
Abstract
Background
The safety profile of COVID-19 vaccines in immunocompromised patients has not been comprehensively evaluated.
Aim
To measure the frequency of patient-reported adverse drug reactions (ADRs) related to the first/second/booster dose of COVID-19 vaccine in immunocompromised subject versus matched cohort. As a secondary objective, the time course, evaluated as time to onset (TTO) and time to recovery (TTR), of COVID-19 vaccine-related ADRs was explored.
Methods
A prospective cohort study, based on electronic questionnaires filled by vaccinees from 11 European countries in the period February 2021 to February 2023 was conducted. All immunocompromised vaccinees who provided informed consent and registered to the project’s web-app within 48 h after first/booster vaccine dose administration of any EMA-authorised COVID-19 vaccine were recruited. Participants filled baseline and up to six follow-up questionnaires (FU-Qs) over 6 months from vaccination, collecting information on suspected COVID-19 vaccine-related ADRs. As a control group, non-immunocompromised vaccinees from the same source population were 1:4 matched by sex, age, vaccine dose, and brand. A descriptive analysis of demographic/clinical characteristics of vaccinees was conducted. Heatmaps of the frequency of solicited ADRs, stratified by gender and vaccine brand, were generated. Median TTO/TTR of reported ADRs were visualised using violin/box-plots.
Results
A total of 773 immunocompromised vaccines were included in the analyses. Most participants were females (F/M ratio: 2.1 and 1.6) with a median age of 56 (43–74) and 51 (41–60) years, at the first vaccination cycle and booster dose, respectively. Injection-site pain and fatigue were the most frequently reported ADRs in immunocompromised vaccinees with higher frequency than matched control, especially after the first dose (41.2% vs 37.8% and 38.2% vs 32.9%, respectively). For both cohorts, all solicited ADRs were more frequently reported in females than males, and in those who had received a first dose of the Vaxzevria vaccine. Dizziness was the most frequently reported unsolicited ADR after the first dose in both groups (immunocompromised subjects: 2.5% and matched controls: 2.1%). At the booster dose, lymphadenopathy (3.9%) and lymphadenitis (1.8%) were the most reported unsolicited ADRs for immunocompromised subjects and matched controls, respectively. A very low number of subjects reported adverse event of special interest (AESI) (2 immunocompromised, 3 matched controls) and serious ADRs (5 immunocompromised, 5 matched controls). A statistically significant difference among study cohorts was observed for median TTO after the booster dose, and for median TTR after the first vaccination cycle and booster dose (p < 0.001).
Conclusion
The overall safety profile of COVID-19 vaccines in immunocompromised people was favourable, with minor differences as compared to non-immunocompromised vaccinees. Participants mostly experienced mild ADRs, mainly reported after the first dose of Vaxzevria and Jcovden vaccines. Serious ADRs and AESI were rare.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study provides a comprehensive evaluation of COVID-19 vaccine safety in immunocompromised subjects versus controls. |
COVID-19 vaccines showed a favourable safety profile in immunocompromised people. |
1 Introduction
Immunocompromised patients are at high risk of developing severe infections, including COVID-19 [1,2,3]. For this reason, during the initial phases of the COVID-19 vaccination campaign, immunocompromised patients, such as organ transplant recipients, HIV patients, and individuals with autoimmune diseases receiving immunosuppressant treatments, were prioritised. In addition, public health agencies proposed more aggressive vaccination strategies in immunocompromised patients with a three-dose schedule (instead of two) at the first vaccination cycle for first marketed vaccines [4, 5].
However, pivotal clinical trials of COVID-19 vaccines did not include vulnerable populations such as immunocompromised patients [6]. Due to impaired immune response to vaccines, concerns about the efficacy of COVID-19 vaccines in these patients have been primarily raised. After COVID-19 vaccine marketing, a few trials were conducted to assess COVID-19 vaccine immunogenicity in people with primary/secondary immunosuppressive disorders [7, 8]. These studies documented that after the third dose, most patients showed a strong serological response, despite a substantial proportion of patients with specific primary immunodeficiencies or with immunosuppressive medication being weak responders to the vaccines (Supplementary Table 1).
Several observational studies exploring the COVID-19 vaccine benefit-risk profile in immunocompromised patients in real-world settings were carried out later on. Findings from these studies showed that immunocompromised people experience mild and transient adverse reactions (e.g., fatigue, myalgia, injection site pain), few vaccine-related symptom exacerbations, and worsening disease following COVID-19 vaccinations [9].
However, overall, those studies primarily investigated the vaccine effectiveness in this vulnerable population, while its safety has not been comprehensively evaluated due to small sample size, selection of immunocompromised patients, and limited study duration, which allowed monitoring of vaccinees only during the first weeks following vaccination (Supplementary Table 1). Therefore, the short-/long-term safety of the first cycle, as well as booster doses, of COVID-19 vaccines in immunocompromised patients require additional investigation.
Active surveillance systems play a crucial role in ensuring safety, maintaining trust, and informing policy, especially in the context of a COVID-19 pandemic. In particular, active cohort event monitoring systems provide the ability to monitor vaccine safety in special cohorts in real time and generate rates with a well-defined denominator. For this reason, the European Medicines Agency (EMA) funded the “Covid Vaccine Monitor” (CVM) active surveillance project, establishing a comprehensive cohort event monitoring of COVID-19 vaccinees from different European countries, belonging to the general population as well as “frail patient” categories [10]. In the context of the CVM project, published studies have explored several relevant research questions, focusing on special populations like children/adolescents or people with prior SARS-CoV-2 infection [11,12,13]. In the current study, the aim was to describe the patient-reported adverse drug reactions (ADRs) following the first COVID-19 vaccination cycle as well as the booster dose of different vaccine brands with a focus on immunocompromised patients, as compared to matched controls with no immunosuppression derived from the same source population.
