
Abstract
Introduction
Due to the complexity of biologics and the inherent challenges for manufacturing, it is important to know the specific brand name and batch number of suspected biologics in adverse drug reaction (ADR) reports.
Objective
The aim of this study was to assess the extent to which biologics are traceable by brand name and batch number in UK hospital practice and in ADRs reported by patients and healthcare professionals.
Methods
We performed an online hospital pharmacist survey to capture information on how specific product details are recorded during the processes of prescribing, dispensing and administration of biologics in routine UK hospital practice. We also assessed the proportion of ADR reports specifying brand name and batch number from electronic ADR reports submitted to the UK national spontaneous reporting database, the Yellow Card Scheme, between 1 January 2009 and 30 September 2017.
Results
Brand name recording in routine hospital processes ranged from 79 to 91%, whereas batch numbers were less routinely recorded, ranging from 38 to 58%. Paper-based recording of product details was more commonly used for recording information. A total of 6108 electronic ADR reports were submitted to the Yellow Card Scheme for recombinant biologics, of which 38% and 15%, respectively, had an identifiable brand name and batch numbers. Whereas batch number traceability in electronic ADR reports improved slightly after the implementation of the European Union pharmacovigilance legislation in 2012, no improvement of brand name traceability was observed.
Conclusion
Brand name and batch number traceability for biologics in UK ADR reports are generally low. Shortcomings in the systematic recording of product details in UK clinical practice may contribute to the limited traceability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Brand name recording for biologics is high in UK hospital practice, while batch numbers are recorded to a lesser extent. |
Brand name and batch number identification is generally low for biologics in UK adverse drug reaction reports, with only minor improvement over time observed for identifiable batch numbers. |
Pharmacovigilance legislation has not resulted in a significant improvement in the traceability of biologics in the UK, and may benefit from improvements of recording and tracing product details throughout the entire supply chain in combination with educational activities on the importance of traceability for biologics. |
1 Introduction
Biological medical products (‘biologics’) are derived from living cells or organisms. Due to their inherent complexity and challenging manufacturing processes, a degree of minor variability may exist between different batches of the same product [1,2,3]. In addition, due to this variability, minor differences may occur between original biologic and follow-on versions, so-called biosimilars [4]. Manufacturing variability has resulted in manufacturing source (product- or batch-)-specific adverse drug reactions (ADRs) [2, 5]. Thus, when ADRs occur, it is important to know the specific product name (i.e. brand name) and batch number of the biologic involved in order to detect potential manufacturing-related safety issues in a timely manner. In order to do so, appropriate systems must be in place to ensure that these details are retrievable in the clinical setting and that they are specified within any ADR report submitted to regulatory authorities or marketing authorisation holders (MAHs). In the UK, this is stipulated in European Union (EU) legislation via Directive 2010/84/EU, which was implemented into national law in August 2012 [6, 7].
Several studies have concluded that specific product details are not always included in ADR reports, in particular when it comes to batch numbers [8,9,10]. In the Netherlands, results from a study in 2014 suggested that brand names were not routinely recorded in Dutch clinical practice and batch numbers were poorly recorded [10]. The main reported bottleneck in the study from the Netherlands was that the current EU barcode standards do not support encoding and thus automatic recording by means of electronic barcode scanning of batch numbers in electronic recording systems. This may, at least in part, explain the limited traceability of biologics found in ADR reports in the Netherlands. With an ever-increasing number of authorised biologics and biosimilars on the market, this topic has become even more relevant in recent years and warrants further research. Thus, to assess whether these issues apply on a wider scale, we assessed the traceability of biologics in the UK.
