
Abstract
Blinatumomab is the first-and-only Food and Drug Administration (FDA)-approved cluster of differentiation (CD) 19-directed CD3 bispecific T-cell engager (BiTE®) immunotherapy. It is currently FDA approved for the treatment of adults and children with Philadelphia chromosome-positive (Ph+) and Philadelphia chromosome-negative (Ph−) relapsed/refractory (R/R) B-cell precursor acute lymphoblastic leukemia (ALL) and B-cell precursor ALL with minimal residual disease. Similarly, initial marketing authorization for blinatumomab in the European Union was granted for the treatment of adults with Ph− R/R B-cell precursor ALL. The benefits of treating R/R B-cell precursor ALL patients with blinatumomab include increased overall survival, more favorable hematologic remission and molecular response rates, and a lower incidence rate of selected adverse events when compared with standard-of-care chemotherapy. The key risks associated with blinatumomab treatment include cytokine release syndrome, neurotoxicity, and medication errors. Here, we review the benefits and risks of blinatumomab treatment and describe how these risks can be mitigated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia treated with blinatumomab have increased overall survival and a lower incidence rate of selected adverse events when compared with standard-of-care chemotherapy. |
Blinatumomab treatment risks include cytokine release syndrome, neurotoxicity, and medication errors. |
1 Introduction
1.1 Acute Lymphoblastic Leukemia (ALL)
Acute lymphoblastic leukemia (ALL) is a malignancy characterized by an abnormal proliferation of lymphoid progenitor cells known as blasts. Leukemic blast cells can accumulate in the bone marrow, peripheral blood, and extramedullary sites. Presenting signs and symptoms of ALL may include signs of bone marrow failure (e.g., anemia, neutropenia, or thrombocytopenia) and nonspecific constitutional symptoms (e.g., fever, pain, or malaise). Diagnosis is based on the microscopic assessment of a bone marrow aspirate and/or biopsy, in which at least 20% of the cells are lymphoid/undifferentiated blasts [1, 2]. Based on data from national cancer registries, approximately 7000 new cases of ALL are diagnosed annually in both the US and European Union (EU) [3, 4]. Although ALL occurs predominantly in children, the incidence of ALL has a bimodal distribution and peaks at approximately 5 and 50 years of age [5].
Classification schemes for ALL are based on cytogenetic and immunophenotypic characteristics of the blasts, and subtypes of ALL are defined based on the B-cell and T-cell lineage. Identification of several genetic alterations, including individual point mutations and structural abnormalities, also allows for genetic classification of ALL [6]. Translocation of chromosome 9 and 22 (Philadelphia [Ph] chromosome) is a common molecular abnormality in ALL, with the Ph chromosome present (Ph+) patients among approximately 25% of the adults with B-cell precursor ALL [7, 8].
Treatment of ALL includes induction therapy to induce clinical remission, followed by consolidation and maintenance therapy. Induction regimens are comprised of multi-agent chemotherapy, which typically includes vincristine, anthracyclines, asparaginase, and cyclophosphamide along with corticosteroids [1, 2]. Approximately 40–60% of the adults with ALL may relapse following initial treatment. Relapsed/refractory (R/R) ALL is defined as reappearance of blasts (> 5%) following complete remission (CR) or failure to achieve a CR at the end of induction [1]. R/R ALL has been found to be associated with poor outcomes and treatment options for R/R ALL are limited. Data from several clinical trials have shown poor median overall survival (OS; range 3–5 months) among R/R ALL patients with standard-of-care chemotherapy (SOC) and failure to achieve CR with second-line therapies [9,10,11,12]. The availability of new targeted therapies for R/R ALL may provide a survival benefit in comparison with SOC in this patient population [13, 14].
1.2 Bispecific T-cell Engager (BiTE®) Immunotherapy
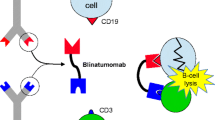
Bispecific antibody constructs enhance tumor killing by targeting T cells to tumor cells [15]. Blinatumomab, developed by Micromet GmbH (Germany) and subsequently acquired by Amgen Inc. in 2012, is a first-in-class bispecific T-cell engager (BiTE®) antibody construct that selectively binds with high affinity to cluster of differentiation (CD) 19 (expressed on tumor cells of B-cell lineage) and CD3 (expressed on T cells). The innovative mechanism of action of blinatumomab utilizes the patient’s own cytotoxic T cells to attack CD19-positive cells, including those represented by B-cell malignancies [16, 17]. When an endogenous T cell is connected via CD3 by blinatumomab to a CD19-expressing B cell, the T cell is activated to kill the B cell and to proliferate, creating more killer T cells (Fig. 1).