2 Methods
2.1 Setting and Study Population
This was a prospective cohort study that was based on electronic questionnaires collecting vaccinee-reported outcomes from 11 European countries (Belgium, France, Italy, Ireland, Portugal, Romania, Slovakia, Spain, Switzerland, the Netherlands, and the UK, in the period February 2021 to February 2023.
All vaccinees aged ≥ 18 years recruited in the CVM project and who were reported to be immunocompromised, provided informed consent, and registered to the project’s web-app within 48 h of receiving the first dose or booster dose of any EMA-authorised COVID-19 vaccine, were included in the study.
In addition, as control group, applying the same inclusion criteria, we selected a random sample of vaccinees participating in the CVM project who did not report being immunocompromised who were matched to immunocompromised vaccinees. The matching was constructed based on a 1:4 ratio using the propensity score values. In particular, the exact matching was applied for gender, dose, and vaccine brand, whereas the nearest neighbour procedure was used for the age variable.
In each country the study protocol was approved by the respective Ethical Committee and the study was carried out in line with the General Data Protection Regulation (GDPR) and the Data protection impact assessment (DPIA). The study protocol is published in the HMA-EMA Catalogue of real-world studies (EUPAS42504) [10].
2.2 Data Collection
Participants from 11 European countries were included and questionnaires were translated into local language [10].
When possible, associations of rheumatic patients were involved to help recruiting specifically immunocompromised patients at the time of vaccination.
Participants could register via a project-specific web-app from February 2021 to November 2022. After registering and providing informed consent, participants were invited via e-mail to fill in the baseline questionnaire, using any of two ad hoc developed and fully harmonised web-based apps: the LAREB-managed Intensive Monitoring (LIM) and Research Online (RO). In particular, the LIM web-app was already developed by the Dutch pharmacovigilance centre Lareb and implemented for the CVM study but could not support additional changes (i.e., to include the booster dose). Therefore, the RO platform was developed by the University Medical Centre Utrecht (UMCU) in the Netherlands, to allow the collection of information for the booster dose. Data from each web-app were converted into a unique common data model and analysed all together.
In the baseline questionnaire, information on vaccinee’s characteristics, including demographics, medical history, concomitant drug use, and administered COVID-19 vaccine brand was collected. Specifically, for immunocompromised vaccinees, detailed information on the cause of immunosuppression in the medical history section was collected (e.g., HIV, transplantation leukaemia/lymphoma, autoimmune disease treated with immunosuppressant drugs).
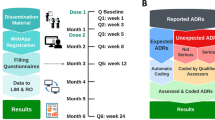
Thereafter, vaccinees completed six follow-up questionnaires (FU-Qs) over 6 months starting from the first dose of COVID-19 vaccine, and 5 follow-up questionnaires over a 3-month period for those who were recruited at the booster dose. The end of the recruitment period was scheduled to have the last FU-Qs completed at latest by February 2023, for both the first and booster doses. Follow-up questionnaires collected information on patient-reported short-/medium-/long-term adverse reaction following the COVID-19 vaccines. Specifically, FU-Qs collected information on solicited local (injection site haematoma, induration, inflammation, pain, pruritus, swelling, and warmth) and systemic ADRs (arthralgia, chills, fatigue, headache, malaise, myalgia, nausea, and fever), based on the most frequently reported adverse events in pivotal trials [14,15,16]. Information on unsolicited ADRs, adverse events of special interest (AESIs), and serious ADRs were also collected. Adverse events of special interest were defined according to a list provided by the Brighton Collaboration (Supplementary Table 2) [17]. Serious ADRs were assessed according to the Council for International Organizations of Medical Sciences (CIOMS) criteria [18]. All patient-reported adverse reactions were coded using the MedDRA coding system (version 24.0) [19].
For each ADR, information on time to onset (TTO), outcome, duration of symptoms (if recovered), severity/impact of the symptoms (including medical assistance & hospitalisation) was collected [20].
2.3 Data Analysis
For all the analyses, we included subjects who filled the baseline questionnaire along with at least the first FU-Q (i.e., Q1). A flowchart of recruited vaccinees and filled questionnaires was reported.
First, a descriptive analysis on demographics and clinical characteristics (e.g., use of any medication, use of painkillers or fever treatment before vaccination, medical history of cardiovascular disorder, diabetes mellitus, hypertension, liver disorder, lung disorder, mental disorder, malignant tumour, nervous system disorder and renal disorder) of immunocompromised vaccinees versus controls was conducted.
Second, the frequency of local and systemic solicited ADRs reported following the first, second and booster dose of any COVID-19 vaccine for both immunocompromised people and controls was measured. Furthermore, the proportion of subjects with at least one ADR overall, and specifically solicited/unsolicited/serious ADR or AESI across different vaccine doses was also evaluated, using as denominator the total number of recruited vaccinees receiving the first, second, or booster dose of any vaccine and who filled the baseline questionnaire and at least the FU-Q1.
Heatmaps of the frequency of vaccinee-reported local and systemic solicited ADRs, stratified by gender and vaccine brand, were also generated.