Hospital pharmacy computer systems in the UK may be used only for pharmacy functions such as dispensing, labelling and stock control, or they may be integrated with hospital-wide systems for electronic recording of prescriptions and administration of medicines. Health professionals are encouraged to report ADRs to the UK Yellow Card Scheme, which is administered by the Medicines and Healthcare products Regulatory Agency (MHRA). In hospitals, ADRs reports may be collated by the local medicines information service (a pharmacy subdepartment) and transmitted electronically to the MHRA, but health professionals may also report to the MHRA directly, independent of this system. The aim of the current study (the UK BIOlogical-TRAraceability in Clinical practice (BIO-TRAC) study) was to assess the extent to which biologics are traceable within the UK hospital setting with regard to brand name and batch number identification and spontaneous ADR reporting in routine clinical practice. The use of recent data also allowed for an assessment of the impact of the EU pharmacovigilance legislation on the traceability of biologics in the UK. The results of this study should inform both national and European policy discussions.
2 Methods
2.1 Data Sources
Data from two sources were evaluated: (1) an online hospital pharmacist survey; and (2) data from electronic ADR reports submitted to the UK national spontaneous reporting database, the Yellow Card Scheme.
The survey was set up for hospital pharmacists working in National Health Service (NHS) hospitals in England, as they represent the most central source of information in the hospital setting. In England, publicly funded healthcare is managed and delivered at a local level by NHS trusts. For the purposes of the survey, only secondary care hospital trusts were included as other types of specialised trusts, such as mental health or community health trusts, may not have their own pharmacy service and are less likely to use biologics. Also, the survey was conducted only within English hospitals in order to minimise the time required to obtain research approvals. After excluding community and mental health trusts, a total of 174 NHS hospital trusts in England were considered eligible for this study and invited to take part in the survey. More than one pharmacist was allowed to participate per NHS trust if they represent different hospital sites or specialties within one hospital site. Pharmacists completed the survey via a secure web platform hosted by the local project partner, the Drug Safety Research Unit (DSRU).
The survey comprised a series of questions on the recording of brand names and batch numbers during the prescribing, dispensing and administration of biologics in the hospital inpatient setting and in outpatient infusion centres (see Electronic Supplementary Material 1). In addition, pharmacists were asked for their views on the current bottlenecks and potential solutions for improving the traceability of biologics. The study was approved by the NHS Health Research Authority and the UK National Institute for Health Research Clinical Research Network (NIHR CRN) provided support in recruiting NHS hospitals and pharmacists.
Anonymised data were analysed from electronic ADR reports submitted directly by patients or healthcare professionals to the UK Yellow Card Scheme between 1 January 2009 and 30 September 2017. ADR reports with a suspected drug from a predefined list of recombinant biologics were included (see Electronic Supplementary Material 2). Non-electronic reports were excluded, because for these reports the original verbatim drug name ‘as reported’ is not feasible to extract from the database. ADR reports submitted via MAHs and literature reports were also excluded.Footnote 1 Access to anonymised Yellow Card data was provided by the MHRA following approval by the Commission on Human Medicines (CHM) Pharmacovigilance Expert Advisory Group.
2.2 Data Analysis
2.2.1 Descriptive Analysis
Simple descriptive statistics from the survey data were used to summarise the level and method of recording of brand name and batch number information in routine clinical practice. Open ‘free-text’ responses were assessed qualitatively by identifying common themes. Pharmacists were asked to indicate if they completed the survey for the hospital as a whole or for their clinical specialty. For the analysis of the hospital survey, the primary unit of analysis was the pharmacist.
We assessed the percentage of suspected biologics from the electronic ADR reports that contained an identifiable brand name (or name of the manufacturer or MAH) from the ‘as-reported’ suspected drug name. For batch number identification, entries were assessed to explore whether reported batch numbers referred to valid entries (e.g. to exclude false entries such as ‘unknown’ or ‘?’). The unit of analysis was the suspected biologic. This means that in case two biologics (from our predefined list) were suspected in one ADR report, then this report contributed to the analyses for each of the two suspect biologics. Data analysis was stratified by year in which the report was received (time trend analysis) and by reporter type.
