
Abstract
Janus kinase inhibitors (JAKi) have enormous appeal as immune-modulating therapies across many chronic inflammatory diseases, but recently this promise has been overshadowed by questions regarding associated cardiovascular and cancer risk emerging from the ORAL Surveillance phase 3b/4 post-marketing requirement randomized controlled trial. In that study of patients with rheumatoid arthritis with existing cardiovascular risk, tofacitinib, the first JAKi registered for chronic inflammatory disease, failed to meet non-inferiority thresholds when compared with tumor necrosis factor inhibitors for both incident major adverse cardiovascular events and incident cancer. While this result was unexpected by many, subsequently published observational data have also supported this finding. Notably, however, such a risk has largely not yet been demonstrated in patients outside the specific clinical situation examined in the trial, even in the face of many studies examining this. Nevertheless, this signal has practically re-aligned approaches to both tofacitinib and other JAKi to varying extents, in other patient populations and contexts: within rheumatoid arthritis, but also in psoriatic arthritis, axial spondyloarthritis, inflammatory bowel disease, atopic dermatitis, and beyond. Application to individual patients can be more challenging but remains important to harness the substantive potential of JAKi to the maximum extent safely possible. This review not only explores the evolution of the regulatory response to the signal, its informing data, biological plausibility, and its impact on guidelines, but also the many factors that clinicians must consider in navigating cardiovascular and cancer risk for their patients considering JAKi as immune-modulating therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
JAK inhibitor therapeutic benefit must be balanced against the potential for harm in the clinical context in which it is being prescribed. |
Cardiovascular and cancer risk from JAK inhibitors appears subject to effect modification dependent on baseline cardiovascular risk. |
Clinical risk for individual patients is dependent on multiple factors, including indication, baseline cardiovascular risk, previous response to therapy, alternative therapies available, type and dose of JAK inhibitor chosen, and other risk–benefit considerations. |
1 Introduction
JAK inhibitors (JAKi) are a novel class of immune-modulating therapies that convey great therapeutic promise [1]. While they were first registered for rheumatoid arthritis (RA) where multiple equally efficacious therapies exist, their potential utility extends across a number of different chronic inflammatory diseases. These not only include other registered indications including psoriatic arthritis (PsA), axial spondyloarthritis, atopic dermatitis, inflammatory bowel diseases (IBD), and alopecia areata, but also diseases with substantive unmet need including systemic lupus erythematosus (SLE), giant cell arteritis, polymyalgia rheumatica, and inflammatory myopathies [2]. Even within RA, while other therapies have similar efficacy, JAKi confer logistical advantages over them, including oral administration and a rapid onset of action.
However, throughout their drug development for chronic inflammatory disease indications, some questions have remained about JAKi safety. The first registered JAKi, tofacitinib, has been subject to enhanced post-marketing surveillance since its registration, culminating in a post-marketing requirement for a phase 4 randomized controlled trial (RCT) identifying safety signals within a patient population with existing cardiovascular risk. Foremost amongst these risks was a potential for increased cardiovascular and cancer events when compared with tumor necrosis factor inhibitors (TNFi), the co-primary endpoints of this study. Safety studies of this fidelity are rare in this therapeutic field, but nevertheless these results have proven challenging to apply in practice.
Clinicians have had to consider multiple different complex interacting factors when applying these results to individual patients, and the implications have differed across the highly varied clinical scenarios where JAKi might be used. In practice, at this point in time, substantial diversity in approach exists between individual clinicians in how these risks are interpreted and to how this might change their prescribing. Among changing data and regulation, and the pharmaceutical industry’s substantial interest in influencing clinical decision-making, it is important to encourage rational decision-making through comprehensive assessment of all factors at play.
In this review, we seek to capture the full complexity of considerations that clinicians face in deriving their clinical approach, as it currently stands. We summarize the current landscape of JAKi for providers, in particular addressing cardiovascular and cancer risk and all factors pertinent to informing clinical decision-making: the history of this safety signal, the broader data informing it, the plausibility behind it, other pertinent individual-level factors, and existing guidance. This information may serve to guide clinicians in managing their patients, as we aim to encapsulate the unique challenges faced by this patient group and detail our approach to managing this risk in practice, acknowledging the inherent associated uncertainty.
2 A Signal Correctly Identified, But with Uncertain Clinical Implications for Patients
Alongside the first US Food and Drug Administration (FDA) approval of tofacitinib in 2012, which was for use in RA, an additional post-marketing clinical trial was mandated due to dyslipidemia observed in the drug’s clinical development program and the extrapolated concern of an increased risk of cardiovascular adverse events [3,4,5,6,7]. Similarly, there was a need to further investigate the incidence of serious infections and malignancy, having seen 11 solid cancers and one lymphoma diagnosed in 3328 patients compared with zero in placebo-receiving patients [8]. These concerns were identified on the background of other known safety signals from tofacitinib’s registration trials, including most notably an increased risk of herpes zoster [4]. Shortly after this time, the European Medicines Agency (EMA) similarly released a notification in 2013 for the refusal of the marketing authorization for tofacitinib [9].
In response to these regulatory actions, the manufacturer of tofacitinib undertook the Oral Rheumatoid Arthritis Trial (ORAL) Surveillance trial, a phase 3b/4, randomized, open-label, non-inferiority, post-marketing study, whose first patient was enrolled in March 2014 [10]. Patients included were those with RA with an inadequate response to methotrexate, aged 50 years or older and with at least one additional cardiovascular risk factor (current cigarette smoker, hypertension, dyslipidemia, diabetes mellitus, family history of premature coronary heart disease, extra-articular RA, or history of coronary artery disease). Included patients were randomized to receive tofacitinib 5 mg BID or 10 mg BID or a TNFi (adalimumab or etanercept), with the trial’s primary endpoints being non-inferiority of tofacitinib compared with TNFi relating to major adverse cardiovascular events (MACE) and malignancies [excluding non-melanoma skin cancer (NMSC)].
Further concerns about tofacitinib’s safety were raised in February 2019 when higher rates of pulmonary embolism and mortality were noted in the 10 mg BID arm in comparison with TNFi but not the 5 mg BID arm. At that time, the other JAKi in clinical use for RA, baricitinib, had similarly shown a signal for increased venous thromboembolism (VTE) risk in the higher 4 mg daily dose, but not the 2 mg daily dose [11], leading the FDA to only register the latter dose for baricitinib’s RA indication the year prior. This combination of events led the ORAL Surveillance study investigators to switch these patients to the lower 5 mg BID dose [10]. In addition, the EMA issued a series of warnings in March 2019 regarding the increased risk of VTE and subsequent restrictions of the use of tofacitinib while this was further assessed [12]. The FDA, too, announced a boxed warning for the 10 mg BID dose in July 2019, with this dose approved only in limited cases of ulcerative colitis (UC) but not RA or PsA [13].
January 2021 saw the announcement of primary endpoint results for ORAL Surveillance, namely the failure to meet prespecified non-inferiority criteria for MACE and malignancy [14]. This prompted further FDA updates to the boxed warning in February 2021 to describe increased risk of cardiovascular disease and cancer with tofacitinib [15], before ultimately placing a broader black box warning to the other JAKi in use for immune-mediated inflammatory diseases—baricitinib and upadacitinib—in September 2021, citing a similar mechanism of action and restricting the class of medications to patients failing TNFi [16].
Following the publication of final results from ORAL Surveillance in January 2022, safety concerns about cardiovascular and cancer risk from JAKi were highly debated among clinicians attempting to evolve their practice to incorporate these new data. Cautionary warnings and advisory statements were evoked by professional organizations, including a statement by the American College of Rheumatology (ACR) recommending shared decision-making between patients and providers about the risks versus benefits of JAKi [17]. Among other actions by key regulatory bodies (Table 1), the EMA issued a direct healthcare professional communication advising against first-line use of JAKi across all approved indications in inflammatory and dermatologic diseases in patients aged 65 or older with smoking history and other cardiovascular or cancer risk factors, explicitly describing MACE and malignancy as class effects [18].
For many clinicians, the implications are still being fully understood. Although rheumatologists frequently manage uncertainty in clinical practice, many remain unclear as to how to best incorporate JAKi into the treatment paradigm of RA and other rheumatologic diseases. For some, these warnings may deter the use of JAKi in a majority of patients, while on the other end of the spectrum, JAKi may endure as a valuable option in the armamentarium for patients without risk factors. For non-rheumatological prescribers who might consider JAKi for autoimmune disease indications, interpreting this safety signal remains even more challenging and currently may not be directly addressed in counseling all patients [19].
The reality is that, while this post-marketing requirement study successfully built on concerns from a signal correctly noted by regulators within registration studies and added certainty within the scope that the research question was asking, its application in clinical practice for individual practice is still subject to multiple factors (Table 2). In this review, we try to address the importance of each of these factors and discuss how this might influence clinical decision-making for individual prescribers.
3 Clinical Data that Inform the Nature and Extent of Risk
Analyses of ORAL Surveillance, registration studies, and real-world data have all informed our understanding of the cardiovascular and cancer risks associated with JAKi (Table 3). To this end, we performed a search of the literature using MEDLINE to capture a broad mix of evidence that might help elucidate these risks in different cohorts. The search strategy is presented in Online Resource 1.
3.1 ORAL Surveillance
Within ORAL Surveillance, there was a trend toward increased incident MACE (HR 1.33, 95% CI 0.91–1.94) and a significant increase in incident cancer (HR 1.48, 1.04–2.09), although neither met the pre-determined non-inferiority threshold [10]. These figures equated to a number needed to harm for tofacitinib 5 mg BID of 567 patient-years for MACE and 276 patient-years for cancer, translating to an additional event for every 113 and 55 patients who were treated within the study, respectively.
3.2 Post Hoc Analyses of ORAL Surveillance
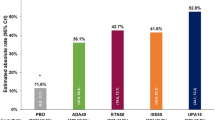
Notably, this effect was enriched in patients aged 65 years or older and was the focus of a post hoc analysis that separated patients into either a high-risk group, defined by age 65 years or older and history of long-term smoking, or low-risk group [20]. Observed in the former was a greater risk of serious adverse events, namely cancer (HR 1.55, 1.05–2.30 versus HR 1.16, 0.53–2.55), MACE (HR 1.41, 0.93–2.15 versus HR 0.98, 0.42–2.31), myocardial infarction (HR 1.92, 0.92–4.00 versus HR 0.78, 0.13–4.65), and all-cause mortality (HR 2.24, 1.20–4.19 versus HR 1.05, 0.36–3.07).
Other post hoc analyses have similarly analyzed the importance of these risk factors. Charles-Schoeman et al. highlighted an increased risk of MACE in patients with a baseline history of atherosclerotic cardiovascular disease (ASCVD) (HR 1.98, 0.95–4.14) compared with those without (HR 1.14, 0.73–1.78), even in association with a high 10-year cardiovascular risk score [21]. A similar post hoc analysis evaluated cancer risk and found a trend to higher incidence in patients with a history of ASCVD or higher cardiovascular risk scores, perhaps reflecting overlapping risk factors for cardiovascular disease and cancer [22]. Interestingly, this study observed similar malignancy risk between tofacitinib and TNFi groups until month 18 (HR 0.93, 0.53–1.62), after which this diverged (HR 1.93, 1.22–3.06), suggesting a possible time-dependent risk with tofacitinib use. It should be noted that ORAL Surveillance demonstrated a greater risk of infections, including opportunistic infections and herpes zoster with tofacitinib [10]. This factor has been consistently demonstrated amongst JAKi to date [23] and may have influenced clinical practice.
These post hoc analyses, while potentially providing important clarity that might help clinicians apply this study in practice, might also be selective and may not be as fair as the pre-specified analysis. Similar to the similar provisos that surround subsequent real-world data analyses, this should be considered when weighing the conclusions that might be drawn from such analyses.