The median (along with 1st quartile and 3rd quartile) TTO and time to recovery (TTR) of all reported ADRs in immunocompromised versus non-immunocompromised vaccinees were computed and visualised using violin plots and box-plots. As participants could report the TTO and the TTR as seconds, minutes, hours, days, weeks, months or indicating a specific calendar date, TTO and TTR were standardised as hours. For this analysis, we included only ADRs for which both TTO and TTR values were available.
Categorical variables were reported as absolute frequencies and percentages, while continuous variables were reported as median (and interquartile range [IQR]) or mean, as appropriate. A Chi-square test or Fisher’s exact test was performed to compare categorical variables as appropriate. A p value < 0.05 denoted the statistical significance.
All analyses were performed using R statistical software (version 4.3.1). For heatmaps, violin plot and box-plot, the ggplot2 R package was used.
3 Results
Overall, a total of 946 recruited immunocompromised vaccinees completed the baseline questionnaire at either first vaccination cycle (653, 69.0%) or booster dose (293, 31.0%). Of these, only 773 (81.7%) (N = 566, 73.2% at the first dose, N = 207, 26.8% at the booster dose) filled also at least the first FU-Q and therefore were finally included in the analysis. Those vaccinees were matched 1:4 to 3092 non-immunocompromised vaccinees by age, gender, dose, and vaccine brand (N = 2264, 73.2% at the first dose; N = 828, 26.8% at the booster dose) from the same source population.
Considering vaccinees recruited at the first vaccination cycle, 59.0% of the immunocompromised subjects and 61.9% of the matched controls completed all six questionnaires; instead, at the booster dose, only 41.5% of the immunocompromised vaccinees versus 48.3% of the matched controls completed all the five questionnaires (Fig. 1).

Flowchart including the number of questionnaires filled by immunocompromised vaccinees and matched control, at the first vaccination cycle and the booster dose
3.1 Demographic and Clinical Characteristics
As shown in Table 1, at the first vaccination cycle, the majority of vaccinees were female (68.0%), and the median age was 56 (IQR: 43–74) years. Regarding vaccine brand, 29.2% of vaccinees included in the study received the Vaxzevria vaccine, 49.1% Comirnaty vaccine, 17.7% Spikevax vaccine and 3.7% Jcovden vaccine.
As for the booster dose, 61.8% of recruited vaccinees were female, and the median age was 51 (IQR: 41–60) years. Almost all the vaccinees received Comirnaty (62.8%) or the Spikevax vaccine (36.2%).
The main cause of immunosuppression reported was immunosuppression due to autoimmune disorder (N = 111, 53.6%). Other causes of immunosuppression reported were related to malignant tumour (13.5%), infection (5.3%), and rheumatic disease (2.9%). In addition, 12.2% of patients did not report this information.
Furthermore, almost half of the subjects enrolled (48.8%) indicated a current use of medications that affect the immune system, particularly the use of immunosuppressants (32.9%) like TNF-alpha inhibitors and selective immunosuppressants.
3.2 Descriptive Analyses of ADR Frequencies
The frequency of immunocompromised people and matched controls reporting at least one ADR in FU-Qs was similar. After the first vaccination cycle and the booster dose, most subjects reported at least one ADR after the FU-Q1 (first vaccination cycle: immunocompromised subjects: 71.4%, matched controls: 65.5%; booster dose: immunocompromised subjects: 57.0%, matched controls: 57.6%) (Supplementary Table 3).
Overall, after all doses, higher frequencies of at least one ADR in the immunocompromised versus matched controls were reported (Table 2). The frequency of vaccinees reporting at least one ADR was higher after the first dose of any COVID-19 vaccine (immunocompromised vaccinees: 82.0%; matched control: 76.1%), as compared to the second (80.7% and 74.0%) and the booster (61.8% and 59.7%) doses.
Overall, when compared to the matched controls, the immunocompromised cohort showed a higher frequency of solicited ADRs after all doses. A statistically significant difference was especially observed for nausea after the first dose (p = 0.003), and for nausea (p = 0.005) and arthralgia (p = 0.005) after the booster dose (Supplementary Table 4).
Injection-site pain was the most frequently reported local solicited ADR following all doses for both groups (immunocompromised subjects vs matched controls: first dose: 41.2% vs 37.8%, second dose: 25.3% vs 24.7%, booster dose: 40.1% vs 35.7%). As for systemic solicited ADR, fatigue was reported most commonly after all doses, for both immunocompromised vaccinees (first dose: 38.2%, second dose: 25.3%, booster dose: 32.9%) and the matched control (first dose: 32.9%, second dose: 23.6%, booster dose: 29,8%) (Supplementary Table 4).
All solicited ADRs were more frequently reported in females, as compared to males, across all doses and study cohorts. Specifically, immunocompromised females showed higher frequencies of local solicited reaction injection-site pain following the first dose (48% vs 27%) as well as the booster dose (52% vs 20%) when compared to males. Additionally, increased frequencies of the systemic solicited reaction fatigue were observed in immunocompromised females vs males (first dose: 47% vs 20%, second dose: 30% vs 17%, booster dose: 38% vs 24%) (Fig. 2 and Supplementary Table 5).