Blinatumomab structure and mode of action. BiTE® bispecific T-cell engager, CD cluster of differentiation
1.3 Blinatumomab Clinical Development Program and Regulatory Approval History in R/R B-cell Precursor ALL
1.3.1 Early Clinical Development Program
Blinatumomab was first evaluated as a short-term continuous intravenous (cIV) infusion in patients with R/R non-Hodgkin’s lymphoma (NHL) and in patients with chronic lymphocytic leukemia. A phase I study (ClinicalTrials.com identifier NCT00274742) in R/R NHL was initiated with blinatumomab administered as cIV infusion at doses of 0.5–90 μg/m2/day; cIV administration is required due to the short half-life of blinatumomab—approximately 2 h in humans [18, 19]. Adverse events reported in the patients included cytokine release syndrome (CRS) and neurologic events [19]. Subsequently, blinatumomab was studied in adults and children with R/R B-cell precursor ALL and in patients with B-cell precursor ALL in complete remission with minimal residual disease (MRD).
1.3.2 Regulatory Approval History
Accelerated regulatory approval is granted for rare and/or serious diseases when there is a need for therapeutics with better efficacy and when a drug with a novel mechanism of action has the potential to significantly improve treatment outcomes. Blinatumomab received multiple designations such as Orphan Drug (May 2008), Breakthrough Therapy (June 2014), and Priority Review (October 2014) in the US, and Orphan Drug (July 2009) in the EU. The initial marketing authorization for blinatumomab for the treatment of adults with Ph− R/R B-cell precursor ALL was obtained using data from two phase II, open-label, single-arm, multicenter clinical trials (MT103-206 [NCT01209286] and MT103-211 [NCT01466179]). Approval in the US was obtained under the accelerated approval pathway in December 2014; the European Commission granted conditional marketing authorization in November 2015 [20, 21]. Regular Food and Drug Administration (FDA) approval was granted when the clinical benefit of blinatumomab was confirmed by the randomized, controlled, phase III TOWER clinical trial (NCT02013167) of blinatumomab versus SOC in heavily pretreated adults with Ph− R/R B-cell precursor ALL [13]. Full FDA approval for the treatment of both Ph− and Ph+ R/R B-cell precursor ALL in adult and pediatric patients was granted in July 2017 following studies of blinatumomab in adults with Ph+ R/R B-cell precursor ALL (ALCANTARA [NCT02000427]) [22] and in children with R/R B-cell precursor ALL (MT103-205 [NCT01471782]) [23, 24]. As of December 2017, blinatumomab has been approved in 53 countries for R/R disease. As of March 2018, blinatumomab has been granted accelerated approval in the US to treat adults and children with B-cell precursor ALL who are in remission but still have MRD based on the phase II BLAST study (NCT01207388).
2 Benefits of Blinatumomab in R/R B-cell Precursor ALL
2.1 Phase II Studies
Data from multiple phase II clinical trials (Table 1) have shown the clinical benefit of blinatumomab in the treatment of R/R B-cell precursor ALL relative to historic cohorts, with the confirmatory phase III TOWER study showing the superior OS of blinatumomab versus SOC. Moreover, additional studies in patients with B-cell precursor R/R ALL are either ongoing or planned (Table 2).
Study MT103-206 (NCT01209286) (N = 36) [25] and study MT103-211 (NCT01466179) (N = 189) [20, 26] were phase II studies evaluating the efficacy, safety, pharmacokinetics, and tolerability of blinatumomab in adults with primarily Ph− R/R B-cell precursor ALL. Patients were treated with blinatumomab by cIV infusion for 28 days followed by a 14-day treatment-free interval (1 cycle = 42 days). Similarly, the phase II ALCANTARA study evaluated blinatumomab in 45 adults with Ph+ R/R B-cell precursor ALL who relapsed or were refractory to prior tyrosine kinase inhibitor therapy [22]. The phase I/II study MT103-205 evaluated the safety, pharmacokinetics, dosage, and efficacy of blinatumomab in children and adolescents with R/R B-cell precursor ALL. In the phase I and II parts of the MT103-205 study, 49 and 44 patients were treated, respectively [23, 24].
Baseline characteristics and treatment exposure of patients in these studies are summarized in Table 1. Among the phase II studies in adult patients, there were large differences in median age (range 32–55 years) and the percentage of patients with two or more prior salvage therapies (range 19–57%), whereas the populations were more similar in the percentage of patients with prior transplant (range 34–44%) and the median percentage of blasts at baseline (range 77–87%) (Table 1).
The efficacy endpoints of CR and CR with partial hematologic recovery (CRh) were considered predictive of clinical benefit. CRh was defined as ≤ 5% blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts with platelets > 50,000/µL and absolute neutrophil counts of > 500/µL. The primary efficacy endpoint in the phase II studies in adults with R/R B-cell precursor ALL was CR/CRh rate within the first two treatment cycles. In these studies, 36–69% of heavily pretreated patients achieved a CR or CRh within two treatment cycles (Table 3) [20, 22, 25, 26]. Among pediatric patients in study MT103-205, the primary efficacy endpoint of CR within the first two cycles was achieved by 39% of the patients (Table 3) [23, 24].
To provide context for the single-arm clinical trials in adults with Ph− R/R B-cell precursor ALL, a historical comparator study (NCT02003612) was conducted among 1139 adults with R/R ALL who received SOC in the US and EU [27]. In the historic SOC cohort, CR was observed in 24% (95% confidence interval [CI] 20–27) of the patients, with a median OS of 3.3 (95% CI 2.8–3.6) months. Although direct comparisons are difficult, CR/CRh in patients treated with blinatumomab ranged from 36 to 69%; median OS in blinatumomab-treated patients was double that of the historic cohort (Table 3). A similar historical comparator study was conducted in children (NCT02303522).
