
Abstract
The FDA guidance for industry in the premarketing clinical evaluation of drug-induced liver injury (DILI) is the most specific regulatory guidance currently available and has been useful in setting standards for the great majority of clinical indications involving subjects with a low risk of liver disorders. However, liver safety assessment faces challenges in populations with underlying liver disease, such as viral hepatitis or metastatic cancer. This is an important issue because there are currently many promising anti-viral and oncologic therapies in clinical development, with a trend toward oral therapies with reduced side effects. Without clearer guidelines, questions regarding liver safety may become a major factor in regulatory approval and ultimately physician uptake of the new treatments. The lack of consensus in defining stopping rules based on serum alanine aminotransferase (ALT) levels underscores the need for precompetitive data sharing to improve our understanding of DILI in these populations and to allow evidence-based rather than empirical definition of stopping rules. A workshop was convened to discuss best practices for the assessment of drug-induced liver injury (DILI) in clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
There is currently a lack of consensus in defining stopping rules based on serum ALT levels in hepatitis B and C and oncology treatment regimens |
Both elevations of baseline ALT as well as ALT elevations during treatment should be considered when assessing the hepatotoxic potential of a candidate drug |
Innovative approaches that combine clinical data from registration trials with biomarker, genetic and metabolomic data in appropriate patient cohorts are urgently required to overcome the limitations of current diagnostic paradigms |
1 Introduction
Timely detection and proper assessment of drug-induced liver injury (DILI) in clinical trials has for decades been one of the key safety challenges for both pharmaceutical industry and regulatory authorities.
A workshop was sponsored and organized jointly by the European Innovative Medicines Initiative (IMI) and the Hamner-UNC Institute for Drug Safety Sciences (IDSS), with the aim of addressing gaps in current guidance and initiating alignment of liver safety assessment on a global scale.
On November 9, 2012, regulatory experts from FDA, EMA, Health Canada, and the Japanese National Institute of Health Sciences discussed in Boston with representatives from industry and academia what could be considered best practices in clinical liver safety assessment, focusing on four key areas: (1) data elements and data standards, (2) methodologies to systematically analyze liver safety data, (3) tools and methods for causality assessment, and (4) liver safety assessment in special populations such as hepatitis and oncology patients.
This section summarizes liver safety assessment challenges in populations with underlying liver disease, such as viral hepatitis or metastatic cancer.
Liver chemistry elevations, typically alanine aminotransferase (ALT) and aspartate aminotransferase (AST), may vary over time in patients with underlying hepatitis B or C, in the presence or absence of treatment. Hepatitis B flares can develop as part of the disease’s natural course or in response to effective treatment, so treatment with candidate antiviral drugs should not be stopped unnecessarily if patients exhibit moderate liver chemistry elevations [1]. Numerous cancers can involve the liver and in oncology trials, patients with elevated pretreatment liver chemistries may require different thresholds for detecting potential drug-induced liver injury (DILI) and considering treatment discontinuation. Moreover, the risk of complications due to DILI in oncology patients needs to be balanced against the potential benefits of novel antineoplastic agents, underscoring the need for safety criteria that reliably define an unacceptable DILI risk in a patient in whom the treatment is effective.