Heatmap for vaccinee-reported local and systemic solicited ADRs following the first, the second and the booster dose of any vaccine, in immunocompromised people and matched controls, stratified by gender
Systemic solicited ADRs were most frequently experienced by participants after the first dose of the Vaxzevria vaccine and the Jcovden vaccine and after the second dose of the Spikevax vaccine. In detail, after the Vaxzevria vaccine, headache was the most reported systemic solicited ADR in both groups (immunocompromised subjects: 57% vs matched control: 60%). Fatigue (67%) was the most frequently reported reaction after the administration of the Jcovden vaccine in immunocompromised vaccinees, while malaise (52%) in the matched control. Regarding the second dose, for immunocompromised vaccinees and matched controls, fatigue (53% vs 46%) and malaise (51% vs 58%), respectively, were the most frequently reported ADRs after receiving the Spikevax vaccine. At the booster dose, the highest frequency was observed for fatigue (37% and 31%) after the Spikevax vaccine for both study groups (Fig. 3 and Supplementary Tables 6, 7, 8).

Heatmap for reported local and systemic solicited ADRs following the first, the second and the booster dose of any vaccine, in immunocompromised people and matched controls, stratified by vaccine brand
The frequency of vaccinees reporting at least one unsolicited ADR was highest after the first dose of COVID-19 in both groups (immunocompromised subjects vs matched controls: first dose: 34.8% vs 30.7%, second dose: 23.1% vs 20.3%, booster dose: 17.4% vs 14.0%). Dizziness was the most frequently reported unsolicited ADR after the first dose of COVID-19 vaccines in both groups (immunocompromised subjects: 2.5% and matched controls: 2.1%) and after the second dose in matched controls (1.0%), while immunocompromised people indicated extensive swelling of the vaccinated limb (1.4%) after the second dose. At the booster dose, lymphadenopathy (3.9%) and lymphadenitis (1.8%) were the most reported unsolicited ADRs for immunocompromised subjects and matched controls, respectively.
Of note, after the first dose, 3 (0.5%) immunocompromised subjects indicated “condition aggravated” and 1 (0.2%) “Crohn’s disease”. At the booster dose, 2 (1.0%) immunocompromised subjects reported “rheumatoid arthritis”, 1 (0.5%) “ankylosing spondylitis”, 1 (0.5%) “polymyalgia rheumatica”, and 1 (0.5%) “psoriasis” (Supplementary Table 9).
For each study cohort, only 1 AESI was reported after the administration of the first dose of any COVID-19 manufacturers. In particular, in the FU-Q1, 1 (0.2%) immunocompromised vaccinee reported “epilepsy”, while 1 (< 0.1%) matched control “facial paralysis”, both with Vaxzevria vaccine.
After the second dose, in the FU-Q3, 1 (0.2%) immunocompromised subject indicated “platelet count decreased” after the administration of Spikevax, while 1 (0.1%) “hypersensitivity” (FU-Q3, Comirnaty) and 1 (0.1%) “facial paralysis” (FU-Q3, Spikevax) were reported for matched controls. No AESIs were reported after the booster dose in either group (Supplementary Table 10).
With regard to serious ADRs, at the first dose, only 3 (0.5%) immunocompromised subjects and 3 (0.1%) matched controls reported at least one serious ADR. At the second dose, only 2 (0.5%) immunocompromised subjects reported serious ADRs, while at the booster dose, serious ADRs were reported by only 2 (0.2%) matched controls. Supplementary Table 10 provides a comprehensive list of reported serious ADRs.
3.3 Analyses of Time-to-Onset and Duration of Events
Overall, after both the first vaccination cycle and the booster dose, participants showed a median TTO within one day. In particular, the median TTO of reported ADRs in the first vaccination cycle was equal to 20.9 h and 17.9 h for immunocompromised vaccinees and matched control, respectively (p = 0.424). For the booster dose, the median TTO for the immunocompromised vaccinees and matched controls was slightly shorter (13.7 h vs 15.4 h, p < 0.001) (Fig. 4).

Combination of violin plot and boxplot of the median time to onset (in h) and time to recovery (in hours) of reported ADRs for the first vaccination cycle and the booster dose, in immunocompromised people and matched control
A statistically significant difference was observed in the median TTR in immunocompromised vaccinees versus matched controls (first vaccination cycle: 40.9 h vs 37.7 h, p < 0.001; booster dose: 40.8 h vs 38.4 h, p < 0.001) (Fig. 4).
4 Discussion
To our knowledge, this is the first active surveillance study from multiple European countries that explored the safety of COVID-19 vaccines among immunocompromised individuals, as compared to matched non-immunocompromised controls, using patient-reported outcomes collected via electronic questionnaires.
Findings from this study showed that, across different vaccine doses and brands, COVID-19 vaccines in immunocompromised people were overall well tolerated, in line with evidence from the general population. Mild ADRs were only marginally increased in the immunocompromised cohort relative to healthy controls. Specifically, injection-site pain and fatigue were the most reported local and systemic solicited ADRs in both cohorts, which is consistent with evidence from the literature and in line with those reported in the Summary of Product Characteristics and RCTs (Supplementary Table 1) [9, 21,22,23,24,25,26,27].
A statistically significant difference was observed for arthralgia in immunocompromised subjects when compared to matched controls. In addition, some unsolicited ADRs, like condition aggravated (0.5%), Crohn’s disease (0.2%), rheumatoid arthritis (0.5%), or psoriasis (0.5%), were reported by immunocompromised subjects. Based on these observations, such ADRs could be related to flares of underlying diseases, thus supporting the low frequency of flares after COVID-19 vaccination, as already described [28, 29].