2.2 Phase III Study in Adults with Ph− R/R B-cell Precursor ALL (TOWER)
Because blinatumomab received an accelerated approval in the US and a conditional marketing authorization in the EU, a phase III confirmatory study was required for both regions. The phase III TOWER study was a randomized, open-label study that evaluated the efficacy of blinatumomab versus SOC among adults with Ph− R/R B-cell precursor ALL [13]. Patients with heavily pretreated B-cell precursor ALL (including intensive combination chemotherapy for initial treatment or subsequent salvage treatment) were randomized in a 2:1 ratio to receive either blinatumomab (n = 271) or SOC (n = 134). The primary endpoint was OS; key secondary endpoints included CR with full hematologic recovery and CR with full, partial, or incomplete hematologic recovery within 12 weeks, and event-free survival. Following a prespecified interim analysis, the TOWER study was stopped early because the primary endpoint of improved OS was met. Patients treated with blinatumomab were found to have superior OS relative to those treated with SOC; median OS was 8 versus 4 months (hazard rate for death: 0.71 [95% CI 0.55–0.93]; p = 0.01). Remission within 12 weeks following initiation of treatment was also significantly higher in the blinatumomab group versus the SOC group: CR with full hematologic recovery (34% vs 16%, p < 0.001) and CR with full, partial, or incomplete hematologic recovery (44% vs 25%; p < 0.001) [13].
2.3 Health-Related Quality of Life
In the phase III TOWER study, the health-related quality of life (HRQL) of patients was assessed using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30). Patients receiving blinatumomab generally reported better HRQL following treatment than did patients receiving SOC. Importantly, the HRQL of patients treated with blinatumomab was stable, showing a delay in deterioration compared with the HRQL of patients treated with SOC [28]. The clinically meaningful benefits of blinatumomab in maintaining the HRQL of patients while significantly improving overall survival support its value in patients with R/R B-cell precursor ALL compared with chemotherapy.
3 Safety Profile of Blinatumomab
The safety profile of blinatumomab has emerged from clinical trials in patients with R/R B-cell precursor ALL and NHL, and from postmarketing data. As of April 2018, 1182 patients have been exposed to blinatumomab in clinical trials and > 1000 from early access programs; in the postmarketing setting, 4483 patients have been exposed. From this exposure, the key risk drivers identified for blinatumomab are CRS, neurotoxicity, and medications errors. The safety and tolerability properties of blinatumomab are summarized below and are generally consistent with the symptoms associated with R/R B-cell precursor ALL. As it is challenging to distinguish between adverse events caused by the drug and the disease, treatment-emergent adverse events (TEAEs), events by severity, and exposure-adjusted events are all presented.
3.1 Clinical Studies
The primary analysis of blinatumomab safety in adults is based on the pooled analyses of the phase II studies MT103-206 (N = 36) and MT103-211 (N = 189). In study MT103-206, patients received doses of 5–30 µg/m2/day as per the protocol dosing schedule. In study MT103-211, blinatumomab was administered as cIV infusion at an initial dose of 9 μg/day for the first 7 days of cycle 1 and at a target dose of 28 μg/day from week 2 in cycle 1 and in the subsequent cycles. The estimated blinatumomab exposure in this study was 25 patient-years, with median treatment exposure of 42 days (Table 1) [20].
In the pooled analysis set (N = 225), essentially all patients developed TEAEs that occurred within the start of the first infusion and 30 days after the end of the last infusion (Table 4). TEAEs that were at least grade 3 in severity were reported for 81% of patients; 65% and 20% of patients had serious adverse events and TEAEs leading to the discontinuation of blinatumomab, respectively. Adverse events considered treatment-related by the investigator were reported for 90% of patients [20]. Among older patients (≥ 65 years), tolerability was similar with TEAEs grade ≥ 3 reported for 31/36 (86%) patients.
Adverse events of interest based on the Council for International Organizations of Medical Sciences definition [29] identified from MT103-211 included events potentially associated with the pharmacologic activity or mechanism of action of blinatumomab—CRS, capillary leak syndrome, and neurologic events. Other adverse events of interest included those associated with disease factors secondary to blinatumomab treatment (e.g., tumor lysis syndrome) and prior therapy (e.g., infections or cytopenias) as well as those related to the preparation, administration, and delivery of blinatumomab (medication errors). The key risk drivers identified were CRS and neurotoxicity, and consequently, the FDA-approved prescribing information for blinatumomab includes a boxed warning for these events [31].