2.2.2 Exploratory Analysis
We assessed a number of possible success factors for facilitating the reporting of product information: (1) whether or not a biosimilar is approved in a certain product class; (2) time to onset of the ADR; and (3) whether biologics required (aseptic) compounding before administration to the patient.
Firstly, a subanalysis was performed with regard to brand name and batch number traceability for biologics for which a biosimilar was available. The year and month of the first EU approval of a biosimilar in a product class was used for including corresponding ADR reports in this subanalysis.
Secondly, brand name and batch number traceability were compared among ADR reports stratified by the time to onset of the suspected ADR: ‘< 1 day’, ‘1–7 days’, ‘7–30 days’, ‘30–180 days’, ‘180–360 days’ and ‘> 360 days’. This analysis was limited to ADR reports for which a single suspected drug was reported, the same time to onset was provided for all reactions reported for the same case and where at least one of the reactions per case was classified as serious,Footnote 2 as time-to-onset data is not routinely captured for non-serious reports. ADR reports were excluded from this analysis if no time-to-onset information was provided.
Thirdly, batch traceability was compared between ADR reports for biologics that do or do not require aseptic compounding prior to administration (e.g. for intravenous formulations) on the basis that good manufacturing practice (GMP) standards recommend that batch number details are recorded within compounding records [11]. This analysis was conducted for the 20 most commonly reported suspect biologics from our sample list. Product information within the electronic Medicines Compendium (eMC) database was used to determine whether compounding was required for each of these 20 biologics [12].
3 Results
3.1 Hospital Pharmacist Survey
Of the 174 trusts that were eligible for this study, we collected a total of 61 responses from hospital pharmacists representing 40 (23%) NHS Trusts and 43 distinct hospital sites. The majority of the survey respondents indicated that biologics were always prescribed by brand name (29; 48%) or very often (19; 31%). Of the latter group, 13 (68%) indicated that brand name prescribing was more likely where a biosimilar (or related product sharing the same International Non-Proprietary Name [INN]) is available.
The majority of survey respondents (≥ 79%) reported that brand names are routinely recorded for biologics during dispensing and administration in both inpatient and outpatient settings (Table 1). Routine batch number recording was reported to be performed less frequently, ranging from 40% during dispensing to 58% during administration of biologics in the inpatient setting and 38% and 57%, respectively, for the outpatient setting.
Paper-based recording of product details was more commonly reported (≥ 84%) than electronic-based recording (≤ 51%) for prescribing and administration of biologics in both inpatient and outpatient settings (Table 1). However, for dispensing, the majority of the respondents (approximately 80%) indicated that electronic-based recording was used.
A total of 54 hospital pharmacists responded to the question about bottlenecks and potential solutions for ensuring adequate brand name and batch number traceability in UK clinical practice. Twenty-six (48%) responses indicated that the lack of electronic recording systems prevent the routine recording of batch number information throughout the supply chain. Moreover, 17 (31%) responders specifically referred to the need for appropriate barcodes and electronic systems to facilitate the automatic scanning of batch number information throughout the supply chain to improve the traceability of biologics. Although many hospitals still have paper-based recording processes in place to some extent, 17 (31%) responses indicated that the manual recording of every batch number of biologics dispensed and administered was considered impractical (i.e. too resource- and time-intensive), hampered adequate retrieval of product information or may be unreliable due to transcription errors or loss of information.
3.2 Analysis of Brand Name and Batch Number Traceability in Spontaneous Adverse Drug Reaction (ADR) Data
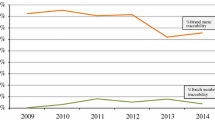
A total of 6108 electronic ADR reports were submitted to the Yellow Card Scheme for recombinant biologics during the study period. However, due to multiple suspect biologics in some ADR reports, the analysis relates to a total of 6219 ADR reports at the level of the individual biologic. Of these, 2358 (38%) had an identifiable brand name, whereas batch numbers were specified for 911 (15%) biologic-level reports. The number of electronic ADR reports received by the MHRA for biologics steadily increased over the study period (Fig. 1). Brand name traceability in electronic ADR reports remained constant, whereas batch number traceability slightly increased from 11% in 2012 to 17% in 2013.