Furthermore, by stratifying analyses per vaccine manufacturer, our study showed that immunocompromised participants, as well as matched controls, who received the first dose of the Vaxzevria or Janssen vaccine, experienced the highest frequency of solicited ADRs. This confirms already published evidence, in which the Vaxzevria vaccine resulted in higher reactogenicity compared to the other vaccines in a cohort of HIV patients [30].
Interestingly, in line with broader studies involving the general population, immunocompromised female vaccinees showed an increased frequency of solicited ADRs as compared to male vaccinees. Differences in response to vaccination between the sexes are widely recognised, with several studies reporting a significantly higher incidence of reported ADRs after COVID-19 vaccination in females than males [31,32,33].
Upon assessing a longer follow-up period compared to published studies, our data suggest a favourable long-term safety profile of COVID-19 vaccines and a minimal risk of delayed ADRs. Notably, despite the long follow-up period available, the majority of ADRs were reported in the FU-Q1 for both study cohorts, as already documented in previous studies [12]. With regard to the timing of ADRs to COVID-19 vaccines, overall, a median TTO of symptoms within one day of vaccination and a TTR within 2 days was observed. Moreover, a slightly longer time to recovery was reported in the immunocompromised cohort when compared with the matched cohort. Knowledge regarding long-term ADRs of COVID-19 vaccines in immunocompromised individuals is sparse. In a prospective observational study with a longer follow-up period than other published studies, in 139 people with HIV, ADRs were mild and resolved spontaneously within 7 days, and until the end of follow-up, no new ADRs occurred [34].
Concerning serious ADRs and AESI, we found a very low rate in both immunocompromised vaccinees and matched controls, following the first vaccination cycle and the booster dose, suggesting that vaccines are safe and reliable. These results appear concordant with previous studies on the safety of the COVID-19 vaccine in immunocompromised subjects, which have reported either no or very low rates of severe adverse reactions after vaccination [21, 35].
Strengths of this study included the international collaboration involving multiple European countries with a common protocol and a common data model to analyse data on individual patient levels in a real-life setting.
The large number of subjects enrolled in this European project allowed us to find four controls for each immunocompromised subject to determine whether there might be relevant differences. Furthermore, another additional value was the possibility to compare different vaccine brands and doses in the same study. Finally, this active surveillance study made it possible to assess the frequency of ADRs due to the presence of a denominator.
Several study limitations warrant a cautious interpretation of the findings. The initial vaccination cycle was expanded for immunocompromised people with an additional third dose administered at least 28 days after the second dose [4]. We could not collect information on the first vaccination cycle’s third dose as the tools we used did not include this information. As such, for the immunocompromised cohort, some ADRs reported in the FU-Q4, 5, and 6 could be related to this additional dose, possibly overestimating the results. However, by exploring the frequencies of at least one ADR per questionnaire, we observed that the trend of the immunocompromised and non-immunocompromised cohorts is very similar, suggesting no marginal influence of the additional third dose on the estimated rates of ADR in immunocompromised vaccinees. Moreover, although study populations were matched using propensity score matching, residual confounding could not be entirely ruled out. Furthermore, stratifying the analyses by type of immunosuppression, or type of immunosuppressive therapy, was not feasible due to the limited number of subjects who provided this information. In addition, the frequency of serious ADRs could have been underestimated due to the exclusion of patients who were unable to return the follow-up questionnaires. Nevertheless, research on surveillance of post-vaccination ADRs reveals that self-reported data document more serious events than those reported by health care professionals [36]. Finally, rare ADRs may not have been captured due to the limited number of participants enrolled.
5 Conclusion
This study documented that the overall safety profile of COVID-19 vaccines in immunocompromised people was favourable, with minor differences in the frequency of ADRs as compared to non-immunocompromised vaccinees. Immunocompromised vaccinees experienced mostly mild and transient adverse reactions, with a median time to onset of symptoms within one day after vaccination and a recovery time of up to 2 days. Vaccinee-reported ADRs were more frequent among females, and the frequency of serious ADRs was extremely low among both groups. Systemic solicited ADRs were most often experienced by participants receiving the first dose of the Vaxzevria and the Janssen vaccines, and the second dose of Spikevax vaccine in both study cohorts.
References
Kuderer NM, Choueiri TK, Shah DP, et al. Clinical impact of COVID-19 on patients with cancer (CCC19): a cohort study [published correction appears in Lancet. 2020 Sep 12;396(10253):758]. Lancet. 2020;395(10241):1907–18. https://doi.org/10.1016/S0140-6736(20)31187-9.
Dai M, Liu D, Liu M, et al. Patients with cancer appear more vulnerable to SARS-CoV-2: a multicenter study during the COVID-19 outbreak. Cancer Discov. 2020;10(6):783–91. https://doi.org/10.1158/2159-8290.CD-20-0422.
Fendler A, de Vries EGE, GeurtsvanKessel CH, et al. COVID-19 vaccines in patients with cancer: immunogenicity, efficacy and safety. Nat Rev Clin Oncol. 2022;19(6):385–401. https://doi.org/10.1038/s41571-022-00610-8.
Centre for Disease Control and Prevention (CDC). COVID-19 Vaccines for People Who Are Moderately or Severely Immunocompromised. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/recommendations/immuno.html. Accessed 20 Sept 2023.
Comirnaty and Spikevax: EMA recommendations on extra doses and boosters. https://www.ema.europa.eu/en/news/comirnaty-and-spikevax-ema-recommendations-extra-doses-and-boosters. Accessed Sept 2023.