The emerging safety profile of blinatumomab was further assessed in a phase III, randomized, open-label TOWER study of blinatumomab (n = 267) versus SOC (n = 109). In the TOWER study, adverse events were consistent with the previous studies (Table 4). The most frequently reported adverse events in the blinatumomab arm, occurring at an incidence of ≥ 5% in comparison with the SOC arm, were pyrexia, CRS, and infusion-site reactions; those occurring at an incidence of ≥ 5% in the SOC arm included neutropenia, anemia, and infections (Table 5). Overall, grade ≥ 3 TEAEs for blinatumomab were higher in cycle 1 than in cycle 2 (76% vs 37%) [30]. There were fewer events related to myelosuppression and associated complications in the blinatumomab arm than the SOC arm. A higher incidence of serious adverse events, including neurologic events, CRS, administration-site conditions, and procedural complications, was reported in the blinatumomab arm. However, after adjusting for treatment exposure, the overall incidence of serious adverse events was lower in the blinatumomab arm (349 vs 642 events per 100 patient-years of exposure) [13]. In an exploratory analysis of the TOWER study data, blinatumomab showed an exposure-adjusted adverse event profile that was well tolerated compared with SOC [30]. After adjusting for time on treatment, the overall grade ≥ 3 exposure-adjusted event rates were significantly lower for blinatumomab compared with SOC (11 vs 45 events per patient-year; p < 0.001). For specific grade ≥ 3 events of clinical interest, the exposure-adjusted event rates for blinatumomab versus SOC were lower for infections (2 vs 6 events per patient-year; p < 0.001), cytopenias (4 vs 20 events per patient-year; p < 0.001), and neurologic events (0.4 vs 1 event per patient-year; p = 0.008), and higher for CRS (0.2 vs 0 events per patient-year; p = 0.038) [30].
Adverse reactions in 45 patients with Ph+ R/R B-cell precursor ALL were assessed in the phase II ALCANTARA study and were consistent with the adverse events observed previously in patients with primarily Ph− R/R B-cell precursor ALL. Grade ≥ 3 TEAEs with an incidence above 15% were febrile neutropenia (27%), thrombocytopenia (22%), and anemia (16%) [22].
Adverse reactions in pediatric patients were consistent with those reported for adults [24]. In the phase I/II study MT103-205, all 93 patients had at least one TEAE (Table 4), with pyrexia reported most frequently. The most frequently occurring grade ≥ 3 adverse events were pyrexia (16%), anemia (15%), and increased alanine transaminase level (13%) (Table 6).
3.2 Key Risk Drivers
Utilizing a structured benefit–risk assessment algorithm [31], the key risk drivers associated with the administration of blinatumomab were identified as CRS, neurotoxicity, and medication errors (Fig. 2). These risk drivers are included in the Warning and Precautions sections of the approved country labels and are the focus of additional risk minimization and pharmacovigilance activities [20, 32]. Other risks included under the Warnings and Precautions sections of labels include infections, tumor lysis syndrome, neutropenia and febrile neutropenia, effects on the ability to drive and use machines, elevated liver enzymes, pancreatitis, leukoencephalopathy, immunization with live viral vaccines, and specifically in pediatric patients, the potential risk of serious adverse reactions due to additional exposure of a benzyl alcohol preservative in the 7-day-bag infusion solutions [33].

Mitigation of key risks of blinatumomab treatment
3.2.1 Cytokine Release Syndrome
IV therapeutics such as blinatumomab and other CD19-directed immunotherapies [34, 35] can trigger the rapid release of inflammatory mediators and cellular cytokines into the blood. Organ toxicity affecting cardiovascular, respiratory, neurologic, and renal function may result. This systematic inflammatory response, referred to as CRS, is often mild, even though it can be severe and life-threatening in some patients. Of note, CRS may be indistinguishable from other infusion-related reactions such as pyrexia and hypotension [20].
In the phase II study MT103-211 in adult patients with R/R B-cell precursor ALL, CRS was reported for 24 (13%) of 189 patients, with median time to event onset of 2 days and most CRS events occurring during cycle 1 from day 1 to 7. Among the 24 patients, CRS resulted in the interruption of blinatumomab infusion for three patients, two patients developed grade 3 CRS (days or weeks after treatment), and one patient developed a grade 3 cytokine storm (minutes to hours after treatment). CRS did not lead to permanent discontinuation of treatment for any of the patients and there were no incidents leading to death [20].
In the phase III randomized TOWER study evaluating blinatumomab compared with SOC, CRS was only reported in the blinatumomab arm, with 16% (43/267) of the patients having a CRS event; most patients (86% [37/43]) had the CRS event in the first cycle of treatment. Of the CRS events, 5% (13/267) of patients had grade ≥ 3. The median time to first onset of any CRS event was 2 (range 1–254) days; for grade ≥ 3 CRS events, the median time to onset was 4 (range 1–246) days. The median time to resolution of any CRS event was 4 (range 1–21) days; of the grade ≥ 3 CRS events, there was one unresolved event. CRS events led to treatment interruption for 13 patients and treatment discontinuation for three patients. None of the CRS events had a fatal outcome.
In the phase I/II study (MT103-205) in children, 11% (8/70) of the patients treated with the step-up dose of 5/15 μg/m2/day had CRS; four (6%) patients had grade ≥ 3 CRS. Of these four patients, two had interrupted treatment and two discontinued the treatment permanently; all four patients achieved CR at response assessment [24].