Time trend analysis of brand and batch traceability in electronic adverse drug reaction reports between 1 January 2009 and 30 September 2017. *End date 30 September 2017. ADR adverse drug reaction
Product traceability was slightly higher for non-serious events, with 43% of the ADR reports having a traceable brand name and 17% a traceable batch number compared with 33% and 14%, respectively, for serious events.
The majority of the electronic ADR reports for biologics were submitted by hospital health professionals, including nurses (33% of the reports), doctors (23%) and pharmacists (14%) (Table 2). Brand name was most frequently specified in reports submitted by patients (76%) and batch numbers were most often provided by hospital nurses (20%) and patients (19%).
3.3 Exploratory Analyses
For 916 ADR reports, a biosimilar was available in the EU for the suspected biologics. Brand name and batch number traceability was higher in this subset of reports than overall traceability: brand name was specified in 510 of these ADR reports (56%), whereas a batch number was provided in 252 ADR reports (28%).
Time to onset of the reported ADR was available in 1629 (26%) of the electronic ADR reports for biologics submitted to the Yellow Card Scheme. The frequency of batch number reporting decreased with increasing time to onset, from 54% for ADRs occurring within 24 h of administration to less than 3% for ADRs occurring more than a year after administration (Fig. 2). This effect was observed to a lesser extent for brand name identification (Fig. 2).

Overview of the correlation between time to onset and likelihood that a brand name or batch number was included in an adverse drug reaction report. ADR adverse drug reaction
For the 20 most commonly reported suspect biologics, a total of 3165 ADR reports were identified for biologics that are supplied in ready-to-use formulations compared with 1339 ADR reports for biologics that required compounding. For the biologics that required compounding, 354 (26%) ADR reports contained an identifiable batch number, which was higher than that observed for the 263 (8%) ADR reports for biologics supplied in ready-to-use formulations.
4 Discussion
This study shows that product details such as brand name or manufacturer were identifiable in less than half of the ADR reports submitted for biologics in the UK between 1 January 2009 and 30 September 2017. Furthermore, only 15% of the ADR reports included a batch number. The results of this study further indicate that the traceability of biologics in UK clinical practice is not always ensured, particularly for batch numbers. Brand name identification for biologics for which a biosimilar has been approved was also limited, with only 56% of these ADR reports having an identifiable brand name. Although some minor improvements of batch traceability in ADR reporting have been observed over the study period, it would appear that the pharmacovigilance legislation introduced in 2012 to strengthen product identification for biologics in ADR reports has not yet achieved its objective. These findings are in line with an earlier study on the traceability of biologics in the Netherlands from 2016 [10].
4.1 Hospital Pharmacist Survey
According to the majority of the respondents to the survey, brand names are routinely recorded during the dispensing process (Table 1). The survey also showed that routine batch numbers recording occurs less frequently during routine clinical practice. With regard to recording practices throughout the hospital supply chain, it was indicated by the respondents that batch number recording is highest at the time of administration. A reason for this could be the prevalent use of paper-based recording for administration that facilitates the manual entry of batch numbers or the use of peel-off stickers (‘flag labels’) containing the batch number. It was indicated that electronic-based recording was primarily used during dispensing, which may hamper the routine recording of batch numbers because the current EU barcode standards do not support automatic recording of batch numbers in electronic systems [13,14,15]. The absence of adequate recording and tracing of batch numbers in UK clinical practice as well as integrated IT systems, for example coupled with the independent Yellow Card Scheme, may contribute to the limited traceability of batch numbers in ADR reporting.