2 Viral Hepatitis
Elevations of ALT >3-fold the upper limit of normal (3× ULN) and ALP >2× ULN are rare in clinical trial populations without underlying liver disease [2] and can thus be considered a safety signal [3]. However, approximately one-third of patients enrolling in chronic viral hepatitis C trials have ALT >3× ULN at baseline [4–6]. An ALT of >3× ULN was even shown to be a favorable prognostic factor in predicting response to peginterferon alpha and ribavirin (OR 1.47 vs. ALT ≤3× ULN, p = 0.003) [7]. The safety of peginterferon based regimens is confounded by the risk of inducing idiopathic autoimmune hepatitis in patients in whom this condition was previously unidentified [8]. Pre-treatment blood samples should be stored to facilitate the retrospective assessment of such cases. Treatment regimens that include ribavirin could confound interpretation of indirect hyperbilirubinemia secondary to hemolysis, as could regimens containing protease-inhibitors that inhibit uridine diphosphate glucurunosyltransferase 1A1 (UGT1A1)—especially in the presence of an underlying UGT1A1 gene variant in Gilbert’s syndrome [6, 9]. Impairment of UGT1A1-mediated bilirubin conjugation, caused by Gilbert’s syndrome or by drugs that inhibit UGT1A1 activity, is associated with >50 % indirect bilirubin [10, 11]. Gilbert’s syndrome is characterized by a concentration of total bilirubin ranging from 20 to 90 μmol/L (1.2–5.3 mg/dL), with a fraction of unconjugated bilirubin ≥80 % [10]. These cases of hyperbilirubinemia would not meet Hy’s law criteria, defined as an elevation of ALT or AST ≥3× ULN in combination with bilirubin >2× ULN, without initial findings of cholestasis (elevated serum alkaline phosphatase). In contrast, an elevation of bilirubin in the context of severe DILI reflects a major impairment of the liver’s excretory capacity for bilirubin and in these instances the fraction of direct bilirubin typically exceeds 35 % [12].
When there are elevations in the pretreatment serum ALT, controversy exists regarding the use of ULN of ALT for the detection of liver injury and definition of stopping rules as compared to elevations relative to baseline values. The ULN has been shown to vary across laboratories according to methodology and the choice of reference population used to define the limits. An approach that examines baseline and change from baseline is believed by some to provide a more quantitative and individualized measure of ALT elevation [13]. This may be true both for healthy populations as well as for study subjects with underlying liver disease. A shortcoming of the ULN is that the reference population used to establish ULN values may include cases of subclinical liver disease, notably non-alcoholic fatty liver disease (NAFLD). To date, changes from baseline have not often been used in clinical trials and therefore it is difficult to define stopping rules based on appropriate cutoffs. Stopping rules in clinical trials should be based on the extent of experience with the investigational drug or drug class, as well as on the background variability of liver tests in the target population. As patients can typically meet inclusion criteria for viral hepatitis trials with ALT values of up to 10× ULN, defining stopping rules based solely on multiples of upper limit of normal can lead to inconsistent stopping rules. For example, patients with a normal ALT at study entry could be allowed to continue in the study until their ALT reached >10× ULN, while patients entering with an ALT of 8× ULN would have to discontinue with an elevation of only 25 % over their baseline value. The use of a combined approach (Table 1) accounts for the level of elevation of ALT in the context of the patient’s baseline ALT level:
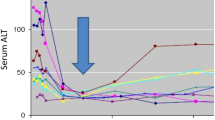
One issue regarding use of the baseline value for normalizing enzyme elevations is that serum ALT will typically fall during effective treatment of hepatitis C (see Fig. 1). Since DILI onset is often delayed by weeks to months, it seems logical that nadir values occurring early in treatment might be the appropriate “baseline” reference point for subsequent elevations. It was pointed out at the workshop that this would be a difficult concept to convey in a study protocol and would probably require real time central monitoring of the data with individualized stopping rules conveyed to the site. Alternatively, decisions regarding treatment modifications would need to be determined centrally and conveyed to the performance site. It was the consensus that systems are not universally in place to allow either approach at this time.