To mitigate the risk of CRS, a step-up dose regimen is indicated for the first cycle of blinatumomab administration. For adults, treatment is initiated with a dose of 9 μg/day in week 1 and then increased through a step-up to 28 μg/day on day 8 and continued at that dose for the subsequent weeks and cycles. For pediatric patients, a step-up dose of 5/15 μg/m2/day is indicated for the first cycle (i.e., 5 μg/m2/day, days 1–7; 15 μg/m2/day, days 8–28). In addition, premedication with dexamethasone is required prior to the initiation of therapy to minimize the occurrence and severity of events that may be associated with CRS and other infusion reactions. Data from in vitro analyses evaluating blinatumomab-mediated cell lysis and cytotoxicity did not demonstrate dexamethasone-related effects on blinatumomab-mediated cytotoxicity [39].
Patients receiving treatment with blinatumomab should be observed closely for CRS and other infusion reactions, which may be fatal or life-threatening. Management of these events may require temporary interruption or discontinuation of blinatumomab treatment. These events can generally be rapidly resolved due to the short half-life of blinatumomab [33].
3.2.2 Neurotoxicity
Neurologic events, including events with a fatal outcome, were reported among patients receiving blinatumomab in clinical trials. One proposed mechanism for neurotoxicity is the release of neurotoxic cytokines and chemokines by blinatumomab-activated T cells en route to the central nervous system, causing inflammation at the neuroendothelium [36]. Neurotoxicity is also seen with the administration of other CD19 immunotherapies [34, 35]. For example, axicabtagene ciloleucel and tisagenlecleucel are two CD19 chimeric antigen receptor T-cell agents that have boxed warnings for neurologic toxicities (e.g., encephalopathy, delirium, or seizures) in their FDA-approved labels [37, 38].
In the phase II study, MT103-211, in adult patients with R/R B-cell precursor ALL, about half of the patients (52% [98/189]) had some sort of neurologic event. Treatment-emergent serious neurologic events were reported for 16% (31/189) of the patients. Confusional state, tremor, and encephalopathy were the most common treatment-emergent serious neurologic events (3% [5/189] for each) [20]. A majority of the events occurred during the first cycle, with median time to onset of the first neurologic event of 9 days following initiation of blinatumomab treatment, and most resolved after interruption of blinatumomab treatment.
In the phase III randomized TOWER study that evaluated blinatumomab compared with SOC, the incidence of treatment-emergent neurologic events was higher in the blinatumomab arm (61% [163/267]) than the SOC arm (50% [54/109]), as was the incidence of treatment-emergent serious neurologic events (7% [18/267] vs 2% [2/109], respectively). As the drug exposure was greater for blinatumomab compared with SOC, the rates of neurologic events adjusted for treatment exposure were found to be lower in the blinatumomab arm. Tremor, somnolence, paresthesia, hypoesthesia, and seizure occurred at higher rates in the blinatumomab arm; all other treatment-emergent neurologic event rates were either higher in the SOC arm or similar between treatment arms.
The neurologic events observed among pediatric patients in study MT103-205 were similar to those reported in adults. Seventeen (24%) of the 70 patients developed neurologic events, mostly tremor (4 [6%]), somnolence (3 [4%]), and dizziness (3 [4%]). Three patients had grade 3 neurologic events, including somnolence (2 patients) and neuralgia (1 patient). No grade 4 or 5 neurologic events were reported, and no pediatric patient permanently discontinued blinatumomab treatment due to neurologic events [23, 24].
Patients receiving blinatumomab should be monitored for signs and symptoms of neurologic toxicities. Recommendations and guidance regarding dosage adjustments following the onset of neurologic events are provided in the prescribing information for blinatumomab.
3.2.3 Medication Errors
Medication errors are an event of interest for blinatumomab due to the complex method of preparation and administration. Treatment with blinatumomab is initiated in the hospital setting and patients are subsequently discharged to the home setting with a portable cIV infusion pump. The types of medication errors have been related to either the preparation or the administration steps of blinatumomab, resulting in overdose or underdose.
In the clinical trial setting, the most commonly reported reason for medication errors was increased flow rate of the infusion pump (through malfunction leading to an accidental increase, including setting of incorrect infusion rate). Overdoses also resulted from preparation errors by pharmacists in calculating the correct concentration of blinatumomab solution to be administered to patients [39].
In the phase II study, MT103-211, in adult patients with R/R B-cell precursor ALL, overdoses were reported for six (3%) of the 189 patients treated with blinatumomab. All overdose events were grade 1 or 2 in severity [20]. In the phase III randomized TOWER study, medication errors including overdose were reported for 12 (4%) of the 267 patients in the blinatumomab arm; 11 of the patients had overdose. No medication errors were reported in the SOC arm. The adverse events associated with the overdoses included events such as fever, tremors, and headache that are consistent with the known safety profile of blinatumomab [33]. For pediatric patients treated with blinatumomab (5/15 µg/m2/day) in study MT103-205, three (4%) of 70 patients had medication errors.