4.2 Product Traceability in ADR Reports
The ADR data analysis showed that the pharmacovigilance legislation of 2012 did not have a marked or sustained effect on brand name and batch number traceability for biologics after its implementation in UK law, even after a number of years. Although the results show a slight increase of batch number traceability initially after 2012, from 11 to 17% in 2013 (Fig. 1), this could also be explained by the automatic batch number prompts that were introduced by the MHRA in the online reporting form of the Yellow Card Scheme in 2013 as a result of the legislation (MHRA, personal communication, 2019). Brand name traceability was slightly increased for biologics for which biosimilars were available. Besides an increased awareness of the legislative requirement to specify the brand where biosimilars are available, e.g. by NHS guidance on biosimilars [16], this effect can be further amplified by the fact that if only one brand is available, the reporter may not consider it necessary to specify a brand when submitting an ADR report. Nonetheless, with only 56% of the ADR reports for biologics for which a biosimilar has been approved having an identifiable brand, traceability needs to be further improved to ensure timely detection of product-specific safety signals.
We observed that patients were most likely to include a brand name in ADR reports (Table 2). This finding is supported by other EU studies [8]. Batch number inclusion was highest for hospital nurses. A reason for this can be found in the findings from the survey indicating that batch number recording is highest during administration (Table 1). This could indicate that the availability of batch number information at the point of administration allows for hospital nurses to have easier access to this information and thus it is more likely to be included in ADR reports submitted by hospital nurses.
The limited traceability of batch numbers in ADR reports due to shortcomings in recording and tracing detailed product information in clinical practice is further supported by the time-to-onset analysis, which shows that batch numbers (and to a lesser extent brand names) are more likely to be included in ADR reports when the time to onset is short, e.g. within 1 day (i.e. acute onset), and consistently decreases with increasing time to onset. This can be explained by the fact that with an acute onset, the medicinal product package or container on which the batch number information is printed may still be present and therefore accessible to retrieve the batch numbers of the biologic that was administered. Obviously, retrieving the batch number from the product package will not be possible once the package has been discarded. However, it is also known that batch-related ADRs, particularly those leading to immune-mediated reactions, have a short latency period between administration and manifestation of the ADR. This could contribute to a greater alertness by the reporter to include the batch number in such cases and, vice versa, reduce the perceived relevance of reporting a batch number for ADRs occurring a longer period after administration.
The assumption that the adequate recording (and therefore availability) of product information is directly related to inclusion of these details in ADR reports is further supported by our subanalysis comparing biologics requiring aseptic compounding, for which batch number recording in aseptic preparation worksheets is a mandatory procedure as part of GMP requirements, with biologics that can readily be distributed to the patient. The results indicate that batch numbers are more likely included in ADR reports for biologics that require aseptic compounding than for biologics that do not require compounding. This suggests that batch numbers are more likely to be traceable in ADR reports in situations where these details are more readily accessible (e.g. if routinely recorded) in clinical practice.
4.3 Comparison to Other Studies
Our findings here support our earlier findings in the Netherlands that the limited traceability of biologics in ADR reports may be primarily caused by shortcomings in the adequate recording and tracing of this information in clinical practice [10]. The finding that the new pharmacovigilance legislation from 2012 has not yet made any substantial improvement with regard to traceability, in particular for batch numbers, is supported in the literature. We found poor batch number traceability (5%) in the Netherlands [10], while another recent study in EudraVigilance estimated batch traceability at 20.5% [17]. However, it needs to be pointed out that the study in EudraVigilance by Vermeer et al. [17] included blood products for which standard protocols for batch number recording have existed for over 25 years, which may have skewed the analysis towards a more positive result for batch number traceability [15, 18].
Discrepancies exist between studies with regard to brand name traceability in the EU. The recent study in EudraVigilance indicated that brand name traceability was 96% for biologics that share the same INN (e.g. if a related biologic or biosimilar is approved) [17]. These findings are, however, not supported in the current UK study nor in our earlier study in the Netherlands. The discrepancies in the findings by other studies that showed a higher traceability of brand names for biologics may be due to inclusion of ADR reports from MAHs. ADR reports originating from MAHs may account for a large portion of total ADR reports found in databases. It has been shown in the scientific literature that many reported adverse events are provided by industry-sponsored programmes, such as PSPs [19, 20]. In these cases, brand names are logically known at the time of reporting.