Early fall in serum ALT during effective treatment of viral hepatitis C. Eight subjects with chronic viral hepatitis started treatment with an investigational treatment on day 0, prompting a rapid fall in serum ALT that coincided with a fall in viral count (not shown). The arrow notes a nadir point that could be used to define the baseline values for the purpose of defining ALT elevations that may be drug induced. ALT alanine aminotransferase
Some participants of the Working Group who convened as a follow-up to the Best Practices Workshop in Boston believe that in viral hepatitis trials an ALT >20× ULN should be defined as the stopping rule for patients whose initial value is <5× ULN. While true for hepatitis B virus, several investigators stated they would not be comfortable allowing a hepatitis C virus (HCV) patient with baseline ALT <5× ULN to progress to 20× ULN on active therapy and to continue treatment with the study drug. This degree of ALT elevation is not normally seen in HCV patients unless they receive interferon treatment. If this occurred while on an investigational drug without interferon, the case should be viewed as suspicious of potential DILI. ALT elevations associated with interferon treatment are frequent, even when viral load is suppressed [14]. It will be interesting to see whether this is observed in the new interferon-free direct acting antiviral regimens. The phase II and III data published for the newer antiviral drugs such as telaprevir, boceprevir or sobosfuvir indicate an acceptable hepatic safety profile, however spontaneous reports on ALT elevations typically about 8 weeks after initiating treatment are emerging. Finally, hepatitis virus titres determined before and during therapy should be taken into consideration when defining stopping rules for antiviral treatment.
3 Chronic Viral Hepatitis and HIV Coinfection
The diagnosis of DILI in patients with HIV infection is challenging because of (i) treatment regimens that include potentially hepatotoxic drugs, (ii) a high incidence of underlying liver disease, including coinfection with hepatitis B or C, (iii) liver injury due to ethanol and illicit drug abuse, (iv) steatohepatitis due to insulin resistance, and (v) dyslipidemia caused by certain HIV medications. Moreover, the immune reconstitution that can result from anti-HIV treatment can also cause a flare of liver injury due to an immune attack on hepatocytes chronically infected with viral hepatitis.
HIV trials have some of the highest rates of liver injury. The reported incidence of liver toxicity in HIV patients after initiating highly active antiretroviral therapy (HAART) ranges from 2 to 18 % [15]. Hepatic profile analyses of ritonavir-boosted tipranavir regimens in phase II and III clinical trials showed grade 3/4 transaminase elevations in 11.1 % of patients, with 2.7 % developing hepatic serious adverse events (SAEs) [16]. The risk was greater in patients with underlying liver disease. However, 84 % of patients with grade 3/4 transaminase elevations only temporarily interrupted treatment or continued, with transaminase levels returning to grade ≤2. The nonnucleoside reverse-transcriptase inhibitor nevirapine leads to ALT elevations >5× ULN in 10 % of treated patients, although 6.3 % remain asymptomatic [17]. Among 8,851 subjects enrolled in 16 adult AIDS Clinical Trial Group studies, hepatitis C coinfection was associated with an increased risk of severe hepatotoxicity (ALT or AST >5× ULN or total bilirubin >2.5× ULN) and baseline elevation in ALT or AST was a significant risk factor for severe hepatotoxicity in all regimens [18]. The protease inhibitor atazanavir is an inhibitor of hepatic UGT activity and hyperbilirubinemia >2.5× ULN was significantly associated with genetic variants of the UGT1A1 gene including the variant associated with Gilbert’s disease [19].
4 Oncology Trials
As with the newer antiviral agents, the introduction of novel anticancer agents into the market poses considerable challenges to the regulators with regard to liver safety. For example, the small molecule tyrosine kinase inhibitors (TKIs) offer great therapeutic potential; however, the risk of hepatotoxicity is considerable. 22 such agents have been approved by the US Food and Drug Administration (FDA), 19 of these also by the European Medicines Agency (EMA), and many more are in development or under regulatory review [20]. The HER2/EGFR dual tyrosine kinase inhibitor lapatinib has been associated with hepatotoxicity (including Hy’s law cases) in patients treated for metastatic breast cancer. A pharmacogenetic association with the HLA allele DQA*02:01 confers negative and positive predictive values of 0.97 and 0.17, respectively [21], and this could potentially allow pre-selection of patients likely to experience hepatotoxicity or could be useful in implicating lapatinib in liver injury where multiple etiologies are possible. In addition to lapatinib, the TKIs pazopanib, ponatinib, regorafenib and sunitinib have a boxed hepatotoxicity label warning. Pazopanib-induced hyperbilirubinemia is associated with the UGT1A1 TA7 polymorphism of Gilbert’s syndrome [11]. The management of TKI-induced hepatotoxicity requires an individually tailored reappraisal of the risk versus the benefit of treatment and cannot be based solely on ALT and bilirubin cutoffs.