To mitigate the risk of medication errors with blinatumomab, comprehensive instructions for preparation (including reconstitution and dilution) and administration are provided in the product labeling. In addition, the product labeling includes a warning advising the importance of strictly following these instructions to minimize the risk of medication errors. Patients also should be advised to notify their doctor or nurse if they notice any problems with their infusion pump. Medication errors are part of the Risk Evaluation and Mitigation Strategy (REMS) in the US and are subject to additional educational materials in the EU and other parts of the world.
In the event of an overdose, blinatumomab infusion should be temporarily interrupted; patients should be monitored, and supportive care should be initiated. Treatment with blinatumomab at the correct therapeutic dose may be reinitiated after all toxicities have resolved but no earlier than 12 hours after the interruption of infusion [33].
3.3 Other Risks
3.3.1 Infections
As anticipated in a patient population with ALL and prior immunosuppressive therapy, serious and sometimes fatal infections such as sepsis and pneumonia were observed in about 25% of the patients in the blinatumomab clinical studies [33].
In the phase II study, MT103-211, TEAE infections were reported in 63% (119/189) of the patients, of which pneumonia was the most commonly reported infectious event [20]. Serious TEAE infections, including sepsis, pneumonia, bacteremia, opportunistic infections, and catheter-site infections were observed in 32% (60/189) of the patients. TEAEs leading to death were reported for 9% (17/189) of the patients; three deaths were identified as related to treatment. Depletion of B cells may be associated with decreases in circulating immunoglobulins; and among patients with cancer, risk factors for infections may include decreased concentrations of serum immunoglobulins [40]. Adverse events of decreased immunoglobulins were reported for 11.1% (21/189) of the patients, with none reported as serious or fatal in the phase II study.
In the phase III randomized TOWER study, fewer infections of grade ≥ 3 were reported in the blinatumomab arm compared with the SOC arm (34% vs 52% of the patients) [13]. As underlying neutropenia may predispose patients to the development of infections, patients in the SOC arm were at a greater risk; 58% of the patients in the SOC arm had grade ≥ 3 adverse events of neutropenia compared with 38% in the blinatumomab arm.
As would be expected in a pediatric patient population with underlying R/R B-cell precursor ALL, infections were reported for nearly half of the patients in study MT103-205. For patients administered the recommended step-up dose of 5/15 μg/m2/day, 21% had serious infections. For patients weighing < 45 kg in study MT103-205 and the expanded access protocol (RIALTO, NCT02187354), the most common serious infections were sepsis (4%) and device-related infection (4%). One case of sepsis and one case of fungal infection were fatal [24].
The prescribing information for blinatumomab includes a warning regarding infections [32, 41]. Patients receiving blinatumomab should be monitored for signs and symptoms of infection and treated as appropriate. Administration of prophylactic anti-infective therapies may also be considered during treatment with blinatumomab [20, 33].
3.3.2 Elevated Liver Enzymes
Transient elevations of liver enzymes, observed primarily in the setting of CRS, were reported among patients receiving blinatumomab, with median time to onset of 3 days. For events in the absence of CRS, the median time to onset was 19 days [21].
In the phase II study, MT103-211, 28% (52/189) of the patients reported elevated liver enzyme adverse events; 15% (29/189) had grade ≥ 3 elevations and 2% (4/189) had serious elevations. No fatal events due to elevation of liver enzymes were reported [20].
In the phase III randomized TOWER study, the patient incidence of elevated liver enzymes during the first cycle of therapy was similar in both treatment groups, with TEAEs reported for 22% of the patients in the blinatumomab arm and 25% in the SOC arm; grade ≥ 3 TEAEs were reported for 13% and 15%, respectively. Three serious elevated liver enzyme events and one treatment discontinuation were reported in the blinatumomab arm. No fatal events due to elevation of liver enzymes were reported during this trial [13].
Among pediatric patients administered blinatumomab with the step-up dose of 5/15 μg/m2/day in the phase I/II MT103-205 study, 31% had elevated liver enzymes, with 16% having grade ≥ 3. No reported events were serious or fatal [24]. Elevated liver enzymes were detected during the first week of infusion and returned to baseline within the first cycle. No clinical symptoms or signs of hepatic toxicity were observed.
The prescribing information for blinatumomab includes a warning regarding elevated liver enzymes following administration of blinatumomab along with appropriate guidance regarding the risks of elevated liver enzymes. Patients receiving blinatumomab should be monitored for elevated liver enzymes (alanine aminotransferase, aspartate aminotransferase, and γ-glutamyl transferase) and total blood bilirubin prior to the start of and during blinatumomab treatment, and especially during the first 48 h of the first two cycles. Management of elevated liver enzymes may require either a temporary interruption or the discontinuation of blinatumomab. Blinatumomab treatment should be interrupted if the transaminases levels rise to > 5 times the upper limit of normal or if bilirubin levels rise to > 3 times the upper limit of normal [20, 33].
4 Risk Management and Risk Minimization Activities
4.1 United States
Key components of any risk management plan include routine pharmacovigilance and an approved product label. When assessing the benefit–risk of new products or additional indications, the FDA mainly focuses on four factors: analysis of condition, unmet medical need, clinical efficacy, and risks. Using this approach, the FDA concluded that the perceived benefit of blinatumomab in the R/R B-cell precursor ALL setting in adults outweighed the potential risks of the product and recommended approval by the accelerated pathway. In addition, using the benefit–risk framework, the FDA concluded that to minimize risks, labeling should include dose modifications and instructions for monitoring, a medication guide created and distributed to the patients, and a Dear Health Care Provider letter distributed to physicians outlining the serious risks of blinatumomab.