3.3 Tofacitinib Clinical Program
Excessive MACE risk was not originally identified in tofacitinib’s clinical trial program that consisted of 21 phase 1–3b/4 and two long-term extension studies [24]. However, a notable difference between registration studies and ORAL Surveillance was the lower proportion of patients with a history of ASCVD (4% versus 15%), and this was explored in a study that emulated the risk-enriched cohort of ORAL Surveillance using 3125 patients from the clinical trial program [25]. Compared with ORAL Surveillance, the MACE IRs were lower overall but comparable in patients without a history of ASCVD.
Integrated safety summaries of prior RCTs and extension studies of tofacitinib have reported similar long-term rates of safety events compared with biologic disease-modifying antirheumatic drugs (bDMARDs). Of these, the largest, longest clinical dataset accrued 22,875 patient-years of exposure pooled from phase 1–3b/4 and long-term extension studies and reported an incidence rate (IR, per 100 patient-years) of 0.4 (0.3–0.5) for MACE and 0.8 (0.7–0.9), 0.6 (0.5–0.7), and 0.1 (0.0–0.1) for malignancies (excluding NMSC), NMSC, and lymphomas, respectively [26]. These figures appeared to be consistent with previous analyses of both MACE and malignancy with tofacitinib treatment [27,28,29,30]. To put these Irs into perspective, long-term safety data for adalimumab in RA with 37,106 patient-years reported an IR of 0.7 for malignancies (excluding lymphoma, skin cancer, leukemia, and hepatosplenic T-cell lymphoma), although such risk was likely historically higher at the time many of these data were collected [31].
All these findings raise the question as to whether the signals seen in ORAL Surveillance arose due to the studied population having a higher risk for cardiovascular events at baseline. Whatever the case, the importance of addressing traditional cardiovascular risk factors in patients with RA has never been clearer, and clinicians should take this as a reminder that risk stratification is a key component of the shared decision-making process.
3.4 Real-World Evidence for Tofacitinib Safety in RA
One must be prudent in generalizing the results of ORAL Surveillance as this population was not inclusive of all patients with RA. The RCT selected older, higher-risk individuals; as a consequence, the external validity and generalizability of the trial needs to be taken into consideration. Real-world evidence serves as an important, complementary form of evidence which may differently represent the effectiveness and safety of therapeutics. One such example is a large observational study (STAR-RA) using US commercial and Medicare claims data that, in a cohort of routine care patients (n = 102,263), did not identify an increased risk of a composite outcome of myocardial infarction and stroke with tofacitinib versus TNFi (pooled weighted HR 1.01, 0.83–1.23) [32]. Meanwhile, a sub-cohort within this study that was designed to mimic the inclusion and exclusion criteria of ORAL Surveillance (n = 35,070) did demonstrate a cardiovascular signal similar to that seen in the reference trial (pooled weighted HR 1.24, 0.90–1.69). While the STAR-RA study served purely for an exploratory analysis and was not powered to compare individual MACE risk factors across each treatment group, the findings seem to support the proposition that increased risk of MACE is particularly strong within the cardiovascular risk-enriched population captured in ORAL Surveillance. The latter group also puts forward the implication that the cardiovascular observations in ORAL Surveillance may have been influenced by effect modification, in that the effect of having a baseline history of increased cardiovascular risk led to a higher risk of developing incident MACE.
The STAR-RA study also investigated malignancy risk in both real-world evidence (n = 83,295) and ORAL Surveillance duplicate (n = 27,035) groups [33]. There was no significant difference between tofacitinib versus TNFi amongst all patients with RA in the real-world setting, but a numerically higher pooled weighted HR was observed in the ORAL Surveillance-matched group (1.01, 0.83–1.22 versus 1.17, 0.85–1.62), again suggesting older patients with cardiovascular risk factors may be at higher risk. However, with a mean follow-up time of just over 10 months and the knowledge that cumulative VTE risk from JAKi increases over time, there are insufficient data to comment on any effect on cardiovascular and cancer risk that a longer duration of tofacitinib use may incur.
The safety profile of tofacitinib observed in real-world settings across the world is also compatible with what has been described during its clinical development program. Cardiovascular and malignancy outcomes from select studies are summarized in Table 2.
Kremer et al. performed a registry study comparing tofacitinib (n = 1999) and biologic DMARD (n = 8358) initiation and found similar MACE and malignancy rates over 5 years [34]. In Europe, a French national health data cohort study of 15,835 patients compared patients with RA who started a JAKi (40% tofacitinib including 5 mg and 10 mg BID, 60% baricitinib) versus adalimumab and found no difference in MACE risk between the groups, even at the higher 10 mg BID dose of tofacitinib, although this dose was only observed in 21 patients [35]. Interestingly, the same outcome was delineated in a risk-enriched subset of patients aged 65 years or older with at least one cardiovascular risk factor, although the study was not powered for this subgroup analysis. Another French registry study of 100 patients reported one patient with SLE who had recurrence of cutaneous T-cell lymphoma after 6 months of tofacitinib but no MACE over the 9-month follow-up period [36]. A prospective Swedish cohort study of 10,447 patients with RA and 4443 patients with PsA compared cancer risk with JAKi (78% baricitinib, 18% tofacitinib, 4% upadacitinib), TNFi, and non-TNFi bDMARDs [37]. After less than 3 years of treatment, increased rates of NMSC were observed in the combined JAKi group in patients with RA (HR 1.39, 1.01–1.91), but the same was not seen for other malignancies (HR 0.94, 0.65–1.38). Of note, analysis of a cardiovascular-enriched subset demonstrated higher incidences but a similar HR of 1.03 (0.58–1.82), in keeping with the main analysis. In a Swiss cohort of 144 patients with RA receiving tofacitinib, no MACE or malignancy was reported after a mean follow-up of 1.22 years [38]. British registry data from a single center comparing tofacitinib 5 mg BID (n = 145) and etanercept (n = 114) identified three and four cases of MACE in the respective groups [IR ratio (IRR) 0.8, 0.2–4.1], with all affected patients having pre-existing cardiovascular risk factors [39]. The number of cancer events were three and one, respectively (IRR 3.5, 0.4–92.2). Notably, patients in the tofacitinib group were older, exposed more to active smoking, had higher odds of hypertension and family history of early coronary heart disease, and had lower odds of cancer history, but still had similar event rates compared with etanercept.
Studies in Asia include a 1-year multicenter study consisting of 370 patients with RA across 12 Japanese sites that compared adverse events with tofacitinib and abatacept and discovered comparable rates of MACE and malignancy [40]. In contrast, a Japanese cohort of 242 patients with RA treated with tofacitinib (n = 161) or baricitinib (n = 81) reported three cases of malignancy (colon, skin, and lung cancer) in the tofacitinib group and one (breast cancer) with baricitinib over a relatively short 24-week period [41]. In a Taiwanese insurance database study of patients with RA treated with tofacitinib (n = 822) with a mean follow-up of 2.02 years versus TNFi (n = 2357) with a mean follow-up of 2.10 years, there was no statistically significant difference in rates of coronary heart disease (HR 1.03, 0.45–2.36), stroke (HR 0.75, 0.29–1.94), or malignancy (HR 1.10, 0.44–2.78) [42].
These real-world experiences are reassuring of similar MACE and malignancy risk from JAKi compared with bDMARD therapy in a general RA population. Simultaneously, they suggest that specific patient populations with RA do hold increased odds of cardiovascular events, namely those who are older or have a pre-existing history of cardiovascular disease, and that caution should be exercised to balance these risks with the potential benefits of tofacitinib in these patients.
3.5 Tofacitinib in the Treatment of Other Diseases
While the present discussion revolves around RA, it would be sensible to explore whether the safety signals seen in ORAL Surveillance extend to tofacitinib use in other diseases, such as UC, where the more routine use of the higher 10 mg BID dose as part of induction therapy is also of note. The underlying molecular targets in different immune-mediated diseases may affect the efficacy and safety of each JAKi, so potential MACE and malignancy risks of tofacitinib may not necessarily be reflected when treating other conditions. An integrated summary of major trials in UC consisting of a total of 1157 patients treated with tofacitinib, predominantly at a dose of 10 mg BID (83%), found IRs of 0.29 (0.13–0.55) for MACE, 0.84 (0.55–1.24) for malignancies excluding NMSC, and 0.73 (0.45–1.10) for NMSC [43], figures not dissimilar to those seen in comparable RA analyses. Interestingly, the signal for VTE at the higher dose seen in ORAL Surveillance was not demonstrated in this UC group, with IRs of 0.03 (0.00–0.18) for deep vein thrombosis and 0.19 (0.07–0.42) for pulmonary embolism, and it is known that RA itself confers an increased risk of VTE [44,45,46,47]. Analysis of the same cohort stratified by age found that those aged 65 years or older had a significantly higher risk of adverse events than those younger than 65: MACE, IR 1.06, 0.13–3.81 versus 0.20, 0.07–0.47; malignancies excluding NMSC, IR 2.05, 0.56–5.25 versus 0.65, 0.37–1.06; and NMSC, IR 3.97, 1.60-8.19 versus 0.37, 0.17–0.70 [48]. These were reminiscent of age being recognized as a risk factor in ORAL Surveillance and STAR-RA, albeit without enrichment for cardiovascular history. These data do not appear to indicate an increased risk of MACE or malignancy with tofacitinib in patients with UC; discrepancies noted here in contrast to RA may be a reflection of how the pathophysiology of different diseases intersects with JAK inhibition and unexplained mechanisms behind cardiovascular disease and malignancy.
3.6 The Role of JAK Specificity
Although the safety of other JAKi has not yet been subject to post-marketing studies, the FDA has applied a blanket warning for the two other JAKi registered for use in autoimmune diseases (baricitinib and upadacitinib) given their similar mechanisms of action [49]. It remains unknown whether JAK specificity contributes to safety profile; while tofacitinib inhibits both JAK1 and JAK3, the other JAKi of interest in RA include baricitinib, which inhibits JAK1 and JAK2, and JAK1-selective upadacitinib and filgotinib [2]. In theory, this selectivity may facilitate more targeted blockade of the JAK cascade and its downstream immunomodulatory effects on particular inflammatory responses, but how this translates clinically to efficacy and safety remains to be seen, especially given the inherent redundancies between the signaling pathways with different JAKs [50, 51].
3.7 Safety of Other JAKi
Real-world data and integrated analyses for baricitinib and upadacitinib in RA have largely demonstrated a consistent safety profile without increased rates of MACE or malignancy over time. An integrated database consisting of nine phase 1b/2/3 trials and one long-term extension study of patients with RA taking either 2 mg or 4 mg of baricitinib investigated the long-term safety of baricitinib through 9.3 years of treatment, amounting to 14,744 patient-years [52]. The IR for MACE in all baricitinib doses was 0.5 (0.40–0.64); rates were similar in the 2 mg (0.42, 0.21–0.74) and 4 mg (0.54, 0.41–0.69) subsets. In a risk-enriched population aged 50 years or older with at least one cardiovascular risk factor, there was a higher incidence of MACE (0.77, 0.56–1.04), though this figure approximated more closely to the TNFi rates (0.73, 0.52–1.01) in ORAL Surveillance than tofacitinib (0.98, 0.79–1.19) [10]. Regarding malignancy (excluding NMSC), the IR stabilized at 0.9 (0.77–1.09) after 48 weeks, with respiratory and mediastinal (n = 26), breast (n = 23), and gastrointestinal (n = 19) cancers being most frequently reported. Compared with the general US population using SEER, a cancer dataset derived from population-based registries, an age-adjusted standardized IR of 1.07 (0.90–1.26) suggested a similar incidence of malignancy as the general population. The IRs for NMSC and lymphoma were 0.3 (0.25–0.44) and 0.06 (0.03–0.11), respectively.