4.4 Limitations
A number of limitations need to be taken into account when interpreting the results of this study. First, the known limitations that apply to surveys in general are also applicable to this study, such as response variability and response rate. This survey tried to capture a very diverse environment with a limited set of simple questions. We believe that the total of 61 responses provided a sufficiently robust overview of the recording practices in the UK hospital setting to support the conclusions from this study. As the survey requested information on recording of biologics in general, it is possible that it did not fully capture variation in recording practices that may occur for different products. For example, recording of the brand name may occur when a biosimilar is available and only a non-proprietary name is recorded if no biosimilar is on the market. However, we believe that this does not impact the overall conclusions of the study. Since this was a cross-sectional survey, pharmacists’ responses at a particular point in time may not reflect the much greater time span covered by the second analysis of electronic ADR reports submitted to the UK national spontaneous reporting database. Thus, any indirect correlation between recording practices and their effect on traceability within ADR reports should be made with caution.
The use of the ‘as-reported’ drug name field to measure brand name recording in ADR reports can have minor implications since this information could have been updated, e.g. during follow-up of spontaneous cases. Such updates were not identifiable in the analysis dataset. However, this was considered to have no meaningful impact on the broader interpretation of the results of the ADR data analysis. For the subanalysis of the brand and batch identification in ADR reports for biologics for which a biosimilar has been approved, we used the date of approval by the European Medicines Agency (EMA). However, this may not reflect the actual date of biosimilar uptake in UK clinical practice, which may slightly influence the interpretation of this analysis.
4.5 Relevance and Recommendations
With clinical healthcare settings in the UK moving from paper-based recording to electronic-based recording, UK hospitals should consider addressing the current shortcomings of appropriate routine recording and tracing of dynamic product information such as batch numbers (and expiry dates). Furthermore, UK hospitals should address how ADRs that occur within their healthcare settings are captured fully and transmitted to the MHRA for patient safety analysis and signal detection. To this end, the MHRA has developed an electronic information standard for clinical IT system suppliers to integrate ADR reporting into their systems, which includes the appropriate fields required [21]. However, until the encoding of batch numbers in barcodes printed on the single unit dose is ensured to facilitate barcode scanning at the administration site, challenges will remain with implementing electronic-based routine recording of dynamic product information throughout the entire supply chain. The recently established Falsified Medicines Directive (FMD), a piece of legislation that aims to prevent the entry of counterfeit medicinal products into the supply chain, could at least partially be deployed for improving the traceability of biologics in the transition towards more routine electronic-based recording in UK healthcare practice [14, 22, 23].
The transition towards electronic-based recording and in tandem addressing of current limitations of traceability should be approached from the broader public health perspective to find cost-effective solutions to improve the traceability. Other initiatives, such as the FMD or ‘Scan4Safety’ for the reduction of medication errors by implementing barcode medication verification technology at the site of administration, could be aligned in a joint effort to improve the traceability of biologics in the wider framework of ultimate patient care and supply chain efficiency [24]. Nonetheless, from a policy perspective, more efforts are needed at the national level to support automated recording of product information with electronic barcode scanning throughout the supply chain. Furthermore, more awareness and additional education about the importance of the traceability of biologics, such as the MHRA’s Drug Safety Updates [25, 26], ADR e-learning modules [27] and a strengthened dialogue between regulators and healthcare professionals via the National Medication Safety Network, could contribute to improving traceability in ADR reports and ultimately guarantee adequate product and batch identification for optimal public health protection. This study tries to contribute to this.