4.1.1 Risk Evaluation and Mitigation Strategy
A REMS can include one or more components that go beyond an approved label to ensure that the benefits outweigh the risks for patients. The FDA-approved REMS for blinatumomab includes a communication plan targeting healthcare providers and hospital and home healthcare pharmacists. The Dear Health Care Provider and Dear Pharmacists letters address three key risk drivers associated with the administration of blinatumomab—CRS, neurotoxicity, and medication errors. This information is available on the FDA REMS website (http://www.blincytorems.com/). Additionally, FDA required the marketing authorization holder to create a blinatumomab REMS Fact Sheet for Healthcare Providers that provides information on the REMS. Outside of the REMS, a medication guide was created to provide patients with information on the risks associated with blinatumomab.
4.2 European Union
4.2.1 Risk Management Plan
In Europe, the Risk Management Plan is intended to prevent or minimize the impact of unintended incidents by identifying and addressing potential risks before significant negative consequences occur. Risk minimization measures include both routine measures (e.g., product labeling) and additional risk minimization measures. In Europe, the additional risk minimization activities for blinatumomab include educational materials for physicians, nurses, pharmacists, and patients (including caregivers) highlighting the risks of neurotoxicity and medication errors. The materials for physicians, nurses, and patients describe neurologic events and medication errors. The educational materials for pharmacists are specific to medication errors.
5 Conclusions
The clinical benefit of blinatumomab for the treatment of patients with R/R B-cell precursor ALL, initially recognized in phase II studies, was confirmed in the phase III TOWER study that showed superior OS in patients treated with blinatumomab relative to SOC. Importantly, molecular remission (MRD negativity defined as blasts < 10−4) was noted in most responders treated with blinatumomab (Table 3). In the pediatric phase I/II trial, efficacy similar to that seen in adults was observed. The key risk drivers of blinatumomab are CRS, neurotoxicity, and medications errors. These risks are communicated via approved product labels and may be mitigated through premedication, step-up dosing, monitoring, and dose adjustment, interruption, or discontinuation.
References
Alvarnas JC, Brown PA, Aoun P, Ballen KK, Barta SK, Borate U, et al. Acute lymphoblastic leukemia, Version 2.2015. J Natl Compr Canc Netw. 2015;13(10):1240–79.
Hoelzer D, Bassan R, Dombret H, Fielding A, Ribera JM, Buske C, et al. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v69–82.
Gatta G, van der Zwan JM, Casali PG, Siesling S, Dei Tos AP, Kunkler I, et al. Rare cancers are not so rare: the rare cancer burden in Europe. Eur J Cancer. 2011;47(17):2493–511.
Howlader N, Noone A, Krapcho M, Miller D, Bishop K, Kosary C, et al. SEER Cancer Statistics Review, 1975–2014, based on November 2016 SEER data submission. National Cancer Institute. April 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed 14 Nov 2018.
Paul S, Kantarjian H, Jabbour EJ. Adult acute lymphoblastic leukemia. Mayo Clin Proc. 2016;91(11):1645–66.
Taylor J, Xiao W, Abdel-Wahab O. Diagnosis and classification of hematologic malignancies on the basis of genetics. Blood. 2017;130(4):410–23.
Faderl S, O’Brien S, Pui CH, Stock W, Wetzler M, Hoelzer D, et al. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer. 2010;116(5):1165–76.
Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109(8):3189–97.
Advani AS, Gundacker HM, Sala-Torra O, Radich JP, Lai R, Slovak ML, et al. Southwest Oncology Group Study S0530: a phase 2 trial of clofarabine and cytarabine for relapsed or refractory acute lymphocytic leukaemia. Br J Haematol. 2010;151(5):430–4.
Gökbuget N, Kneba M, Raff T, Trautmann H, Bartram CR, Arnold R, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–76.
Kantarjian HM, Thomas D, Ravandi F, Faderl S, Jabbour E, Garcia-Manero G, et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer. 2010;116(24):5568–74.
O’Brien S, Thomas D, Ravandi F, Faderl S, Cortes J, Borthakur G, et al. Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer. 2008;113(11):3186–91.
Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Gokbuget N, Dombret H, Ribera JM, Fielding AK, Advani A, Bassan R, et al. International reference analysis of outcomes in adults with B-precursor Ph-negative relapsed/refractory acute lymphoblastic leukemia. Haematologica. 2016;101(12):1524–33.
Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8:130.
Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100(6):690–7.
Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer. 2005;115(1):98–104.
Zhu M, Wu B, Brandl C, Johnson J, Wolf A, Chow A, et al. Blinatumomab, a bispecific T-cell engager (BiTE®) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271–88.
Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol. 2016;34(10):1104–11.
Blincyto: European Public Assessment Report (24 Sep 2015). European Medicines Agency. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003731/WC500198227.pdf. Accessed 14 No 2018.