A large US study of registry and insurance data propensity score-matched 7606 baricitinib-treated patients with RA with patients who received TNFi, amounting to 5879 and 6512 person-years of exposure, respectively [53]. A numerically higher but non-statistically significant IRR was observed for baricitinib for MACE (1.54, 0.93–2.54), with a difference in incidence rates of 0.22 (0.07–0.52) per 100 person-years, translating to an additional two MACE per year for every 1000 patients treated with baricitinib rather than TNFi. In a 12-week phase 2b RCT with a 52-week extension period in a Japanese cohort, 145 patients with active RA despite methotrexate therapy were randomized initially to placebo or 1 mg, 2 mg, 4 mg, or 8 mg of baricitinib during the first 12 weeks, then re-randomized to 4 mg or 8 mg during the extension period for those on other doses [54]. While MACE were not specifically described, hyperlipidemia and hypercholesterolemia were commonly reported. Malignancy occurred in one patient in each arm of the extension period, a case of rectal cancer and chondrosarcoma. Also in Japan, a post-marketing surveillance study examined the safety of baricitinib for RA over 24 weeks [55]. Out of 4731 patients over 1863 patient-years, MACE occurred in seven patients (0.15%) and malignancy in 17 (0.36%). A British observational study that included 69 patients treated with baricitinib, 54 with tofacitinib, and 8 with both found one case of malignancy in the tofacitinib arm but no MACE or malignancy events with baricitinib [56].
In relation to upadacitinib, an international RCT spanning across China, Brazil, and South Korea that randomized 338 patients with RA on stable conventional synthetic DMARDs (csDMARDs) to upadacitinib 15 mg daily or placebo reported no MACE or NMSC in either group after 12 weeks (although one patient in the upadacitinib arm was diagnosed with breast cancer on the day of randomization) [57].
Integrated analyses of existing phase 3 trials of upadacitinib have also been encouraging. Data from five randomized trials of upadacitinib 15 mg or 30 mg compared with methotrexate or adalimumab with 4020 patient-years of exposure revealed similar rates of MACE and malignancy across treatment groups [58]. There was a trend to higher IRs with 30 mg compared with 15 mg for both MACE (1.0, 0.5–1.6 versus 0.6, 0.4–1.0), malignancies (excluding NMSC) (1.4, 0.8–2.2 versus 0.9, CI 0.5–1.3), and NMSC (1.1, 0.6–1.8 versus 0.3, 0.1–0.6), suggesting a dose-dependent effect. In PsA, an analysis of one RCT evaluating upadacitinib 15 mg and 30 mg and another that included adalimumab as well pooled 2257 upadacitinib-treated patients for 2505 patient-years of exposure found similar safety profiles with regard to MACE and cancer [59]. Differing from the aforementioned RA analysis, numerically higher IRs were observed for the 15 mg dose in comparison with 30 mg for NMSC (0.8, 0.4–1.5 versus 0.7, 0.3–1.4) and MACE (0.3, 0.1–0.8 versus 0.2, 0.0–0.7), while the opposite was true for malignancies (excluding NMSC) (0.7, 0.3–1.4 versus 0.9, 0.4–1.6).
The long-term safety for upadacitinib is also a matter of interest in atopic dermatitis and UC, where it is sometimes given in higher doses than RA. A 2-year interim analysis of a phase 3 RCT (n = 272) of upadacitinib 15 mg versus 30 mg versus placebo in moderate-to-severe atopic dermatitis with 1:1 re-randomization of placebo patients to either upadacitinib dose at week 16 described one case of rectal cancer and one of cerebellar hemorrhage in the upadacitinib 15 mg group, representing an IR of 0.3 for both malignancy and MACE [60]. A clinical program consisting of two phase 3 multicenter RCTs of induction therapy in patients with moderately to severely active UC with upadacitinib 45 mg versus placebo for 8 weeks (n = 989), followed by one maintenance RCT with re-randomization to upadacitinib 15 mg versus 30 mg versus placebo for 52 weeks (n = 451) [61]. In the induction studies, no events of cancer or MACE were reported. In the maintenance trial, one case of malignancy (excluding NMSC) was reported in both the placebo (breast cancer) and upadacitinib 15 mg (breast cancer) groups and two in the upadacitinib 30 mg group (colon and prostate cancer); two cases of NMSC in the upadacitinib 30 mg group; and one MACE (myocardial infarction) in the placebo group.
Filgotinib, another JAK1-selective JAKi, is not marketed in the USA after the FDA rejected the initial filing in 2020 in light of concerns about male reproductive safety, but data from Europe and Japan regarding MACE and malignancy have been consistent with other JAKi. An integrated analysis of safety data from seven trials of filgotinib 100 mg or 200 mg in moderately to severely active RA included 3691 patients with 6081 patient-years and a median of 1.6 years and maximum 5.6 years of exposure [62]. Exposure-adjusted IRs for MACE for filgotinib 100 mg versus 200 mg were 0.6 (0.4–1.1) versus 0.4 (0.2–0.7); for malignancies (excluding NMSC), 0.5 (0.3–1.0) versus 0.6 (0.4–0.9); and for NMSC, 0.1 (0.0–0.5) versus 0.2 (0.1–0.4), respectively. In UC, preliminary findings from an ongoing long-term extension study of a phase 2b/3 trial of filgotinib 200 mg with 971 patient-years of exposure included exposure-adjusted IRs of 0.3 (0.1–0.9) for MACE and 0.8 (0.4–1.6) for malignancies (excluding NMSC) [63].
In summary, registration trial extension studies and real-world evidence examining non-cardiovascular risk-enriched patient populations have repetitively failed to demonstrate any clear increase in cardiovascular and cancer risk with any JAKi over what might be expected with TNFi. In contrast, patients with RA with existing cardiovascular risk seem to incur increased rates of both incident cardiovascular events and cancer with tofacitinib when compared with TNFi in the context of both RCT and real-world evidence, although fewer comparable data for other JAKi or for other indications exist.
4 Risk and Its Plausibility
While these data for other JAKi appear to dispel major fears about cardiovascular and cancer safety, is there any biological plausibility of JAKi giving rise to these adverse events in the first instance? Broadly speaking, side effects observed with DMARD therapy can often be mechanistically explained by the drug’s pharmacodynamics. For instance, type I interferons are key antiviral cytokines, and JAK inhibition has been linked to a heightened risk of herpes zoster through an effect on downstream signaling from type I interferons via TYK2 and JAK1 [64]. Similarly, JAKi have been labeled with a special concern for small bowel perforation [8, 65], which could be explained by the effect of JAK inhibition on IL-6 receptor signaling through JAK1, JAK2, and TYK2 [64].
In contrast, the mechanisms behind the potential risk for VTE, MACE, and cancer are still unclear.
4.1 VTE
While not the focus of this review, VTE was the first clinically important adverse event subject to substantial regulatory warnings for JAKi. As a consequence, it has been investigated for a longer period of time, and an exploration of whether this phenomenon can be mechanistically explained is worthwhile in trying to understand whether the processes that may underpin MACE and malignancy may be explainable from a mechanistic point of view.
Inflammatory disease activity in RA is associated with increased risk of VTE [66], whereby inflammation causes immunothrombosis, characterized by alterations in the endothelium surface [67]. Numerous cells, cytokines, chemokines, and adhesion molecules work together to generate a specific environment that leads to thrombus formation [68]. The effects on thrombus development have been elucidated for many cytokines [69]. Therefore, blocking the JAK-signal transducer and activator of transcription (STAT) pathway that supports the action of a particular cytokine may result in either a decrease or increase in the risk of VTE depending on the specific function of that cytokine [70].
From a hematopoiesis perspective, blocking JAK2 should suppress platelet production by the inhibition of thrombopoietin (TPO) signaling [71]. The direct effect of changes in JAK–STAT signaling and risk of VTE can also be informed by the phenotype of patients who have JAK2 genetic variants, which through gain of function may lead to thrombocytosis and VTE [72, 73]. In line with this, inhibition of JAK2 by ruxolitinib has been associated with significantly lower rates of thrombosis in patients with polycythemia vera, suggesting that JAK2 inhibition can decrease thrombosis risk in a myeloproliferative milieu [74].
However, clinical trials with JAKi in patients with inflammatory arthritis have shown increases in platelet counts. One study observed transient increases in platelet counts after initiation of JAKi therapy without association with VTE [75]. Another suggested that a higher risk of VTE with JAKi was associated with an elevation in platelet counts, but this elevation returned to baseline with long-term follow-up [76]. This underpins the complexity of the JAK–STAT signaling pathway that involves a wide range of activating ligands and downstream signaling effects.
Few studies have directly explored the association between JAK inhibition and risk components of VTE. A sub-analysis of the ORAL Surveillance study analyzed 291 protein biomarkers and three genetic markers such as C-reactive protein, d-dimer, TPO, factor VIII, thrombin–antithrombin complex, tissue factor pathway inhibitor, plasminogen activator inhibitor-1, protein C, antithrombin, apolipoprotein C-III, and leptin, along with 276 markers from a high-throughput proteomic assay. d-Dimer levels were weakly linked to a higher risk of developing VTE in this RA population, but the authors concluded that more data are necessary to inform the proper clinical application of this test. The study did not find associations between any of the other analytes and higher rates of VTE with tofacitinib compared with TNF inhibition [77]. Preliminary investigations have explored the putative convergence of genomic VTE risk factors on JAK–STAT signaling, including STAT target genes [78]. An overlap between differentially expressed genes and STAT target genes includes cyclins, an interesting observation given that VTE with cyclin-dependent kinase (CDK) 4/6 inhibitors used in oncology occurs in up to 5% of clinical trials and 10% in real-world settings [79, 80]. Cyclin D and CDK4/6 are downstream STAT target genes that regulate cell progression in early gap 1 phase in the cell cycle [81]. This suggests that factors separate to blockade of cytokine signaling and dampening of inflammation may also be involved in increasing VTE risk with JAK inhibition.
4.2 MACE
While it is recognized that inflammatory disease activity in RA is associated with increased risk of MACE [82], the consequence of dysregulated JAK–STAT signaling has not been as clearly elucidated. STAT signaling has been generally associated with harmful effects on the heart; in contrast, a cardioprotective effect with STAT3 has been observed [83,84,85,86]. Only a few studies have directly explored the mechanistic associations between JAK inhibition and risk of MACE. JAKi in the treatment of RA induce a significant increase in high-density and low-density lipoprotein levels [87], and dyslipidaemia has been reported for both non-selective JAKi [88,89,90,91,92] and JAK1-selective inhibition [93]. Inhibition of the JAK–STAT pathway by tofacitinib contributes to lipid release via enhanced expression of cellular liver X receptor alpha and ABCA1 synthesis [94]. How an increase in lipid levels with JAKi use translates to clinical outcomes, however, is not fully understood.
Conventionally, hyperlipidemia is a risk factor for MACE, but this is not necessarily the case for patients with RA, whereby an elevated risk of MACE may exist despite relatively low cholesterol levels. This is the so-called “lipid paradox,” with changes in lipid levels representing a response to attenuation of inflammation [95]. Data are conflicting about the effect that tofacitinib treatment exerts on some parameters associated with lowered MACE risk; decreased carotid intima-media thickness (IMT) [96] and attenuated vascular inflammation as determined by fluorodeoxyglucose-positron emission tomography/computed tomography (CT) [97] have been described with tofacitinib, although another study found that carotid IMT increased despite treatment [98].
4.3 Malignancy
The ramifications of inflammatory disease activity in RA also extend to an increased risk of cancer [99]. Given the established role of JAKi in the treatment paradigm of hematopoietic malignancies [100], the potential for JAK inhibition in managing solid tumors has been of interest. Collectively, data from preclinical cancer models of solid tumors demonstrate that JAK inhibition can prevent tumor proliferation and growth [101]. However, most solid tumors that exhibit increased JAK–STAT signaling lack somatic JAK mutations, thus differing from myeloproliferative diseases.
