, controlled trial (ESTEEM 1) [oral presentation]. Annual meeting of the American Academy of Dermatology, Miami; 2013.
Papp K, Reich K, Leonardi C et al. Apremilast, an oral phosphodiesterase 4 inhibitor in nail and scalp psoriasis: 52-week results from the ESTEEM 1 study [abstract P1601]. Annual Congress of the European Academy of Dermatology and Venereology, Istanbul; 2013.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: results of a phase 3, randomized, controlled trial [abstract L13]. Arthritis Rheum. 2012;64(12):4172–3.
Celgene International Sarl. Apremilast achieves statistical significance for the primary endpoint of the first phase III study (PALACE-1) in patients with psoriatic arthritis [press release]; 2012. http://ir.celgene.com/phoenix.zhtml?c=111960&p=irol-newsArticle&ID=1714113&highlight= (Accessed 6 Aug 2013).
van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–19.
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367(6):495–507.
Fleischmann R, Cutolo M, Genovese MC, et al. Phase 2B dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to DMARDs. Arthritis Rheum. 2012;64(3):617–29.
Boy MG, Wang C, Wilkinson BE, et al. Double-blind, placebo-controlled, dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patients with psoriasis. J Invest Dermatol. 2009;129(9):2299–302.
Papp KA, Menter A, Strober B, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol. 2012;167(3):668–77.
Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. 2013;168(2):412–21.
Rich P, Sigurgeirsson B, Thaci D, et al. Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled, phase II regimen-finding study. Br J Dermatol. 2013;168(2):402–11.
McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73(2):349–56.
Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–9.
Mease PJ, Genovese MC, Greenwald MW, et al. Efficacy of brodalumab, an anti-IL-17R antibody, in subjects with psoriatic arthritis [abstract OP0103]. Ann Rheum Dis. 2013;72(Suppl 3):85.
Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366(13):1190–9.
Skvara H, Dawid M, Kleyn E, et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J Clin Invest. 2008;118(9):3151–9.
David M, Akerman L, Ziv M, et al. Treatment of plaque-type psoriasis with oral CF101: data from an exploratory randomized phase 2 clinical trial. J Eur Acad Dermatol Venereol. 2012;26(3):361–7.
Papp KA. Secukinumab efficacy and safety preliminary results from a phase II subcutaneous dose-ranging study in the treatment of moderate-to-severe plaque psoriasis [abstract FC01.5]. Congress of the European Academy of Dermatology and Venereology, Lisbon; 2011.
Papp KA. Secukinumab, a novel fully human antibody to interleukin-17A, in the treatment of moderate-to-severe plaque psoriasis: efficacy and safety interim results from a phase II intravenous induction dose-ranging study [abstract FC01.7]. Congress of the European Academy of Dermatology and Venereology, Lisbon; 2011.
Rich PA. Secukinumab, a new fully human monoclonal anti-Interleukin-17A antibody, in the treatment of moderate-to-severe plaque psoriasis: interim efficacy and safety data from a phase II regimen-finding trial [abstract FC01.6]. Congress of the European Academy of Dermatology and Venereology, Lisbon; 2011.
Mease PJ, Fleischmann R, Deodhar A, et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24 week results of a phase 3 double blind randomized placebo-controlled study (RAPID-PSA) [abstract LB0001]. Ann Rheum Dis. 2012;71(Suppl 3):150.
Papp K, Bissonnette R, Rosoph L, et al. Efficacy of ISA247 in plaque psoriasis: a randomised, multicentre, double-blind, placebo-controlled phase III study. Lancet. 2008;371(9621):1337–42.
Waller M, Toone EC. Normal individuals with positive tests for rheumatoid factor. Arthritis Rheum. 1968;11(1):50–5.
McInnes IB, Ritchlin CT, Rahman P, et al. Ustekinumab is effective radiographic progression in patients with active psoriatic arthritis: integrated data analysis of two phase 3, randomized, placebo-controlled studies [abstract 1695]. Arthritis Rheum. 2013;65(10 Suppl):S718.
Acknowledgments
This review was supported by Celgene Corporation (Summit, NJ, USA). The authors received editorial support in the preparation of this manuscript from Amy Zannikos, PharmD, and Karen Dougherty, PhD, of Peloton Advantage, LLC (Parsippany, NJ, USA), funded by Celgene Corporation. The authors, however, directed and are fully responsible for all content and editorial decisions involved in this article.
Conflict of interest
P.J.M. has received grant support and honoraria as a consultant for AbbVie, Amgen, Biogen Idec, Bristol-Myers Squibb, Celgene, Crescendo, Genentech, Janssen, Eli Lilly, Merck, Novartis, Pfizer, UCB and Vertex. He has also received honoraria as a lecturer or member of speakers’ bureaux for AbbVie, Amgen, Biogen Idec, Bristol-Myers Squibb, Crescendo, Genentech, Janssen, Eli Lilly, Pfizer and UCB.
A.W.A. has received grant support from AbbVie, Amgen, Janssen and Novartis, and has received honoraria as a consultant for Merck and Pfizer. She has also received honoraria for participation as a member of data monitoring boards for AbbVie, Amgen, Janssen and Pfizer, and as a lecturer or member of speakers’ bureaux for AbbVie and Janssen.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mease, P.J., Armstrong, A.W. Managing Patients with Psoriatic Disease: The Diagnosis and Pharmacologic Treatment of Psoriatic Arthritis in Patients with Psoriasis. Drugs 74, 423–441 (2014). https://doi.org/10.1007/s40265-014-0191-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0191-y