Luxi N, Giovanazzi A, Capuano A, et al. COVID-19 Vaccination in pregnancy, paediatrics, immunocompromised patients, and persons with history of allergy or prior SARS-CoV-2 infection: overview of current recommendations and pre- and post-marketing evidence for vaccine efficacy and safety. Drug Saf. 2021;44(12):1247–69. https://doi.org/10.1007/s40264-021-01131-6.
Lee ARYB, Wong SY, Chai LYA, et al. Efficacy of covid-19 vaccines in immunocompromised patients: systematic review and meta-analysis. BMJ. 2022;376: e06863. https://doi.org/10.1136/bmj-2021-068632.
Maneikis K, Šablauskas K, Ringelevičiūtė U, et al. Immunogenicity of the BNT162b2 COVID-19 mRNA vaccine and early clinical outcomes in patients with haematological malignancies in Lithuania: a national prospective cohort study. Lancet Haematol. 2021;8(8):e583–92. https://doi.org/10.1016/S2352-3026(21)00169-1.
Geisen UM, Berner DK, Tran F, et al. Immunogenicity and safety of anti-SARS-CoV-2 mRNA vaccines in patients with chronic inflammatory conditions and immunosuppressive therapy in a monocentric cohort. Ann Rheum Dis. 2021;80(10):1306–11. https://doi.org/10.1136/annrheumdis-2021-220272.
European Network of Centres for Pharmacoepidemiology and Pharmacovigilance. https://catalogues.ema.europa.eu/node/3177/administrative-details. Accessed Dec 2023.
Ahmadizar F, Luxi N, Raethke M, et al. Safety of COVID-19 vaccines among the paediatric population: analysis of the european surveillance systems and pivotal clinical trials. Drug Saf. 2023;46(6):575–85. https://doi.org/10.1007/s40264-023-01304-5.
Raethke M, van Hunsel F, Thurin NH, et al. Cohort event monitoring of adverse reactions to COVID-19 vaccines in seven European countries: pooled results on first dose. Drug Saf. 2023;46(4):391–404. https://doi.org/10.1007/s40264-023-01281-9.
Ciccimarra F, Luxi N, Bellitto C, L’Abbate L, Raethke M, van Hunsel F, Lieber T, Mulder E, Riefolo F, Dureau-Pournin C, et al. Safety monitoring of COVID-19 vaccines in persons with prior SARS-CoV-2 infection: a European multi-country study. Vaccines. 2024;12(3):241. https://doi.org/10.3390/vaccines12030241.
Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15. https://doi.org/10.1056/NEJMoa2034577.
Sadoff J, Gray G, Vandebosch A, Cárdenas V, Shukarev G, Grinsztejn B, et al. Final analysis of efficacy and safety of single-dose Ad26COV2.S. N Engl J Med. 2022;386(9):847–60.
Kant A, Jansen J, van Balveren L, van Hunsel F. Description of frequencies of reported adverse events following immunization among four different COVID-19 vaccine brands. Drug Saf. 2022;45(4):319–31. https://doi.org/10.1007/s40264-022-01151-w.
Brighton Collaboration. COVID-19 AESI list. 2020. https://brightoncollaboration.us/wp-content/uploads/2021/01/COVID-19-updated-AESI-list.pdf. Accessed May 2023.
VIII CWG. Practical Aspects of Signal Detection in Pharmacovigilance: Report of CIOMS Working Group VIII. Geneva; 2010. Report No.: 9290360828.
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human U. Welcome to MedDRA 2021 [updated 2017/06/16/]. https://www.meddra.org/.
Raethke M, van Hunsel F, Luxi N, et al. Frequency and timing of adverse reactions to COVID-19 vaccines; a multi-country cohort event monitoring study. Vaccine. 2024. https://doi.org/10.1016/j.vaccine.2024.03.001.
Han S, Yang Y, Wang T, et al. Safety and immunogenicity of the third (booster) dose of inactivated and recombinant protein SARS-CoV-2 vaccine for patients with endocrine-related cancer. Front Public Health. 2023;11:1086872. https://doi.org/10.3389/fpubh.2023.1086872.
Monin L, Laing AG, Muñoz-Ruiz M, et al. Safety and immunogenicity of one versus two doses of the COVID-19 vaccine BNT162b2 for patients with cancer: interim analysis of a prospective observational study. Lancet Oncol. 2021;22(6):765–78. https://doi.org/10.1016/S1470-2045(21)00213-8.
Connolly CM, Ruddy JA, Boyarsky BJ, et al. Safety of the first dose of mRNA SARS-CoV-2 vaccines in patients with rheumatic and musculoskeletal diseases. Ann Rheum Dis. 2021;80(8):1100–1. https://doi.org/10.1136/annrheumdis-2021-220231.
https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf.
Picchianti Diamanti A, Navarra A, Cuzzi G, et al. The third dose of BNT162b2 COVID-19 vaccine does not "boost" disease flares and adverse events in patients with rheumatoid arthritis. Biomedicines. 2023;11(3):687. https://doi.org/10.3390/biomedicines11030687.
Furer V, Eviatar T, Zisman D, et al. Immunogenicity and safety of the BNT162b2 mRNA COVID-19 vaccine in adult patients with autoimmune inflammatory rheumatic diseases and in the general population: a multicentre study [published correction appears in Ann Rheum Dis. 2022 Jul;81(7):e133]. Ann Rheum Dis. 2021;80(10):1330–1338. https://doi.org/10.1136/annrheumdis-2021-220647.