A preclinical study demonstrated that JAK inhibition negatively affects the phenotype and function of natural killer (NK) cells [102]. In support of these findings, NK cells with acquired STAT5 deficiency have reduced antitumor cytotoxicity in melanoma mice models [103]. Furthermore, tofacitinib decreases NK cell activation and lymphoma cell-killing efficacy by decreasing their capacity for degranulation and cytokine secretion [104]. Interestingly, CD8 T cells have also been shown to be affected by JAKi [105, 106].
In summary, whilst a likely mechanism exists to explain herpes zoster reactivation from JAKi, and plausible pathways may link VTE to JAKi, explanatory pathophysiology for both cardiovascular and cancer risk from JAKi remains speculative.
5 Individual-Level Factors Influencing Cardiovascular and Cancer Risk in Patients with Autoimmune Disease
At a molecular level, the risk that JAKi contribute to MACE and malignancy is clearly an intricate affair. The same can be said of an individual patient’s degree of risk when considering how their underlying disease and other treatments contribute to this risk.
In addition to traditional cardiovascular risk factors, it is understood that chronic inflammatory diseases inherently pose an increased risk of cardiovascular disease [107,108,109,110]. This notion has been particularly well established in RA, in which it is recognized that inflammatory processes are pivotal to atherosclerotic processes through endothelial dysfunction and maladaptive vessel wall remodeling [111], and that the disease may confer an additional 50% risk over and above other risk factors [109, 112, 113]. There is less supportive evidence in patients with PsA [114, 115], but an increased cardiovascular risk is also suspected. The magnitude of risk conferred by a wide range of autoimmune diseases was recently assessed in a large epidemiological study using data from 22 million people in the UK [116]. Among patients with rheumatic diseases, namely RA, SLE, systemic sclerosis (SSc), polymyalgia rheumatica, axial spondyloarthritis, and Sjögren’s syndrome, there was an overall 68% higher risk for cardiovascular disease, with the greatest associations observed in SSc (HR 3.59, 2.81–4.59) and SLE (HR 2.82, 2.38–3.33). Beyond rheumatic autoimmune diseases, the association between non-rheumatic autoimmune conditions and cardiovascular risk has also been acknowledged. In a meta-analysis of ten cohort studies, Feng et al. observed that patients with inflammatory bowel disease had an increased risk of ischemic heart disease (RR 1.24, 1.14–1.36) [117]. Meanwhile, no increased risk of cardiovascular disease has been shown among patients with alopecia areata [118, 119].
Furthermore, disease activity in RA correlates with the cardiovascular risk in this patient population, adding another layer of complexity. To this end, a study by Solomon et al. examined the relationship between RA disease activity measured longitudinally and the risk of cardiovascular events [82]. Among a large cohort followed for a median of 2.7 years, they found a 21% reduction in cardiovascular risk for each 10-point lowering of the clinical disease activity index, while achieving remission from a state of high disease activity saw a 53% reduction. These data suggest that controlling disease activity in RA might be important, not only to lower pain and improve joint health, but also to attenuate the risk of adverse cardiovascular outcomes. Future research may look to expound on the role of gut dysbiosis, a recognized hallmark of RA [120], in the development of inflammation, and in turn, cardiovascular risk; this has already been hypothesized to play a potential role in IBD [121, 122].
In terms of cancer outcomes, RA is associated with an increased risk of some malignancies, as demonstrated in a meta-analysis that indicated highest risk for lymphoma and lung cancer [123]. This association is explained in part by certain shared risk factors, such as smoking, as well as underlying inflammation. The best data supporting the role of chronic inflammation in the pathogenesis of lymphoma in RA is derived from a seminal case-control study of Swedish patients with RA in whom the risk of lymphoma substantially increased in a subset with very severe disease, leading to the conclusion that high inflammatory activity is a major risk determinant for lymphoma [124].
The association between other immune-mediated diseases and malignancy risk is more controversial. Skin cancer, particularly NMSC, is among the most commonly reported cancers in PsA [125, 126]. In ankylosing spondylitis, evidence remains inconclusive, with possible increased risk of hematological, skin, and other solid cancers reported in a number of studies [127,128,129,130]. IBD has been associated with an increased risk of colorectal cancer, small bowel cancer, intestinal lymphoma, and cholangiocarcinoma [131].
Further complicating matters is the modulatory effect that various therapies may have on cardiovascular risk. TNF is a pro-inflammatory cytokine involved in inflammation and lipid metabolism, and higher levels have been associated with cardiovascular disease in epidemiological studies [132]. Inhibition of TNF has accordingly been shown to improve markers for atherosclerosis such as carotid IMT and pulse wave velocity [133], and decreases the incidence of cardiovascular events in RA and PsA [134]. This is important to consider when interpreting ORAL Surveillance: were both JAKi and TNFi actually cardioprotective but TNFi more so? Both csDMARDs and non-TNFi biologics have also been shown to reduce cardiovascular risk in RA and PsA, while glucocorticoids, even at prednisone-equivalent doses of less than 5 mg daily, and non-steroidal anti-inflammatory drugs (NSAIDs) likely confer an increased risk in these patients [107, 134, 135].
Regarding cancer risk, TNF plays a key role in apoptosis and cell survival and has been demonstrated to exert an antitumor effect [136, 137]. As such, safety concerns about the risk of cancer have been raised with TNFi, although there is an overall preponderance of evidence that is reassuring that this class of medications does not increase the risk of most solid tumors, except for NMSC [138,139,140,141,142,143]. Malignancy risk is less well established with other therapies, although methotrexate may cause a small increased risk of lymphoproliferative disorders [144, 145].
6 Guidance on JAKi Use from Regulation and Guidelines
Although falling short of explicitly calling cardiovascular and malignancy risks a class effect, the FDA has placed labels on all JAKi, warning about safety signals. For example, while tofacitinib gets its own specific warning about observed risks, the label for upadacitinib references increased rates of MACE, lymphoma, and lung cancer with “another JAK inhibitor” [146]. Despite avoiding terminology that would unambiguously describe this as a class effect, the presence of a black box warning describing increased MACE and malignancies serves as a clear warning of risk for prescribers of all JAKi. At the same time, the FDA revised the indication for all JAKi for inflammatory arthritis, such that they are approved only for use after inadequate response to or intolerance of at least one TNFi. Beyond this, the FDA has not given specific guidance about use in special populations, leaving a void to be filled by professional body guidelines [17].
In contrast, the EMA’s Pharmacovigilance Risk Assessment Committee, prompted by a multi-database observational cohort study on baricitinib [53], has produced specific guidance designed to “minimise the risk of serious side effects associated with Januse kinase (JAK) inhibitors,” which include “cardiovascular conditions, blood clots, cancer and serious infections” [147]. Although yet to reach a final decision by the European Commission, the wording makes it clear that the EMA considers these to be class effects and that safety findings relating to tofacitinib “apply to all approved uses of JAK inhibitors in chronic inflammatory disorders” [147]. In the same vein, the Medicines and Healthcare products Regulatory Agency in the United Kingdom released a drug safety update highlighting the same risks and explicitly referring to these as “class effects across JAK inhibitors used for chronic inflammatory disorders” [148].
Although no JAK inhibitors have been removed from the market for any indication, the EMA has produced a series of recommendations designed to curtail their use in populations considered at higher risk. In particular, the EMA has recommended that, where alternative treatment options exist, JAKi should be avoided in patients with an extensive smoking history, baseline increased risk of cancer, increased cardiovascular risk, or age greater than 65. Further guidance has been given for healthcare professionals to conduct regular skin checks in patients prescribed a JAKi.
In response to these regulatory updates, several learned organizations have published statements and guideline updates. The 2022 European Alliance of Associations for Rheumatology guidelines for the management of RA provide the most comprehensive response and broadly reflect the EMA guidance, with caution recommended in the same risk groups as the EMA [149]. However, after extensive deliberation, the authors elected to keep JAKi on the same line of therapy as bDMARDs, thus deviating from the FDA-revised indication that necessitates trial of a TNFi. Published guidelines for RA management from other professional associations including ACR and the National Institute for Health and Care Excellence in England have yet to be updated to reflect these safety signals [150, 151]. Going forward, threading the needle between rigid rules and clinician discretion will be the great balancing act, particularly when many patients might derive great therapeutic value from being prescribed a JAKi.
The future landscape of JAKi is uncertain and will likely be shaped by the ongoing discussions surrounding their safety profile. While some guidelines have been established, further studies are necessary to give clinicians clear, evidence-based guidance. The delicate balance between patient empowerment and responsible prescription will remain a challenge for healthcare professionals. To mitigate risks, clear and evidence-based guidance on safe use will be crucial. As more data become available, it will be important for regulatory agencies and professional organizations to stay vigilant and continue to revise their recommendations accordingly. Ultimately, the goal must be to provide patients with the most effective treatments that are safe and well-tolerated, and this will require continued collaboration and communication between the medical community, regulatory agencies, and pharmaceutical companies.
7 Practical Approach for Clinicians
The cardiovascular and cancer risks attributable to JAKi, as demonstrated incrementally comparing tofacitinib versus TNFi in patients with RA with baseline cardiovascular risk in a rigorous phase 4 RCT, have proven challenging for many to interpret and act upon. This is largely because of the complexity of the context in which it sits. Data informing this risk have evolved substantially over the course of 10 years of registered use, but understanding relevant clinical implications requires an appreciation of the magnitude of risk in different diseases, against different comparators, with multiple other factors frequently contributing to that risk. In addition, there is an inherent stochasticity, not only in the development of cancer and cardiovascular adverse events following JAKi use, but also in the capacity of JAKi to achieve a clinical response, a clinical state which itself mitigates cancer and cardiovascular risk in many diseases, and there are few known predictive factors for either. It is unsurprising that the broader implications of this signal have been hard to interpret.
Clinicians have the advantage of considering single patients at a time but must ensure that rigor trumps exception in decision-making, and we propose a number of mitigating strategies useful in clinical practice (Box 1). This is necessary but challenging, as clinical scenarios quickly become complicated, even when considering the at-risk population with baseline cardiovascular risk. Patients with RA, the prototypic disease in which JAKi risk has mainly been studied, have multiple options at a similar stage of the disease algorithm, all with varying contraindications but equivalent efficacy. Among them, TNFi have a far greater depth of collective experience over 25 years, with a comparatively well-understood risk profile, and remain a hard standard to surpass. Perhaps, if JAKi for RA were considered in isolation without the context of a plethora of choice of effective RA therapies, the magnitude of risk would much less frequently outweigh potential efficacy gains, but for this indication JAKi are now held to the standard set by TNFi.

Nevertheless, as for all bDMARDs, predicting TNFi clinical response in individual patients remains challenging. Among many choices, some patients may only clinically respond to a single therapeutic agent. Even once a response is achieved, it may dissipate over time despite optimal use, often due to the development of anti-drug immunogenicity. As a consequence, prescribers are wary of switching away from effective therapies on the basis of an incremental risk, particularly when such a switch might lead to a loss of response and a consequent increased cancer and cardiovascular risk. Additionally, a return to therapies previously effective in an individual patient may not confer the same clinical response as it did initially. It may therefore be inevitable that, despite known risk, some patients with RA will need a JAKi on a risk–benefit balance, and for some, exploring other therapeutic options after a JAKi response may be considered too risky in terms of losing clinical response. For other patients with known risk, if these caveats do not apply, JAKi use as a first-line therapy should be unpalatable on the basis of the patient’s known risks. Categorizing patients appropriately remains a substantial challenge in clinical practice.
JAKi beyond RA may be less frequently employed, but often will have fewer therapeutic alternatives, and fewer data inform both benefit and risk in these situations. For some indications such as PsA, higher baseline risks and safer alternative options make such extrapolation across indications straightforward. For other indications, particularly for off-label use within orphan diseases, the few alternative therapeutic agents may be substantially less appealing than TNFi and may confer greater cancer or cardiovascular risks themselves. For this reason, it is critical that regulation does not confer undue practical impairment of access to such therapies among attempts to inform prescribers and patients of attendant risks.