There has been a major effort within GlaxoSmithKline to mine their aggregate clinical trial data to provide data driven cutoffs for liver safety concern [22, 23]. The aggregated dataset consisted of 3,998 patients identified from 31 phase II and III oncology trials (the GSK historical oncology patient data, GSK-HOPD), and a second dataset of 18,672 patients without liver disease from 28 GSK phase II-IV trials (the generally healthy patient data, GSK-GHPD). Truncated robust multivariate outlier detection (TRMOD) was used to identify thresholds that define outliers for peak serum ALT and bilirubin levels. A false detection probability of 0.001 was used, meaning that 99.9 % of the subjects from an underlying normal distribution are expected to be within the decision boundary, or only 0.1 % of the patients are expected to fall outside of the decision boundary. When this statistical approach was applied to the 18,672 subjects without liver disease (GSK-GHPD), threshold values obtained were 3.4× ULN for ALT and 2.1× ULN for total bilirubin [22]. It is interesting that the thresholds that are proposed in the FDA guidance as “Hy’s Law” criteria (ALT >3× ULN and Bili >2× ULN), which were empirically determined, are essentially identical to the data derived threshold. Applying the same TRMOD approach to liver chemistry data obtained from 3998 subjects in oncology trials [24] resulted in considerably higher thresholds: ALT >5× ULN and total bilirubin >2.7× ULN defined outliers in oncology patients. These thresholds were therefore proposed as suitable limits to define the four quadrants of the eDISH (evaluation of drug-induced serious hepatotoxicity) plot, termed mDISH [24]. When the TRMOD approach was applied to fold baseline ALT and bilirubin data, an ALT limit of 6.9× baseline and a bilirubin limit of 6.5× baseline was calculated from oncology clinical trials (see figure 13 in [25]). Parks and colleagues from GSK emphasize the weakness of employing fold ULN, since only peak values are considered, whereas any information regarding baseline values is disregarded [24]. In their view, fold elevation of baseline rather than ULN provides more sensitivity when identifying liver safety signals.
The mDISH approach has been criticized [23] because the authors may have implied that the modified thresholds alone could be used to define a “Hy’s Law Case” whereas individual case causality assessment is critically important. In addition, the approach relies on all cancers being equivalent whereas differences in subgroups are likely. Nonetheless, all at the workshop agreed that guidelines for liver safety assessment in special populations should be data driven and that the approach taken by Parks and colleagues supported a large scale, precompetitive effort to aggregate the relevant historical and prospectively collected data across the industry and apply innovative statistical approaches to the problem.
5 Conclusions
New approaches are urgently needed to identify liver safety signals in patient populations that exhibit baseline liver chemistry elevations. The use of standardized ALT and bilirubin cutoffs cannot account for the complex pathophysiology that determines phenotype in patients with underlying liver disease. Statistical approaches such as TRMOD can generate hypotheses that require validation in prospective datasets correlating new liver chemistry thresholds with clinical outcomes such as progression to serious liver injury. The limitations of ALT and bilirubin become all the more evident in patients with underlying liver disease, in whom deranged signaling mechanisms require innovative biomarkers for the assessment of prognosis. All these requirements underline the need for creating a novel liver safety research consortium, which combines clinical data from registration trials with biomarker, genetic and metabolomic data from appropriate patient cohorts. This will pave the way for defining the next-generation liver safety criteria required for accurate assessment of DILI in special populations.
References
Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125(6):1714–22.