5 Conclusion
Brand name and batch number traceability are generally low for biologics in UK ADR reports, being 38% and 15%, respectively. Improvements of batch number traceability have been observed after the implementation of the pharmacovigilance legislation in 2012; however, no major improvements were identified with regard to brand name traceability. Shortcomings in the systematic recording of product details in UK clinical practice, particularly for batch numbers, may contribute to the limited traceability of this information in ADR reporting. Pharmacovigilance legislation has not resulted in a significant improvement in the traceability of biologics. This study tries to contribute to increasing the understanding that the legislation can only be effective for these products if a robust traceability of product information in clinical practice is ensured. Improvements of recording and tracing of product details throughout the entire supply chain are therefore needed and should be aligned with other drug safety objectives. Education and training on the importance of traceability for biologics in ADR reporting can further support improving the traceability. These efforts will eventually help to build a robust pharmacovigilance system that allows for the timely identification of any product-related issues.
Notes
Industry reports were excluded as adverse event reports from industry-sponsored programmes, such as patient support programmes (PSPs), may influence the findings from routine clinical practice in the UK, which was the focus of this study.
Based on classification of seriousness at source by MHRA according to an internal reference list of dictionary terms from the Medical Dictionary for Regulatory Activities (MedDRA®).
References
Sharma B. Immunogenicity of therapeutic proteins. Part 3: impact of manufacturing changes. Biotechnol Adv. 2007;25:325–31.
Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22:1383–91.
Schiestl M, Stangler T, Torella C, Čepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2.
European Medicines Agency and the European Commission. Biosimilars in the EU—information guide for healthcare professionals. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 30 July 2019.
Louët S. Lessons from Eprex for biogeneric firms. Nat Biotechnol. 2003;21:956–7.
Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010, amending as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use. http://ec.europa.eu/health/files/eudralex/vol-1/dir_2010_84/dir_2010_84_en.pdf. Accessed 30 July 2019.
Statutory Instruments 2012 No. 1916—The Human Medicines Regulations 2012. http://www.legislation.gov.uk/uksi/2012/1916/made. Accessed 20 April 2019.
Vermeer NS, Straus SMJM, Mantel-Teeuwisse AK, Domergue F, Egberts TCG, Leufkens HGM, et al. Traceability of biopharmaceuticals in spontaneous reporting systems: a cross-sectional study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance Databases. Drug Saf. 2013;36:617–25.
Cutroneo PM, Isgrò V, Russo A, Ientile V, Sottosanti L, Pimpinella G, et al. Safety profile of biological medicines as compared with non-biologicals: an analysis of the Italian spontaneous reporting system database. Drug Saf. 2014;37:961–70.
Klein K, Scholl JHG, Vermeer NS, Broekmans AW, Van Puijenbroek EP, De Bruin ML, et al. Traceability of biologics in The Netherlands: an analysis of information-recording systems in clinical practice and spontaneous ADR reports. Drug Saf. 2016;39:185–92.
Royal Pharmaceutical Society. Quality assurance of aseptic preparation services: standards. Part A. Fifth edition. https://www.rpharms.com/Portals/0/RPS%20document%20library/Open%20access/Professional%20standards/Quality%20Assurance%20of%20Aseptic%20Preparation%20Services%20%28QAAPS%29/rps—qaaps-standards-document.pdf. Accessed 19 April 2019.
Electronic Medicines Compendium (eMC). https://www.medicines.org.uk/emc. Accessed 19 April 2019.
European Association of Hospital Pharmacists (EAHP). EAHP statement on the need for barcoding of the single dose administered in hospitals. http://www.eahp.eu/sites/default/files/files/Barcode_2012%20pdf.pdf. Accessed 20 April 2019.
Klein K, Stolk P. Challenges and opportunities for the traceability of (biological) medicinal products. Drug Saf. 2018;41:911–8.
Vermeer NS, Spierings I, Mantel-Teeuwisse AK, Straus SM, Giezen TJ, Leufkens HG, et al. Traceability of biologicals: present challenges in pharmacovigilance. Expert Opin Drug Saf. 2015;14:63–72.