Bieńkowski C, Skrzat-Klapaczyńska A, Firląg-Burkacka E, Horban A, Kowalska JD. The clinical effectiveness and safety of vaccinations against COVID-19 in HIV-positive patients: data from observational study in Poland. Vaccines (Basel). 2023;11(3):514. https://doi.org/10.3390/vaccines11030514
Duijster JW, Lieber T, Pacelli S, et al. Sex-disaggregated outcomes of adverse events after COVID-19 vaccination: a Dutch cohort study and review of the literature. Front Immunol. 2023;14:1078736. https://doi.org/10.3389/fimmu.2023.1078736.
Green MS, Peer V, Magid A, Hagani N, Anis E, Nitzan D. Gender differences in adverse events following the Pfizer-BioNTech COVID-19 vaccine. Vaccines (Basel). 2022;10(2):233. https://doi.org/10.3390/vaccines10020233.
Rivera-Izquierdo M, Soler-Iborte E, de Rojas JP, et al. Factors associated with adverse reactions to BNT162b2 COVID-19 vaccine in a cohort of 3969 hospital workers. Vaccines (Basel). 2021;10(1):15. https://doi.org/10.3390/vaccines10010015.
Ao L, Lu T, Cao Y, et al. Safety and immunogenicity of inactivated SARS-CoV-2 vaccines in people living with HIV. Emerg Microbes Infect. 2022;11(1):1126–34. https://doi.org/10.1080/22221751.2022.2059401.
Giuliano A, Kuter B, Pilon-Thomas S, et al. Safety and immunogenicity of a third dose of mRNA-1273 vaccine among cancer patients. Cancer Commun (Lond). 2023;43(7):749–64. https://doi.org/10.1002/cac2.12453.
Clothier HJ, Selvaraj G, Easton ML, Lewis G, Crawford NW, Buttery JP. Consumer reporting of adverse events following immunization [published correction appears in Hum Vaccin Immunother. 2015;11(5):1294]. Hum Vaccin Immunother. 2014;10(12):3726–3730. https://doi.org/10.4161/hv.34369.
Acknowledgements
The authors acknowledge Cecile Droz-Perroteau, Estelle Guiard, Pauline Bosco, Pauline Diez and Stéphanie Lamarque (University of Bordeaux); Brigitte Keller-Stanislawski and Dirk Mentzer (Paul-Ehrlich Institut); Fergal O’Shaughnessy, Brian Cleary (Rotunda Hospital Dublin); Ana Paula Martins, Ana Penedones, Ana Silva, Carla Torre, Carlos Miguel Costa Alves, Diogo Mendes, Inês Vaz, Jorge Polonia, Paula Barao and Renato Ferreira da Silva (CLPP Vaccines Network); Camelia Busca (University of Medicine and Pharmacy ‘Iuliu Hatieganu’ Cluj-Napoca); Beata Čečetková, Daniel Pella and Radka Troníčková (SLOVACRIN, Medical faculty of Pavol Jozef Šafárik University in Košice); Alice Panchaud, Émeline Maisonneuve and Guillaume Favre (University of Bern); Agnes Kant, Jasper Schmitz and Loes Ruijs (Lareb); Hester de Melker (RIVM); Derek Hall, Elizabeth Lynn, Saad Shakir, Vicki Osborne, Samantha Lane and Shane Freemantle (UK DSRU); the Primary Care Centres from the Catalan Institute of Health (for their support and the recruitment of vaccines), namely, Amposta, Tortosa, Barberà del Vallés, Mataró, Pineda de Mar and Salou; and “ilmiovaccinoCOVID19 collaborating group”. This study has been labelled as a National Research Priority by the National Orientation Committee for Therapeutic Trials and other researchers on Covid-19 (CAPNET). The investigators would like to acknowledge ANRS, Emerging Infectious Diseases for their scientific support, the French Ministry of Health and Prevention and the French Ministry of Higher Education, Research and Innovation for their funding and support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
This document expresses the opinion of the authors of the paper, and may not be understood or quoted as being made on behalf of, or reflecting the position of the European Medicines Agency or one of its Committees or Working Parties. Miriam Sturkenboom is head of a department that conducts studies for regulatory agencies and pharmaceutical companies, with research grants to the institution; this includes Pfizer, Janssen and AstraZeneca. All studies are conducted using the ENCePP code of conduct. Gianluca Trifirò: Participation in advisory boards and seminars on topics not related to this paper and sponsored by the following pharmaceutical companies in the last two years: UCB, MSD, Eli Lilly; Sanofi; Amgen; Novo Nordisk; Sobi; Gilead; Celgene; Daikii Sankyo. Scientific coordinator of the UNIVR academic spin-off INSPIRE that in the last two years carried out observational studies/systematic reviews on topics not related to the content of this paper and which were funded by Novo Nordisk, Kiowa Kirin, Shionogi, and Daiichi Sankyo, Consultant for Viatris in a judicial proceeding, the subject of which is not related to the topic of this paper. Professor Gianluca Trifirò is an Editorial Board member of Drug Safety. Gianluca Trifirò was not involved in the selection of peer reviewers for the manuscript nor any of the subsequent editorial decisions. Chiara Bellitto, Nicoletta Luxi, Francesco Ciccimarra, Luca L’Abbate, Monika Raethke, Florence van Hunsel, Thomas Lieber, Erik Mulder, Fabio Riefolo, Felipe Villalobos, Nicolas H. Thurin, Francisco B. Marques, Kathryn Morton, Fergal O'Shaughnessy, Simona Sonderlichová, Andreea Farcas, Giele-Eshuis Janneke have no conflicts of interest that are directly relevant to the content of this article. The research leading to these results was conducted as part of the activities of the EU PE&PV (Pharmacoepidemiology and Pharmacovigilance) Research Network which is a public academic partnership coordinated by the Utrecht University, The Netherlands. Scientific work for this project was coordinated by the University Medical Center Utrecht in collaboration with the Vaccine Monitoring Collaboration for Europe network (VAC4EU). The project has received support from the European Medicines Agency under the Framework service contract nr EMA/2018/23/PE. The content of this paper expresses the opinion of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties.