Furthermore, the unknown capacity to extrapolate this risk within class creates greater uncertainty. This risk has only clearly been demonstrated for tofacitinib, and it remains plausible that some component of this risk is agent specific. Future data may help to inform this, particularly from post-marketing requirement studies for baricitinib currently underway, but such questions will likely remain incompletely answered with respect to other JAKi such as upadacitinib and filgotinib. In the absence of clear alternative explanations or meaningful points of differentiation within class beyond the theoretical benefits of JAK selectivity [51], it has to be considered that these agents cannot be completely exonerated and that some possibility of risk exists.
In fact, we may have to accept that we may never have data sufficient to inform every situation, and that RCTs may not be the most effective and informative tool in this respect. To some extent, we will need to accept some uncertainty, and that, at least within RA, calls for more data being required should carry limited weight. Uncertainty will not only need to be communicated with patients, but clinicians will also need to learn how to navigate uncertainty. To this end, guidance is useful, and if regulatory warnings are adopted with appropriate caveats and do not stymie personalized care for patients, then they can also play an important role in communicating risk to patients and prescribers.
This signal and the attendant changes it has already brought should rightfully have impacted clinician decision-making [152]. Regulation should drive awareness and lead concern among clinicians, but given competing clinical needs in practice, also rightfully does not appear to have substantially inhibited funding of JAKi in major jurisdictions, either when governed by health technology assessment (HTA) bodies or private insurance. It is therefore incumbent on clinicians to not become complacent about either the risk that JAKi might confer on relevant patients, or the impact of the natural history of disease and the capacity of therapeutic agents to modify this. Other clinical factors may also already be relevant in therapeutic agent selection, including the expense of JAKi in contrast to price reductions in TNFi driven by patent expiry and biosimilar uptake in many jurisdictions. Therapeutic algorithms in the context of some HTA-based systems have already practically pushed JAKi to later lines of therapy. Other safety risks may also affect JAKi use in similar patient populations, including increased risk of general infection, herpes zoster, and VTE, and further therapeutic selection has likely occurred from this as well. Nevertheless, to help navigate this uncertainty, further practical guidance will be useful, particularly in high-risk groups such as those with a past history of cancer or cardiovascular disease. Learned bodies addressing practice in such areas will need to provide frameworks to help clinicians make responsible decisions as much as they possibly can. Constructive scrutiny of clinical prescribing practice in a collaboratively supportive manner, through peer or self-review, may also guard against aberrant practice and add to a milieu where clinicians can be confident of making better decisions.
Regulatory attention to cancer and cardiovascular risk from JAKi in such patients may be leading clinicians to be more conscious of managing and preventing these risks more broadly. A focus on safety should drive increased diligence for dedicated counseling and age-appropriate screening, and in some high-risk subpopulations there may be a role for engagement with dedicated programs or screening techniques such as CT coronary angiogram. Many other factors such as smoking and obesity may realistically confer higher magnitudes of risk, and risk factor modification remains of great importance, particularly given advances in their non-pharmacological and pharmacological management. This could, and should, signal a broader holistic approach to medicines’ safety amongst sub-specialty prescribers, who may otherwise be tempted to neglect the broader health needs of their patients. Better strategic health service interventions are also likely to be needed to improve screening and implement appropriate preventative medicine in patients [153].
Practically, navigating this process for individual patients necessitates some element of joint decision-making, and in this, patient preferences matter. Many different individual patient factors will be important. Not all will be of equal weight: even when an oral mode of action might influence patient choices, its importance is likely to be contextual to other risks and benefits for most patients. Key contributors to patient impression include the primary diagnosis and its functional impact, previous cancer and cardiovascular disease experience (both personal and sociocultural), and previous JAKi experience. Some patients will be able to contextualize their own risk, but others will need assistance, and a lack of unbiased patient-facing information remains an unmet need. Ultimately, however, most patients will seek substantive guidance from their treating clinicians, and it behooves clinicians who might prescribe JAKi to become more skilled at such counseling.
Thankfully, such conversations are not without precedent, albeit in different contexts [154]. Rheumatology prescribers frequently counsel regarding NSAIDs and cardiovascular disease and concerns previously held about TNFi and cancer risk, although these therapeutic scenarios are largely more straightforward in terms of efficacy in context. Furthermore, expectations for therapeutic outcomes have changed across autoimmune diseases, particularly regarding quality of life and disease-related disability, and there is a greater appetite for therapeutic action in a treat-to-target manner across many relevant diseases than previously. It is also plausible that changes in understanding, therapeutic agent cost, or baseline risk may change the considered risk–benefit balance as it stands. A variety of different responses might be appropriate, but if JAK inhibitors are to be prescribed at all, and certainly if they are to be prescribed contrary to regulatory guidance, all clinicians considering JAKi will need to make, communicate, and document decisions on the basis of the complex needs of individual patients, dynamically responding to changes in risk–benefit analysis.
At present, this essential compound risk calculation will require the clinician to consider many factors in tandem [152]. Baseline risk and baseline benefit from JAKi differ between patients. Given disease-related factors already are hard to categorize and quantify, and clinical scenarios are often complex, this calculation will often rely solely on rough mental calculus. Having said this, a diversity of clinical scenarios and a need to consider individual patient scenarios should not be a veil for ignoring risk. It is undoubtedly true that there are other therapies and disease states that have greater cancer or cardiovascular risk quotients but have unsolved risk calculation, although this is arguably true of some metabolic syndrome risk factors as well. Nevertheless, the knowledge that long-term therapeutic choices might lead to unnecessary iatrogenic changes in risk should mean that improving decision-making remains a priority. This remains a gray area in which the development of new technologies may lead to better risk–benefit calculation to inform both clinicians and patients as one possibility, and while this process of shared-decision making is complex, machine-learning-driven recommendations and dynamic decision support for optimal therapeutic agent selection in the future could improve both safety and efficacy [155, 156]. Until then, patients will rely on clinicians to consider many factors relevant to their individual situation, communicate those factors clearly to them, mitigate risk wherever possible, and make the best shared decisions they can.
8 Conclusion
Many questions remain around the safety of JAKi and how they fit in the landscape of RA treatment as well as other inflammatory diseases. From a mechanistic perspective, further research is required to explain the cardiovascular and cancer risks observed, and clinically, comparisons among the different JAKi will help to determine if a class effect is indeed at play. We clinicians are no strangers to uncertainty, and it is prudent that we continue to share the decision-making process with our patients at this time.
References
Nash P. JAK inhibitors: new indication and emerging safety data in 2022. Nat Rev Rheumatol. 2023;19:72–3. https://doi.org/10.1038/s41584-022-00891-4.
Tanaka Y, Luo Y, O’Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol. 2022;18:133–45. https://doi.org/10.1038/s41584-021-00726-8.
Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–60. https://doi.org/10.1016/S0140-6736(12)61424-X.
Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. https://doi.org/10.1056/NEJMoa1109071.
van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–70. https://doi.org/10.1002/art.37816.
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19. https://doi.org/10.1056/NEJMoa1112072.
Kremer J, Li Z-G, Hall S, Fleischmann R, Genovese M, Martin-Mola E, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–61. https://doi.org/10.7326/0003-4819-159-4-201308200-00006.
US Food and Drug Administration. Xeljanz (tofacitinib) prescribing information. n.d.
European Medicines Agency. Refusal of the marketing authorisation for Xeljanz (tofacitinib). 2013. https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-refusal-marketing-authorisationxeljanz_en.pdf. Accessed 21 Jul 2023.
Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–26. https://doi.org/10.1056/NEJMoa2109927.
Taylor PC, Weinblatt ME, Burmester GR, Rooney TP, Witt S, Walls CD, et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 2019;71:1042–55. https://doi.org/10.1002/art.40841.
European Medicines Agency. Restrictions in use of Xeljanz while EMA reviews risk of blood clots in lungs. 2019. https://www.ema.europa.eu/en/news/restrictions-use-xeljanz-while-ema-reviews-risk-blood-clots-lungs. Accessed 21 Jul 2023.
US Food and Drug Administration. FDA approves boxed warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR). 2019. https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-bloodclots-and-death-higher-dose-arthritis-and. Accessed 21 Jul 2023.
Pfizer. Pfizer Shares Co-Primary Endpoint Results from Post-Marketing Required Safety Study of XELJANZ® (tofacitinib) in subjects with rheumatoid arthritis (RA). 2021. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-co-primary-endpoint-results-post-marketing. Accessed 21 Jul 2023.
US Food and Drug Administration. Initial safety trial results find increased risk of serious heart-related problems and cancer with arthritis and ulcerative colitis medicine Xeljanz, Xeljanz XR (tofacitinib). 2021. https://www.fda.gov/drugs/fda-drug-safety-podcasts/initial-safety-trial-results-find-increased-risk-serious-heart-related-problems-and-cancer-arthritis. Accessed 21 Jul 2023.
US Food and Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. 2021. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death. Accessed 21 Jul 2023.
American College of Rheumatology. Janus kinase inhibitor boxed warning. 2022. https://rheumatology.org/api/asset/bltd58974433694d161. Accessed 21 Jul 2023.
European Medicines Agency. Cibinqo (abrocitinib), Jyseleca (filgotinib), Olumiant (baricitinib), Rinvoq (upadacitinib) and Xeljanz (tofacitinib) – Updated recommendations to minimise the risks of malignancy, major adverse cardiovascular events, serious infections, venous thromboembolism and mortality with use of Janus kinase inhibitors (JAKi). 2023. https://www.ema.europa.eu/en/medicines/dhpc/updated-recommendations-minimise-risks-malignancy-major-adverse-cardiovascular-events-serious. Accessed 21 Jul 2023.
JAK inhibitors: What your dermatologist wants you to know n.d. https://www.aad.org/public/diseases/a-z/jak-inhibitors. Accessed 26 Mar 2023.
Kristensen LE, Danese S, Yndestad A, Wang C, Nagy E, Modesto I, et al. Identification of two tofacitinib subpopulations with different relative risk versus TNF inhibitors: an analysis of the open label, randomised controlled study ORAL Surveillance. Ann Rheum Dis. 2023. https://doi.org/10.1136/ard-2022-223715.
Charles-Schoeman C, Buch MH, Dougados M, Bhatt DL, Giles JT, Ytterberg SR, et al. Risk of major adverse cardiovascular events with tofacitinib versus tumour necrosis factor inhibitors in patients with rheumatoid arthritis with or without a history of atherosclerotic cardiovascular disease: a post hoc analysis from ORAL Surveillance. Ann Rheum Dis. 2023;82:119–29. https://doi.org/10.1136/ard-2022-222259.
Curtis JR, Yamaoka K, Chen Y-H, Bhatt DL, Gunay LM, Sugiyama N, et al. Malignancy risk with tofacitinib versus TNF inhibitors in rheumatoid arthritis: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheum Dis. 2023;82:331–43. https://doi.org/10.1136/ard-2022-222543.
Adas MA, Alveyn E, Cook E, Dey M, Galloway JB, Bechman K. The infection risks of JAK inhibition. Expert Rev Clin Immunol. 2022;18:253–61. https://doi.org/10.1080/1744666X.2022.2014323.
Burmester GR, Nash P, Sands BE, Papp K, Stockert L, Jones TV, et al. Adverse events of special interest in clinical trials of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis and psoriasis with 37 066 patient-years of tofacitinib exposure. RMD Open. 2021. https://doi.org/10.1136/rmdopen-2021-001595.
Dougados M, Charles-Schoeman C, Szekanecz Z, Giles JT, Ytterberg SR, Bhatt DL, et al. Impact of cardiovascular risk enrichment on incidence of major adverse cardiovascular events in the tofacitinib rheumatoid arthritis clinical programme. Ann Rheum Dis. 2023. https://doi.org/10.1136/ard-2022-223406.
Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open. 2020. https://doi.org/10.1136/rmdopen-2020-001395.
Charles-Schoeman C, Wicker P, Gonzalez-Gay MA, Boy M, Zuckerman A, Soma K, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum. 2016;46:261–71. https://doi.org/10.1016/j.semarthrit.2016.05.014.
Curtis JR, Lee EB, Kaplan IV, Kwok K, Geier J, Benda B, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. 2016;75:831–41. https://doi.org/10.1136/annrheumdis-2014-205847.
Curtis JR, Lee EB, Martin G, Mariette X, Terry KK, Chen Y, et al. Analysis of non-melanoma skin cancer across the tofacitinib rheumatoid arthritis clinical programme. Clin Exp Rheumatol. 2017;35:614–22.
Charles-Schoeman C, DeMasi R, Valdez H, Soma K, Hwang L-J, Boy MG, et al. Risk factors for major adverse cardiovascular events in phase III and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019;71:1450–9. https://doi.org/10.1002/art.40911.
Burmester GR, Gordon KB, Rosenbaum JT, Arikan D, Lau WL, Li P, et al. Long-term safety of adalimumab in 29,967 adult patients from global clinical trials across multiple indications: an updated analysis. Adv Ther. 2020;37:364–80. https://doi.org/10.1007/s12325-019-01145-8.
Khosrow-Khavar F, Kim SC, Lee H, Lee SB, Desai RJ. Tofacitinib and risk of cardiovascular outcomes: results from the Safety of TofAcitinib in Routine care patients with Rheumatoid Arthritis (STAR-RA) study. Ann Rheum Dis. 2022;81:798–804. https://doi.org/10.1136/annrheumdis-2021-221915.
Khosrow-Khavar F, Desai RJ, Lee H, Lee SB, Kim SC. Tofacitinib and risk of malignancy: results from the safety of tofacitinib in routine care patients with rheumatoid arthritis (STAR-RA) study. Arthritis Rheumatol. 2022;74:1648–59. https://doi.org/10.1002/art.42250.
Kremer JM, Bingham CO 3rd, Cappelli LC, Greenberg JD, Madsen AM, Geier J, et al. Postapproval comparative safety study of tofacitinib and biological disease-modifying antirheumatic drugs: 5-year results from a United States-based rheumatoid arthritis registry. ACR Open Rheumatol. 2021;3:173–84. https://doi.org/10.1002/acr2.11232.
Hoisnard L, Pina Vegas L, Dray-Spira R, Weill A, Zureik M, Sbidian E. Risk of major adverse cardiovascular and venous thromboembolism events in patients with rheumatoid arthritis exposed to JAK inhibitors versus adalimumab: a nationwide cohort study. Ann Rheum Dis. 2023;82:182–8. https://doi.org/10.1136/ard-2022-222824.
Gottenberg J-E, Chaudier A, Allenbach Y, Mekinian A, Amoura Z, Cacoub P, et al. Tolerance and efficacy of targeted therapies prescribed for off-label indications in refractory systemic autoimmune diseases: data of the first 100 patients enrolled in the TATA registry (TArgeted Therapy in Autoimmune Diseases). RMD Open. 2022. https://doi.org/10.1136/rmdopen-2022-002324.
Huss V, Bower H, Hellgren K, Frisell T, Askling J, Behalf of the ARTIS Group, et al. Cancer risks with JAKi and biological disease-modifying antirheumatic drugs in patients with rheumatoid arthritis or psoriatic arthritis: a national real-world cohort study. Ann Rheum Dis. 2023. https://doi.org/10.1136/ard-2022-223636.
Mueller RB, Hasler C, Popp F, Mattow F, Durmisi M, Souza A, et al. Effectiveness, tolerability, and safety of tofacitinib in rheumatoid arthritis: a retrospective analysis of real-world data from the St Gallen and Aarau cohorts. J Clin Med Res. 2019. https://doi.org/10.3390/jcm8101548.
Bilgin E, Duran E, Ünaldı E, Kalyoncu U, Kiraz S, Ertenli İ. Comparison of cardiovascular, cancer and herpes zoster risk of tofacitinib versus etanercept: single-centre observational study. Rheumatology. 2022;61:e267–9. https://doi.org/10.1093/rheumatology/keac226.
Hirose W, Harigai M, Amano K, Hidaka T, Itoh K, Aoki K, et al. Real-world effectiveness and safety of tofacitinib and abatacept in patients with rheumatoid arthritis. Rheumatol Adv Pract. 2022;6:rkac090. https://doi.org/10.1093/rap/rkac090.
Iwamoto N, Sato S, Kurushima S, Michitsuji T, Nishihata S, Okamoto M, et al. Real-world comparative effectiveness and safety of tofacitinib and baricitinib in patients with rheumatoid arthritis. Arthritis Res Ther. 2021;23:197. https://doi.org/10.1186/s13075-021-02582-z.
Fang Y-F, Liu J-R, Chang S-H, Kuo C-F, See L-C. Comparative safety of Janus kinase inhibitors and tumor necrosis factor inhibitors in patients undergoing treatment for rheumatoid arthritis. Int J Rheum Dis. 2022;25:1254–62. https://doi.org/10.1111/1756-185X.14414.
Sandborn WJ, D’Haens GR, Sands BE, Panaccione R, Ng SC, Lawendy N, et al. Tofacitinib for the treatment of ulcerative colitis: an integrated summary of up to 7.8 years of safety data from the global clinical program. J Crohns Colitis. 2022. https://doi.org/10.1093/ecco-jcc/jjac141.
Cohen SB. JAK inhibitors and VTE risk: how concerned should we be? Nat Rev Rheumatol. 2021;17:133–4. https://doi.org/10.1038/s41584-021-00575-5.
Lee JJ, Pope JE. A meta-analysis of the risk of venous thromboembolism in inflammatory rheumatic diseases. Arthritis Res Ther. 2014;16:435. https://doi.org/10.1186/s13075-014-0435-y.
Holmqvist ME, Neovius M, Eriksson J, Mantel Ä, Wållberg-Jonsson S, Jacobsson LTH, et al. Risk of venous thromboembolism in patients with rheumatoid arthritis and association with disease duration and hospitalization. JAMA. 2012;308:1350–6. https://doi.org/10.1001/2012.jama.11741.
Kim SC, Schneeweiss S, Liu J, Solomon DH. Risk of venous thromboembolism in patients with rheumatoid arthritis. Arthritis Care Res. 2013;65:1600–7. https://doi.org/10.1002/acr.22039.
Lichtenstein GR, Bressler B, Francisconi C, Vermeire S, Lawendy N, Salese L, et al. Assessment of safety and efficacy of tofacitinib, stratified by age, in patients from the ulcerative colitis clinical program. Inflamm Bowel Dis. 2023;29:27–41. https://doi.org/10.1093/ibd/izac084.
Center for Drug Evaluation, Research. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. US Food and Drug Administration n.d. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death. accessed 23 Feb 2023.
Choy EH. Clinical significance of Janus kinase inhibitor selectivity. Rheumatology. 2019;58:953–62. https://doi.org/10.1093/rheumatology/key339.
Mortezavi M, Martin DA, Schulze-Koops H. After 25 years of drug development, do we know JAK? RMD Open. 2022. https://doi.org/10.1136/rmdopen-2022-002409.
Taylor PC, Takeuchi T, Burmester GR, Durez P, Smolen JS, Deberdt W, et al. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: final results from long-term extension study and integrated database. Ann Rheum Dis. 2022;81:335–43. https://doi.org/10.1136/annrheumdis-2021-221276.
Salinas CA, Louder A, Polinski J, Zhang TC, Bower H, Phillips S, et al. Evaluation of VTE, MACE, and serious infections among patients with RA treated with baricitinib compared to TNFi: a multi-database study of patients in routine care using disease registries and claims databases. Rheumatol Ther. 2023;10:201–23. https://doi.org/10.1007/s40744-022-00505-1.
Tanaka Y, Ishii T, Cai Z, Schlichting D, Rooney T, Macias W. Efficacy and safety of baricitinib in Japanese patients with active rheumatoid arthritis: a 52-week, randomized, single-blind, extension study. Mod Rheumatol. 2018;28:20–9. https://doi.org/10.1080/14397595.2017.1307899.
Takagi M, Atsumi T, Matsuno H, Tamura N, Fujii T, Okamoto N, et al. Safety and effectiveness of baricitinib for rheumatoid arthritis in Japanese clinical practice: 24-week results of all-case post-marketing surveillance. Mod Rheumatol. 2022. https://doi.org/10.1093/mr/roac089.
Fitton J, Melville AR, Emery P, Nam JL, Buch MH. Real-world single centre use of JAK inhibitors across the rheumatoid arthritis pathway. Rheumatology. 2021;60:4048–54. https://doi.org/10.1093/rheumatology/keaa858.
Zeng X, Zhao D, Radominski SC, Keiserman M, Lee CK, Meerwein S, et al. Upadacitinib in patients from China, Brazil, and South Korea with rheumatoid arthritis and an inadequate response to conventional therapy. Int J Rheum Dis. 2021;24:1530–9. https://doi.org/10.1111/1756-185X.14235.
Cohen SB, van Vollenhoven RF, Winthrop KL, Zerbini CAF, Tanaka Y, Bessette L, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheum Dis. 2021;80:304–11. https://doi.org/10.1136/annrheumdis-2020-218510.
Burmester GR, Winthrop K, Blanco R, Nash P, Goupille P, Azevedo VF, et al. Safety profile of upadacitinib up to 3 years in psoriatic arthritis: an integrated analysis of two pivotal phase 3 trials. Rheumatol Ther. 2022;9:521–39. https://doi.org/10.1007/s40744-021-00410-z.
Katoh N, Ohya Y, Murota H, Ikeda M, Hu X, Ikeda K, et al. Safety and efficacy of upadacitinib for atopic dermatitis in Japan: 2-year interim results from the phase 3 Rising Up Study. Dermatol Ther. 2023;13:221–34. https://doi.org/10.1007/s13555-022-00842-7.
Danese S, Vermeire S, Zhou W, Pangan AL, Siffledeen J, Greenbloom S, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022;399:2113–28. https://doi.org/10.1016/S0140-6736(22)00581-5.
Winthrop KL, Tanaka Y, Takeuchi T, Kivitz A, Matzkies F, Genovese MC, et al. Integrated safety analysis of filgotinib in patients with moderately to severely active rheumatoid arthritis receiving treatment over a median of 1.6 years. Ann Rheum Dis. 2022;81:184–92. https://doi.org/10.1136/annrheumdis-2021-221051.
Feagan Senior Scientific Officer BG, Matsuoka K, Rogler G, Faes M, Oortwijn A, de Haas A, et al. P491 efficacy and safety outcomes of long-term treatment with filgotinib 200 mg among patients with Ulcerative Colitis: an interim analysis of SELECTIONLTE. J Crohns Colitis. 2022;16:i456–7. https://doi.org/10.1093/ecco-jcc/jjab232.618.
Garrido-Trigo A, Salas A. Molecular structure and function of Janus kinases: implications for the development of inhibitors. J Crohns Colitis. 2020;14:S713–24. https://doi.org/10.1093/ecco-jcc/jjz206.
European Medicines Agency. Xeljanz Product Information. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/xeljanz. Accessed 21 Jul 2023.
Molander V, Bower H, Frisell T, Askling J. Risk of venous thromboembolism in rheumatoid arthritis, and its association with disease activity: a nationwide cohort study from Sweden. Ann Rheum Dis. 2021;80:169–75. https://doi.org/10.1136/annrheumdis-2020-218419.
Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. 2021;32:314–24. https://doi.org/10.1080/09537104.2020.1817360.