NHS England. What is a biosimilar medicine? https://www.england.nhs.uk/wp-content/uploads/2019/05/what-is-a-biosimilar-medicine-guide-v2.pdf. Accessed 30 July 2019.
Vermeer NS, Giezen TJ, Zastavnik S, Wolff-Holz E, Hidalgo-Simon A. Identifiability of biologicals in adverse drug reaction reports received from European clinical practice. Clin Pharmacol Ther. 2019;105:962–9.
European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on plasma-derived medicinal products. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plasma-derived-medicinal-products_en.pdf. Accessed 20 April 2019.
Klein K, Scholl JHG, De Bruin ML, van Puijenbroek EP, Leufkens HGM, Stolk P. When more is less: an exploratory study of the precautionary reporting bias and its impact on safety signal detection. Clin Pharmacol Ther. 2018;103:296–303.
Sookoo A. An inspector’s perspective – considerations for patient support and reimbursement programmes (Stakeholder Meeting, 7th June 2013). http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2013/06/WC500144661.pdf. Accessed 20 April 2019.
NHS Digital. DCB1582: Electronic Yellow Card Reporting. https://digital.nhs.uk/data-and-information/information-standards/information-standards-and-data-collections-including-extractions/publications-and-notifications/standards-and-collections/dcb1582-electronic-yellow-card-reporting. Accessed 30 July 2019.
European Commission. Commission Delegated Regulation (EU) 2016/161 of 2 October 2015 supplementing Directive 2001/83/EC of the European Parliament and of the Council by laying down detailed rules for the safety features appearing on the packaging of medicinal products for human use. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2016_161/reg_2016_161_en.pdf. Accessed 30 July 2019.
European Commission. Directive 2011/62/EU of the European Parliament and of the Council of 8 June 2011 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use, as regards the prevention of the entry into the legal supply chain of falsified medicinal products. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2011_62/dir_2011_62_en.pdf. Accessed 30 July 2019.
Scan4Safety. https://www.scan4safety.nhs.uk/. Accessed 20 April 2019.
Medicines and Healthcare products Regulatory Agency (MHRA). Drug safety update. 2012;6(4). https://webarchive.nationalarchives.gov.uk/20141206194247. http://www.mhra.gov.uk/home/groups/dsu/documents/publication/con207196.pdf. Accessed 30 July 2019.
Medicines and Healthcare products Regulatory Agency (MHRA). Reporting suspected adverse drug reactions to vaccines and biological medicines. https://www.gov.uk/drug-safety-update/reporting-suspected-adverse-drug-reactions-to-vaccines-and-biological-medicines. Accessed 30 July 2019.
Strengthening Collaboration for Operating Pharmacovigilance in Europe (SCOPE) Joint Action project ADR e-learning module. Adverse drug reactions: reporting makes medicines safer. http://www.scopejointaction.eu/outputsandresults/adr-collection/awareness-levels/story_html5.html?lms = 1. Accessed 30 July 2019.
Acknowledgements
We acknowledge the support of the National Institute for Health Research Clinical Research Network (NIHR CRN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kevin Klein, Lorna Hazell, Pieter Stolk and Saad Shakir have no conflicts of interest that are directly relevant to the content of this study.
Funding
The UK BIO-TRAC study is conducted by Drug Safety Research Unit (DSRU) Education and Research Ltd (https://www.dsru.org) in collaboration with Lygature (https://www.lygature.org) and was funded by a an unrestricted grant from the Association of the British Pharmaceutical Industry (ABPI). This study is based in part on data obtained under licence from the UK Medicines and Healthcare products Regulatory Agency. However, the interpretation and conclusions contained in this study are those of the author/s alone.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Klein, K., Hazell, L., Stolk, P. et al. The UK BIO-TRAC Study: A Cross-Sectional Study of Product and Batch Traceability for Biologics in Clinical Practice and Electronic Adverse Drug Reaction Reporting in the UK. Drug Saf 43, 255–263 (2020). https://doi.org/10.1007/s40264-019-00891-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-019-00891-6