Blincyto: Drug Approval Package. United States Food and Drug Administration. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000TOC.cfm. Accessed 14 Nov 2018 (Updated 16 March 2017).
Martinelli G, Boissel N, Chevallier P, Ottmann O, Gokbuget N, Topp MS, et al. Complete hematologic and molecular response in adult patients with relapsed/refractory philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol. 2017;35(16):1795–802.
von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett T, Rizzari C, et al. Phase 1/2 study in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL) receiving blinatumomab treatment. Blood. 2014;124:2292.
von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381–9.
Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–40.
Topp MS, Gokbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66.
Gökbuget N, Kelsh M, Chia V, Advani A, Bassan R, Dombret H, et al. Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2016;6(9):e473.
Topp MS, Zimmerman Z, Cannell P, Dombret H, Maertens J, Stein A, et al. Health-related quality of life in adults with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab. Blood. 2018;131:2906–14.
Council for International Organization of Medical S. Definitions and basic requirements for the use of terms for reporting adverse drug reactions (XIII): clinical pathology and general disorders. Pharmacoepidemiol Drug Saf. 1999;8(3):217–24.
Stein AS, Larson RA, Schuh AC, Stevenson W, Lech-Maranda E, Tran Q, et al. Exposure-adjusted adverse events comparing blinatumomab with chemotherapy in advanced acute lymphoblastic leukemia. Blood Adv. 2018;2(13):1522–31.
Yan CH, Wang JZ, Liu DH, Xu LP, Chen H, Liu KY, et al. Chemotherapy followed by modified donor lymphocyte infusion as a treatment for relapsed acute leukemia after haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion: superior outcomes compared with chemotherapy alone and an analysis of prognostic factors. Eur J Haematol. 2013;91(4):304–14.
Blincyto—Summary of Product Characteristics (SmPC) (eMC). Available from: https://www.medicines.org.uk/emc/product/5064/smpc. Last updated on eMC 31 Aug 2018. Accessed 6 Dec 2018.
BLINCYTO (blinatumomab) [package insert on the internet]. Thousand Oaks, CA: Amgen Inc; 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125557lbl.pdf. Accessed 6 Dec 2018.
Annesley CE, Summers C, Ceppi F, Gardner RA. The evolution and future of CAR T cells for B-cell acute lymphoblastic leukemia. Clin Pharmacol Ther. 2018;103(4):591–8.
Foster JB, Maude SL. New developments in immunotherapy for pediatric leukemia. Curr Opin Pediatr. 2018;30(1):25–9.
Benjamin JE, Stein AS. The role of blinatumomab in patients with relapsed/refractory acute lymphoblastic leukemia. Ther Adv Hematol. 2016;7(3):142–56.
YESCARTA (axicabtagene ciloleucel) [package insert on the internet]. Santa Monica, CA: Kite Pharma, Inc.; 2017. Available from: https://www.fda.gov/downloads/UCM581226.pdf. Accessed 6 Dec 2018.
KYMRIAH (tisagenlecleucel) [package insert on the internet]. East Hanover, New Jersey: Novartis Pharmaceuticals Corporation; 2018. Available from: https://www.fda.gov/downloads/UCM573941.pdf. Accessed 6 Dec 2018.
FDA Briefing Document. Oncologic Drugs Advisory Committee MeetingBLA 125557 S-013. Blincy to (blinatumomab). Available from: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM599298.pdf. Accessed 6 Dec 2018.
Furst DE. Serum immunoglobulins and risk of infection: how low can you go? Semin Arthritis Rheum. 2009;39(1):18–29.
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–42.
Clinical Study Report MT103-205. A Single-Arm Multicenter Phase II Study preceded by Dose Evaluation to Investigate the Efficacy, Safety, and Tolerability of the BiTE® Antibody Blinatumomab (MT103) in Pediatric and Adolescent Patients with Relapsed/Refractory B-Precursor Acute Lymphoblastic Leukemia (ALL). 2015.
Acknowledgements
Medical writing assistance was provided by Elizabeth Leight (Leight Medical Communications, LLC), and Julie Gegner (Amgen Inc.). Editorial support was provided by Cactus Communications. Graphics were provided by Allison Saviano of Sephirus communications, Inc.
Author information
Authors and Affiliations
Contributions
All authors participated in data interpretation and drafting of the manuscript, and read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Anthony Stein is on the speakers’ bureau for Amgen Inc. and Celgene. Janet L. Franklin, Victoria M. Chia, Deborah Arrindell, William Kormany, Jacqueline Wright, Mandy Parson, Hamid R. Amouzadeh, Jessica Choudhry, and Guiandre Joseph are employees and stockholders of Amgen, Inc.
Funding
This work was supported by Amgen Inc.
Ethical Approval and Patient Consent
The research reviewed in this benefit–risk assessment complies with ethical standards, including informed consent and ethics approval.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stein, A., Franklin, J.L., Chia, V.M. et al. Benefit–Risk Assessment of Blinatumomab in the Treatment of Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Saf 42, 587–601 (2019). https://doi.org/10.1007/s40264-018-0760-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-018-0760-1