Weil JG, Bains C, Linke A, Clark DW, Stirnadel HA, Hunt CM. Background incidence of liver chemistry abnormalities in a clinical trial population without underlying liver disease. Regul Toxicol Pharmacol. 2008;52(2):85–8. doi:10.1016/j.yrtph.2008.06.001.
Zimmerman HJ. Drug-induced liver disease. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver. 1st ed. New York: Appleton-Century-Crofts; 1978. p. 351–3.
Torriani FJ, Rodriguez-Torres M, Rockstroh JK, Lissen E, Gonzalez-Garcia J, Lazzarin A, et al. Peginterferon Alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med. 2004;351(5):438–50. doi:10.1056/NEJMoa040842.
Hadziyannis SJ, Sette H Jr, Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140(5):346–55.
Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–82. doi:10.1056/NEJMoa020047.
Shiffman ML, Suter F, Bacon BR, Nelson D, Harley H, Sola R, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med. 2007;357(2):124–34. doi:10.1056/NEJMoa066403.
Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335–74. doi:10.1002/hep.22759.
Rotger M, Taffe P, Bleiber G, Gunthard HF, Furrer H, Vernazza P, et al. Gilbert syndrome and the development of antiretroviral therapy-associated hyperbilirubinemia. J Infect Dis. 2005;192(8):1381–6. doi:10.1086/466531.
Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333(18):1171–5. doi:10.1056/NEJM199511023331802.
Xu CF, Reck BH, Xue Z, Huang L, Baker KL, Chen M, et al. Pazopanib-induced hyperbilirubinemia is associated with Gilbert’s syndrome UGT1A1 polymorphism. Br J Cancer. 2010;102(9):1371–7. doi:10.1038/sj.bjc.6605653.
Green RM, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123(4):1367–84.
Cai Z, Christianson AM, Stahle L, Keisu M. Reexamining transaminase elevation in Phase I clinical trials: the importance of baseline and change from baseline. Eur J Clin Pharmacol. 2009;65(10):1025–35. doi:10.1007/s00228-009-0684-x.
Basso M, Giannini EG, Torre F, Blanchi S, Savarino V, Picciotto A. Elevations in alanine aminotransferase levels late in the course of antiviral therapy in hepatitis C virus RNA-negative patients are associated with virological relapse. Hepatology. 2009;49(5):1442–8. doi:10.1002/hep.22810.
Nunez M. Hepatotoxicity of antiretrovirals: incidence, mechanisms and management. J Hepatol. 2006;44(1 Suppl):S132–9. doi:10.1016/j.jhep.2005.11.027.
Mikl J, Sulkowski MS, Benhamou Y, Dieterich D, Pol S, Rockstroh J, et al. Hepatic profile analyses of tipranavir in Phase II and III clinical trials. BMC Infect Dis. 2009;9:203. doi:10.1186/1471-2334-9-203.
Dieterich DT, Robinson PA, Love J, Stern JO. Drug-induced liver injury associated with the use of nonnucleoside reverse-transcriptase inhibitors. Clin Infect Dis. 2004;38(Suppl 2):S80–9. doi:10.1086/381450.
Servoss JC, Kitch DW, Andersen JW, Reisler RB, Chung RT, Robbins GK. Predictors of antiretroviral-related hepatotoxicity in the adult AIDS Clinical Trial Group (1989–1999). J Acquir Immune Defic Syndr. 2006;43(3):320–3. doi:10.1097/01.qai.0000243054.58074.59.
Lankisch TO, Moebius U, Wehmeier M, Behrens G, Manns MP, Schmidt RE, et al. Gilbert’s disease and atazanavir: from phenotype to UDP-glucuronosyltransferase haplotype. Hepatology. 2006;44(5):1324–32. doi:10.1002/hep.21361.
Shah DR, Dholakia S, Shah RR. Effect of tyrosine kinase inhibitors on wound healing and tissue repair: implications for surgery in cancer patients. Drug Saf. 2014;37(3):135–49. doi:10.1007/s40264-014-0139-x.