Funding
Open access funding provided by Università degli Studi di Verona within the CRUI-CARE Agreement. This research was supported by the European Medicines Agency.
Data availability statement
The study reports with results are available in Supplementary Materials and in reference 10, section “Methodological aspects” (https://catalogues.ema.europa.eu/node/3177/methodological-aspects. The datasets for this manuscript are not publicly available. Requests to access the datasets should be directed and motivated to the first author and will be granted upon reasonable request and permission from partners.
Code availability
Code availability will be granted upon reasonable request and permission from partners.
Ethics approval
Netherlands: EC ref number NW2021-04; France: CNIL ref. no. MLD/MFI/AR218727, EC ref. no. 2020-A03554-35. Italy: PARERE N. 463 del Registro delle Sperimentazioni 2020/2021. Portugal: EC ref. no. 74/2021. Romania: EC ref. no. AVZ6; 08/11/2021 and 246; 30/06/2021. Slovakia: Trieda SNP 1,040 11 Kosice; 1/12/2021. Spain: 21/208-PCV; 29/09/2021. The UK: EC ref. no. 21/HRA/2077.
Additional information
The members of the “ilmiovaccinoCOVID19 collaborating group” are listed in the Supplementary Materials.
Consent to participate (include appropriate statements)
Informed consent was obtained from all subjects involved in the study.
Informed consent statements
In order to participate in this study, we need your consent. Furthermore, you will be asked to provide some general details. As soon as the informed consent has been sent, you will receive an e-mail with an activation link. Once this link has been clicked, the participation is confirmed and definitive. You will receive the first questionnaire, which can be filled in immediately. The activation link is valid for a maximum of 48 h. For questions, please contact the study team at [name organisation] via [email organisation] or [telephone number organisation]. I have read the privacy statement and the information regarding this research. Any and all questions I had were answered by contacting [name organisation]. I understand that participation is voluntary. Furthermore, I understand that I can decide at any moment to stop my participation in this research and do not need to give a reason for my decision. I understand that all information will be treated with strict confidentiality. I give permission for my data to be used for the purpose of this research; namely, to gather information and expand knowledge on possible symptoms which can occur after receiving the corona vaccine. It is important for [name organisation] to know precisely which vaccine was given in order to compare the reported symptoms between the given corona vaccines. Gaining more insight in the relevant medical history of participants, reported symptoms, the nature of these symptoms, the course of these symptoms, possible risk factors and the consequences related to health (Available at https://catalogues.ema.europa.eu/node/3177/administrative-details Annex 2: example of informed consent).
Consent for publication (include appropriate statements)
Consent for publication was obtained from all subjects involved in the study.
Consent for publication statements
“I give permission for my data to be used for the purpose of this research, namely, to gather information and expand knowledge on possible symptoms which can occur after receiving the corona vaccine.” (Available at https://catalogues.ema.europa.eu/node/3177/administrative-details Annex 2: example of informed consent)
Data contribution
Conceptualisation: Gianluca Trifirò. Formal analysis: Chiara Bellitto, Luca L’Abbate, Thomas Lieber, Erik Mulder; Investigation: Chiara Bellitto, Nicoletta Luxi, Francesco Ciccimarra, Monika Raethke, Florence van Hunsel, Fabio Riefolo, Felipe Villalobos, Nicolas H. Thurin, Francisco B. Marques, Kathryn Morton, Fergal O'Shaughnessy, Simona Sonderlichová, Andreea Farcas, Giele-Eshuis Janneke, Miriam C. Sturkenboom. Data curation: Monika Raethke, Florence van Hunsel, Fabio Riefolo, Felipe Villalobos, Nicolas H. Thurin, Francisco B. Marques, Kathryn Morton, Fergal O'Shaughnessy, Simona Sonderlichová, Andreea Farcas, Giele-Eshuis Janneke, Miriam C. Sturkenboom. Writing, original draft: Chiara Bellitto, Nicoletta Luxi, Gianluca Trifirò. Writing, review & editing: Francesco Ciccimarra, Monika Raethke, Florence van Hunsel, Fabio Riefolo, Felipe Villalobos, Nicolas H. Thurin, Francisco B. Marques, Kathryn Morton, Fergal O'Shaughnessy, Simona Sonderlichová, Andreea Farcas, Giele-Eshuis Janneke, Miriam C. Sturkenboom. Visualisation: Chiara Bellitto, Francesco Ciccimarra. Supervision: Miriam C. Sturkenboom, Gianluca Trifirò. All authors have read and agreed to the published version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bellitto, C., Luxi, N., Ciccimarra, F. et al. What is the Safety of COVID-19 Vaccines in Immunocompromised Patients? Results from the European “Covid Vaccine Monitor” Active Surveillance Study. Drug Saf (2024). https://doi.org/10.1007/s40264-024-01449-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s40264-024-01449-x