Gaertner F, Massberg S. Blood coagulation in immunothrombosis—at the frontline of intravascular immunity. Semin Immunol. 2016;28:561–9. https://doi.org/10.1016/j.smim.2016.10.010.
Mosevoll KA, Johansen S, Wendelbo Ø, Nepstad I, Bruserud Ø, Reikvam H. Cytokines, adhesion molecules, and matrix metalloproteases as predisposing, diagnostic, and prognostic factors in venous thrombosis. Front Med. 2018;5:147. https://doi.org/10.3389/fmed.2018.00147. https://www.ema.europa.eu/en/news/ema-confirms-measures-minimise-risk-serious-side-effects-janus-kinase-inhibitors-chronic
Kotyla PJ, Engelmann M, Giemza-Stokłosa J, Wnuk B, Islam MA. Thromboembolic adverse drug reactions in Janus kinase (JAK) inhibitors: does the inhibitor specificity play a role? Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22052449.
Pecquet C, Diaconu CC, Staerk J, Girardot M, Marty C, Royer Y, et al. Thrombopoietin receptor down-modulation by JAK2 V617F: restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood. 2012;119:4625–35. https://doi.org/10.1182/blood-2011-08-372524.
Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. https://doi.org/10.1056/NEJMoa051113.
Mead AJ, Rugless MJ, Jacobsen SEW, Schuh A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med. 2012;366:967–9. https://doi.org/10.1056/NEJMc1200349.
Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, Garcia D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis. 2016;27:648–52. https://doi.org/10.1097/MBC.0000000000000446.
Kremer J, Huizinga TWJ, Chen L, Saifan CG, Issa M, Witt SL, et al. FRI0090 Analysis of neutrophils, lymphocytes, and platelets in pooled phase 2 and phase 3 studies of baricitinib for rheumatoid arthritis. In: Poster presentations, BMJ Publishing Group Ltd and European League Against Rheumatism; 2017. https://doi.org/10.1136/annrheumdis-2017-eular.1325.
Rajasimhan S, Pamuk O, Katz JD. Safety of Janus kinase inhibitors in older patients: a focus on the thromboembolic risk. Drugs Aging. 2020;37:551–8. https://doi.org/10.1007/s40266-020-00775-w.
Weitz JI, Szekanecz Z, Charles-Schoeman C, Vranic I, Sahin B, Paciga SA, et al. Biomarkers to predict risk of venous thromboembolism in patients with rheumatoid arthritis receiving tofacitinib or tumour necrosis factor inhibitors. RMD Open. 2022. https://doi.org/10.1136/rmdopen-2022-002571.
Haysen S, Nielsen ALL, Qvist P, Kragstrup TW. POS0038 genomics of JAK–STAT signaling in venous thromboembolism. Ann Rheum Dis. 2022;81:234–234. https://doi.org/10.1136/annrheumdis-2022-eular.2593.
West MT, Kartika T, Paquin AR, Liederbauer E, Zheng TJ, Lane L, et al. Thrombotic events in patients using cyclin dependent kinase 4/6 inhibitors, analysis of existing ambulatory risk assessment models and the potential influences of tumor specific risk factors. Curr Probl Cancer. 2022;46:100832. https://doi.org/10.1016/j.currproblcancer.2021.100832.
West MT, Smith CE, Kaempf A, Kohs TCL, Amirsoltani R, Ribkoff J, et al. CDK 4/6 inhibitors are associated with a high incidence of thrombotic events in women with breast cancer in real-world practice. Eur J Haematol. 2021;106:634–42. https://doi.org/10.1111/ejh.13590.
Łukasik P, Załuski M, Gutowska I. Cyclin-dependent kinases (CDK) and their role in diseases development—review. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22062935.
Solomon DH, Reed GW, Kremer JM, Curtis JR, Farkouh ME, Harrold LR, et al. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015;67:1449–55. https://doi.org/10.1002/art.39098.
Kishore R, Verma SK. Roles of STATs signaling in cardiovascular diseases. JAKSTAT. 2012;1:118–24. https://doi.org/10.4161/jkst.20115.
Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104:e9-18. https://doi.org/10.1161/CIRCRESAHA.108.188243.
Bolli R, Stein AB, Guo Y, Wang O-L, Rokosh G, Dawn B, et al. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–97. https://doi.org/10.1016/j.yjmcc.2011.01.002.
Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286:29610–20. https://doi.org/10.1074/jbc.M111.226209.
Li N, Gou Z-P, Du S-Q, Zhu X-H, Lin H, Liang X-F, et al. Effect of JAK inhibitors on high- and low-density lipoprotein in patients with rheumatoid arthritis: a systematic review and network meta-analysis. Clin Rheumatol. 2022;41:677–88. https://doi.org/10.1007/s10067-021-06003-z.
Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. 2014;41:837–52. https://doi.org/10.3899/jrheum.130683.
Wolk R, Armstrong EJ, Hansen PR, Thiers B, Lan S, Tallman AM, et al. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J Clin Lipidol. 2017;11:1243–56. https://doi.org/10.1016/j.jacl.2017.06.012.
Souto A, Salgado E, Maneiro JR, Mera A, Carmona L, Gómez-Reino JJ. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta-analysis. Arthritis Rheumatol. 2015;67:117–27. https://doi.org/10.1002/art.38894.
Kremer JM, Genovese MC, Keystone E, Taylor PC, Zuckerman SH, Ruotolo G, et al. Effects of baricitinib on lipid, apolipoprotein, and lipoprotein particle profiles in a phase IIb study of patients with active rheumatoid arthritis. Arthritis Rheumatol. 2017;69:943–52. https://doi.org/10.1002/art.40036.
Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety Profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. 2019;46:7–18. https://doi.org/10.3899/jrheum.171361.
Smolen JS, Pangan AL, Emery P, Rigby W, Tanaka Y, Vargas JI, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393:2303–11. https://doi.org/10.1016/S0140-6736(19)30419-2.
Pérez-Baos S, Barrasa JI, Gratal P, Larrañaga-Vera A, Prieto-Potin I, Herrero-Beaumont G, et al. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: understanding the lipid paradox associated with rheumatoid arthritis. Br J Pharmacol. 2017;174:3018–31. https://doi.org/10.1111/bph.13932.
Meune C, Touzé E, Trinquart L, Allanore Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology. 2009;48:1309–13. https://doi.org/10.1093/rheumatology/kep252.
Kume K, Amano K, Yamada S, Kanazawa T, Ohta H, Hatta K, et al. Tofacitinib improves atherosclerosis despite up-regulating serum cholesterol in patients with active rheumatoid arthritis: a cohort study. Rheumatol Int. 2017;37:2079–85. https://doi.org/10.1007/s00296-017-3844-9.
Hamar A, Hascsi Z, Pusztai A, Czókolyová M, Végh E, Pethő Z, et al. Prospective, simultaneous assessment of joint and vascular inflammation by PET/CT in tofacitinib-treated patients with rheumatoid arthritis: associations with vascular and bone status. RMD Open. 2021. https://doi.org/10.1136/rmdopen-2021-001804.
Soós B, Hamar A, Pusztai A, Czókolyová M, Végh E, Szamosi S, et al. Effects of tofacitinib therapy on arginine and methionine metabolites in association with vascular pathophysiology in rheumatoid arthritis: a metabolomic approach. Front Med. 2022;9:1011734. https://doi.org/10.3389/fmed.2022.1011734.
Strangfeld A, Hierse F, Rau R, Burmester G-R, Krummel-Lorenz B, Demary W, et al. Risk of incident or recurrent malignancies among patients with rheumatoid arthritis exposed to biologic therapy in the German biologics register RABBIT. Arthritis Res Ther. 2010;12:R5. https://doi.org/10.1186/ar2904.
Senkevitch E, Durum S. The promise of Janus kinase inhibitors in the treatment of hematological malignancies. Cytokine. 2017;98:33–41. https://doi.org/10.1016/j.cyto.2016.10.012.
Qureshy Z, Johnson DE, Grandis JR. Targeting the JAK/STAT pathway in solid tumors. J Cancer Metastasis Treat. 2020. https://doi.org/10.20517/2394-4722.2020.58.
Meudec L, Richebé P, Pascaud J, Mariette X, Nocturne G. Janus kinase inhibitors alter NK cells phenotype and inhibit their antitumor capacity. Rheumatology. 2022. https://doi.org/10.1093/rheumatology/keac710.
Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. 2016;6:414–29. https://doi.org/10.1158/2159-8290.CD-15-0732.
Nocturne G, Pascaud J, Ly B, Tahmasebi F, Mariette X. JAK inhibitors alter NK cell functions and may impair immunosurveillance against lymphomagenesis. Cell Mol Immunol. 2020;17:552–3. https://doi.org/10.1038/s41423-019-0320-3.
Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheum Dis. 2021;80:865–75. https://doi.org/10.1136/annrheumdis-2020-219012.
Weinhold KJ, Bukowski JF, Brennan TV, Noveck RJ, Staats JS, Lin L, et al. Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Immunol. 2018;191:10–20. https://doi.org/10.1016/j.clim.2018.03.002.
Agca R, Smulders Y, Nurmohamed M. Cardiovascular disease risk in immune-mediated inflammatory diseases: recommendations for clinical practice. Heart. 2022;108:73–9. https://doi.org/10.1136/heartjnl-2019-316378.
Bengtsson K, Forsblad-d’Elia H, Lie E, Klingberg E, Dehlin M, Exarchou S, et al. Are ankylosing spondylitis, psoriatic arthritis and undifferentiated spondyloarthritis associated with an increased risk of cardiovascular events? A prospective nationwide population-based cohort study. Arthritis Res Ther. 2017;19:102. https://doi.org/10.1186/s13075-017-1315-z.
Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524–9. https://doi.org/10.1136/annrheumdis-2011-200726.
Solomon DH, Karlson EW, Rimm EB, Cannuscio CC, Mandl LA, Manson JE, et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107:1303–7. https://doi.org/10.1161/01.cir.0000054612.26458.b2.
Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev Rheumatol. 2015;11:390–400. https://doi.org/10.1038/nrrheum.2015.40.
Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJL, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17–28. https://doi.org/10.1136/annrheumdis-2016-209775.
Lindhardsen J, Ahlehoff O, Gislason GH, Madsen OR, Olesen JB, Torp-Pedersen C, et al. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: a Danish nationwide cohort study. Ann Rheum Dis. 2011;70:929–34. https://doi.org/10.1136/ard.2010.143396.
Coumbe AG, Pritzker MR, Duprez DA. Cardiovascular risk and psoriasis: beyond the traditional risk factors. Am J Med. 2014;127:12–8. https://doi.org/10.1016/j.amjmed.2013.08.013.
Dowlatshahi EA, Kavousi M, Nijsten T, Arfan Ikram M, Hofman A, Franco OH, et al. Psoriasis is not associated with atherosclerosis and incident cardiovascular events: the Rotterdam study. J Investig Dermatol. 2013;133:2347–54. https://doi.org/10.1038/jid.2013.131.
Conrad N, McInnes IB, Mcmurray JJV, Sattar N. Patients with a range of rheumatic diseases are at increased risk of cardiovascular disorders towards a re-evaluation of the European League against Rheumatism (EULAR)’s recommendations for cardiovascular risk management? Ann Rheum Dis. 2023;82:457–9. https://doi.org/10.1136/ard-2022-223315.
Feng W, Chen G, Cai D, Zhao S, Cheng J, Shen H. Inflammatory bowel disease and risk of ischemic heart disease: an updated meta-analysis of cohort studies. J Am Heart Assoc. 2017. https://doi.org/10.1161/JAHA.117.005892.
Lee H, Kim YC, Choi JW. Alopecia areata is not a risk factor for heart diseases: a 10-year retrospective cohort study. PLoS ONE. 2021;16:e0250216. https://doi.org/10.1371/journal.pone.0250216.