Spraggs CF, Budde LR, Briley LP, Bing N, Cox CJ, King KS, et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol. 2011;29(6):667–73. doi:10.1200/JCO.2010.31.3197.
Lin X, Parks D, Painter J, Hunt CM, Stirnadel-Farrant HA, Cheng J, et al. Validation of multivariate outlier detection analyses used to identify potential drug-induced liver injury in clinical trial populations. Drug Saf. 2012;35(10):865–75. doi:10.2165/11632670-000000000-00000.
Senior JR. Why the threshold criteria should not be modified for detection of possibly serious drug-induced hepatotoxicity in special groups of trial subjects. Pharmacoepidemiol Drug Saf. 2013;22(6):579–82. doi:10.1002/pds.3435.
Parks D, Lin X, Painter JL, Cheng J, Hunt CM, Spraggs CF, et al. A proposed modification to Hy’s law and Edish criteria in oncology clinical trials using aggregated historical data. Pharmacoepidemiol Drug Saf. 2013;22(6):571–8. doi:10.1002/pds.3405.
Merz M, Lee K, Kullak-Ublick GA, Brueckner A, Watkins P. Methodology to assess clinical liver safety data. Drug Saf. 2014 (this issue).
Acknowledgements
The Innovative Medicines Initiative and the Hamner-University of North Carolina Institute for Drug Safety Sciences sponsored the workshop, part of which is summarized in this article. This article is part of a supplement entitled Liver Safety Assessment in Clinical Drug Development: A Best Practices Workshop report, which was guest edited by Drs. Paul B. Watkins, Michael Merz and Mark I. Avigan. The guest editing by Dr. Avigan does not reflect the position of, nor imply endorsement from, the US Food and Drug Administration or the US Government. Drs. Watkins, Merz and Avigan did not receive any honoraria for guest editing the supplement. All manuscripts were peer reviewed by Dr. Rolf Teschke. Dr. Rolf Teschke has no conflicts of interest to declare and did not receive any honoraria for peer reviewing the supplement; however, he received a free yearly online subscription to the journal Drug Safety.
The Innovative Medicines Initiative (http://www.imi.europa.eu/) is a public-private partnership set up by the European Commission in 2008 to relieve the bottlenecks in drug development and to provide economic stimulus. With a €2 billion commitment, the IMI now has an important portfolio of projects where experts from academia, industry and regulatory bodies collaborate on an unprecedented scale and at a non-competitive level to develop tools and technologies. Drug-induced liver injury has been a focus of several projects including the SAFE-T (Safer and Faster Evidence-based Translation) consortium, which is working on clinical qualification of new biomarkers to better detect and characterize liver toxicity, and MIP-DILI, which is working to determine the optimal preclinical testing to detect potential of liver injury in patients.
The Hamner-University of North Carolina Institute for Drug Safety Sciences (IDSS—http://www.thehamner.org/idss/), located in Research Triangle Park, NC, is dedicated to solving drug safety challenges through a variety of innovative approaches including mouse genetics, mechanistic biomarkers, and culture models derived from induced pluripotent stem cells. Efforts in drug-induced liver injury include the DILI-sim Initiative, a public-private partnership developing computer models to explain and predict drug-induced liver injury.
Authors thank Dr. Michele Bortolini, Dr. Jerry O. Stern, Dr. Christine Hunt, Dr. Yongyu Wang and Dr. Dominique Larrey for important contributions and discussions.
Conflict of interest
Authors Gerd A. Kullak-Ublick, Michael Merz, Louis Griffel, Neil Kaplowitz and Paul B. Watkins declare no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Kullak-Ublick, G.A., Merz, M., Griffel, L. et al. Liver Safety Assessment in Special Populations (Hepatitis B, C, and Oncology Trials). Drug Saf 37 (Suppl 1), 57–62 (2014). https://doi.org/10.1007/s40264-014-0186-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0186-3