Huang KP, Joyce CJ, Topaz M, Guo Y, Mostaghimi A. Cardiovascular risk in patients with alopecia areata (AA): a propensity-matched retrospective analysis. J Am Acad Dermatol. 2016;75:151–4. https://doi.org/10.1016/j.jaad.2016.02.1234.
Horta-Baas G, Romero-Figueroa MDS, Montiel-Jarquín AJ, Pizano-Zárate ML, García-Mena J, Ramírez-Durán N. Intestinal dysbiosis and rheumatoid arthritis: a link between gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res. 2017;2017:4835189. https://doi.org/10.1155/2017/4835189.
Bigeh A, Sanchez A, Maestas C, Gulati M. Inflammatory bowel disease and the risk for cardiovascular disease: does all inflammation lead to heart disease? Trends Cardiovasc Med. 2020;30:463–9. https://doi.org/10.1016/j.tcm.2019.10.001.
Ahmadmehrabi S, Tang WHW. Gut microbiome and its role in cardiovascular diseases. Curr Opin Cardiol. 2017;32:761–6. https://doi.org/10.1097/HCO.0000000000000445.
Simon TA, Thompson A, Gandhi KK, Hochberg MC, Suissa S. Incidence of malignancy in adult patients with rheumatoid arthritis: a meta-analysis. Arthritis Res Ther. 2015;17:212. https://doi.org/10.1186/s13075-015-0728-9.
Baecklund E, Iliadou A, Askling J, Ekbom A, Backlin C, Granath F, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54:692–701. https://doi.org/10.1002/art.21675.
Karmacharya P, Shahukhal R, Ogdie A. Risk of malignancy in spondyloarthritis: a systematic review. Rheum Dis Clin North Am. 2020;46:463–511. https://doi.org/10.1016/j.rdc.2020.04.001.
Luo X, Deng C, Fei Y, Zhang W, Li Y, Zhang X, et al. Malignancy development risk in psoriatic arthritis patients undergoing treatment: a systematic review and meta-analysis. Semin Arthritis Rheum. 2019;48:626–31. https://doi.org/10.1016/j.semarthrit.2018.05.009.
Chan T-M, Luo S-F, Yu K-H, See L-C, Huang L-H, Kuo C-F. Risk of cancer in patients with ankylosing spondylitis: a nationwide cohort study in Taiwan. Scand J Rheumatol. 2021;50:132–8. https://doi.org/10.1080/03009742.2020.1804612.
Chang C-C, Chang C-W, Nguyen P-AA, Chang T-H, Shih Y-L, Chang W-Y, et al. Ankylosing spondylitis and the risk of cancer. Oncol Lett. 2017;14:1315–22. https://doi.org/10.3892/ol.2017.6368.
Bittar M, Merjanah S, Alkilany R, Magrey M. Malignancy in ankylosing spondylitis: a cross-sectional analysis of a large population database. BMC Rheumatol. 2022;6:44. https://doi.org/10.1186/s41927-022-00275-x.
Deng C, Li W, Fei Y, Li Y, Zhang F. Risk of malignancy in ankylosing spondylitis: a systematic review and meta-analysis. Sci Rep. 2016;6:32063. https://doi.org/10.1038/srep32063.
Laredo V, García-Mateo S, Martínez-Domínguez SJ, López de la Cruz J, Gargallo-Puyuelo CJ, Gomollón F. Risk of cancer in patients with inflammatory bowel diseases and keys for patient management. Cancers. 2023. https://doi.org/10.3390/cancers15030871.
Yuan S, Carter P, Bruzelius M, Vithayathil M, Kar S, Mason AM, et al. Effects of tumour necrosis factor on cardiovascular disease and cancer: a two-sample Mendelian randomization study. EBioMedicine. 2020;59:102956. https://doi.org/10.1016/j.ebiom.2020.102956.
Abdulmajid B, Blanken AB, van Geel EH, Daams JG, Nurmohamed MT. Effect of TNF inhibitors on arterial stiffness and intima media thickness in rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol. 2023;42:999–1011. https://doi.org/10.1007/s10067-023-06505-y.
Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74:480–9. https://doi.org/10.1136/annrheumdis-2014-206624.
Pujades-Rodriguez M, Morgan AW, Cubbon RM, Wu J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immune-mediated inflammatory diseases: a population-based cohort study. PLoS Med. 2020;17:e1003432. https://doi.org/10.1371/journal.pmed.1003432.
van Horssen R, ten Hagen TLM, Eggermont AMM. TNF-α in Cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. https://doi.org/10.1634/theoncologist.11-4-397.
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci. 1975;72:3666–70. https://doi.org/10.1073/pnas.72.9.3666.
Askling J, Fahrbach K, Nordstrom B, Ross S, Schmid CH, Symmons D. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119–30. https://doi.org/10.1002/pds.2046.
Thompson AE, Rieder SW, Pope JE. Tumor necrosis factor therapy and the risk of serious infection and malignancy in patients with early rheumatoid arthritis: a meta-analysis of randomized controlled trials. Arthritis Rheum. 2011;63:1479–85. https://doi.org/10.1002/art.30310.
Solomon DH, Mercer E, Kavanaugh A. Observational studies on the risk of cancer associated with tumor necrosis factor inhibitors in rheumatoid arthritis: a review of their methodologies and results. Arthritis Rheum. 2012;64:21–32. https://doi.org/10.1002/art.30653.
Askling J, van Vollenhoven RF, Granath F, Raaschou P, Fored CM, Baecklund E, et al. Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor alpha therapies: does the risk change with the time since start of treatment? Arthritis Rheum. 2009;60:3180–9. https://doi.org/10.1002/art.24941.
Dreyer L, Mellemkjær L, Andersen AR, Bennett P, Poulsen UE, Ellingsen TJ, et al. Incidences of overall and site specific cancers in TNFα inhibitor treated patients with rheumatoid arthritis and other arthritides—a follow-up study from the DANBIO Registry. Ann Rheum Dis. 2013;72:79–82. https://doi.org/10.1136/annrheumdis-2012-201969.
Lopez-Olivo MA, Tayar JH, Martinez-Lopez JA, Pollono EN, Cueto JP, Rosa Gonzales-Crespo M, et al. Risk of malignancies in patients with rheumatoid arthritis treated with biologic therapy: a meta-analysis. JAMA. 2012;308:898–908. https://doi.org/10.1001/2012.jama.10857.
Wolfe F, Michaud K. Lymphoma in rheumatoid arthritis: the effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum. 2004;50:1740–51. https://doi.org/10.1002/art.20311.
Mariette X, Cazals-Hatem D, Warszawki J, Liote F, Balandraud N, Sibilia J, et al. Lymphomas in rheumatoid arthritis patients treated with methotrexate: a 3-year prospective study in France. Blood. 2002;99:3909–15. https://doi.org/10.1182/blood.v99.11.3909.
Full Prescribing Information (Upadacitinib) n.d. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/211675s003lbl.pdf. Accessed 12 Feb 2023.
European Medicines Agency. EMA confirms measures to minimise risk of serious side effects with Janus kinase inhibitors for chronic inflammatory disorders. 2023. https://www.ema.europa.eu/en/news/ema-confirms-measures-minimise-risk-serious-side-effects-janus-kinase-inhibitors-chronic. Accessed 21 Jul 2023.
Medicines and Healthcare products Regulatory Agency. Janus kinase (JAK) inhibitors: new measures to reduce risks of major cardiovascular events, malignancy, venous thromboembolism, serious infections and increased mortality. 2023. https://www.gov.uk/drug-safety-update/janus-kinase-jak-inhibitors-new-measures-to-reduce-risks-of-major-cardiovascular-events-malignancy-venous-thromboembolism-serious-infections-and-increased-mortality. Accessed 21 Jul 2023.
Smolen JS, Landewé RBM, Bergstra SA, Kerschbaumer A, Sepriano A, Aletaha D, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82:3–18. https://doi.org/10.1136/ard-2022-223356.
Fraenkel L, Bathon JM, England BR, St Clair EW, Arayssi T, Carandang K, et al. 2021 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2021;73:924–39. https://doi.org/10.1002/acr.24596.
National Institute for Health and Care Excellence. Tofacitinib for moderate to severe rheumatoid arthritis. 2017. https://www.nice.org.uk/guidance/ta480. Accessed 21 Jul 2023.
Kragstrup TW, Glintborg B, Svensson AL, McMaster C, Robinson PC, Deleuran B, et al. Waiting for JAK inhibitor safety data. RMD Open. 2022. https://doi.org/10.1136/rmdopen-2022-002236.
Liew DFL, Robinson PC. Preventive medicine in rheumatology: COVID-19 and its lessons for better health outcomes. Lancet Rheumatol. 2022;4:e743–5. https://doi.org/10.1016/S2665-9913(22)00229-6.
Winthrop KL, Cohen SB. Oral surveillance and JAK inhibitor safety: the theory of relativity. Nat Rev Rheumatol. 2022;18:301–4. https://doi.org/10.1038/s41584-022-00767-7.
McMaster C, Bird A, Liew DF, Buchanan RR, Owen CE, Chapman WW, et al. Artificial intelligence and deep learning for rheumatologists: a primer and review of the literature. Arthritis Rheumatol. 2022. https://doi.org/10.1002/art.42296.
Singh N, Grivas P, Makris UE, Suarez-Almazor ME, O’Hare AM, Barton JL. Use of disease-modifying antirheumatic drugs in rheumatoid arthritis: supporting shared decision-making between patients with cancer and clinicians. ACR Open Rheumatol. 2023. https://doi.org/10.1002/acr2.11552.
Health Canada. Janus kinase inhibitors and the risk of major adverse cardiovascular events, thrombosis (including fatal events) and malignancy. 2022. https://recalls-rappels.canada.ca/en/alert-recall/janus-kinase-inhibitors-and-risk-major-adverse-cardiovascular-events-thrombosis. Accessed 21 Jul 2023.
Therapeutic Goods Administration. Important safety information for Janus kinase (JAK) inhibitors. 2023. https://www.tga.gov.au/news/safety-updates/important-safety-information-janus-kinase-jak-inhibitors. Accessed 21 Jul 2023.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflicts of interests
TWK declares speaking fees from Pfizer, Bristol-Myers Squibb, Eli Lilly, Novartis, UCB, and Abbvie; consultancy fees from Bristol-Myers Squibb, UCB, Gilead, and Eli-Lilly; and research grants from Gilead, and is co-founder and clinical developer in Aptol Pharma. PR declares support from the University of Chicago Institute of Translational Medicine Clinical and Translational Science Award K12/KL2 grant 5KL2TR002387-05 and COVID-19 Funds to Retain Clinical Scientists by the SECURED (Supporting Early Career University Researchers to Excel through Disruptions) Steering Committee. NS declares support from the National Institute Of Arthritis And Musculoskeletal And Skin Diseases of the National Institutes of Health under award no. K23AR079588. PCR declares personal fees from Abbvie, Atom Biosciences, Eli Lilly, Gilead, GlaxoSmithKline, Janssen, Kukdong, Novartis, UCB, Roche, and Pfizer; meeting attendance support from Bristol Myers Squibb, Roche, Pfizer, and UCB; and grant funding from Janssen, Novartis, Pfizer, and UCB. DFLL declares membership in the Drug Utilisation Subcommittee of the Pharmaceutical Benefits Advisory Committee, Australian Government. VY, CM, and SRH declare no competing interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Author contributions
DFLL and PCR conceptualized the structure of the review. VY, DFLL, TWK, CM, PR, NS, and SRH drafted and revised the paper. All authors read and approved the final version.
Additional information
Philip C. Robinson: Deceased.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yang, V., Kragstrup, T.W., McMaster, C. et al. Managing Cardiovascular and Cancer Risk Associated with JAK Inhibitors. Drug Saf 46, 1049–1071 (2023). https://doi.org/10.1007/s40264-023-01333-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-023-01333-0