
Abstract
In multiple sclerosis (MS) persisting disability can derive from acute relapses or, alternatively, from slow and steady deterioration, termed chronic progression. Emerging data suggest that the latter process occurs largely independent from relapse activity or development of new central nervous system (CNS) inflammatory lesions. Pathophysiologically, acute relapses develop as a consequence of de novo CNS infiltration of immune cells, while MS progression appears to be driven by a CNS-trapped inflammatory circuit between CNS-established hematopoietic cells as well as CNS-resident cells, such as microglia, astrocytes, and oligodendrocytes. Within the last decades, powerful therapies have been developed to control relapse activity in MS. All of these agents were primarily designed to systemically target the peripheral immune system and/or to prevent CNS infiltration of immune cells. Based on the above described dichotomy of MS pathophysiology, it is understandable that these agents only exert minor effects on progression and that novel targets within the CNS have to be utilized to control MS progression independent of relapse activity. In this regard, one promising strategy may be the inhibition of the enzyme Bruton's tyrosine kinase (BTK), which is centrally involved in the activation of B cells as well as myeloid cells, such as macrophages and microglia. In this review, we discuss where and to what extent BTK is involved in the immunological and molecular cascades driving MS progression. We furthermore summarize all mechanistic, preclinical, and clinical data on the various BTK inhibitors (evobrutinib, tolebrutinib, fenebrutinib, remibrutinib, orelabrutinib, BIIB091) that are currently in development for treatment of MS, with a particular focus on the potential ability of either drug to control MS progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Multiple sclerosis (MS) progression is assumed to be driven by a central nervous system (CNS)-intrinsic inflammatory interplay of chronically activated CNS-resident cells and CNS-trapped hematopoietic immune cells. |
This process substantially differs from MS relapse biology, and, accordingly, all agents designed to control relapses basically failed to control MS progression independent of acute inflammation. |
New targets within the CNS driving MS progression have to be discovered and harnessed therapeutically. |
Inhibition of Bruton's tyrosine kinase (BTK) may be one promising approach to control MS progression. BTK is centrally involved in the activation of immune cells such as B cells, but also in the chronic activation of microglia. |
1 Introduction
Multiple sclerosis (MS) is the most common chronic inflammatory demyelinating disease of the central nervous system (CNS). The majority of patients present a relapsing-remitting (RRMS) course of the disease and, over time, many transition to a progressive disease, termed secondary progressive MS (SPMS). Some patients develop slow and continuous neurological deterioration from onset without definable relapses, termed primary progressive MS (PPMS). The course of progressive MS is defined by steadily increasing neurological disability independent of relapses, with further classification of activity and progression. According to this classification, activity is determined by clinical relapses, assessed by magnetic resonance imaging (MRI), whereas progression is defined as an increase in clinical deterioration independent of ongoing inflammation [1].
While there are a high number of disease-modifying drugs approved for the treatment of RRMS, they remain partially or completely ineffective in SPMS or PPMS, collectively referred as progressive MS (PMS).
The first approved therapy for SPMS in 2000 by the US Food and Drug Administration (FDA) was mitoxantrone, a DNA-intercalating agent used in cancer treatment [2, 3]. However, use of mitoxantrone is limited due to cardiotoxicity and increased rates of hematological malignancies [4, 5]. In 2019 the FDA approved cladribine and siponimod for the treatment of active SPMS [6]. Cladribine is a DNA synthesis inhibitor that selectively reduces lymphocytes in the periphery through induction of cell death [7]. Since it is a small molecule, it is able to cross the blood-brain barrier (BBB) and is detectable in the cerebrospinal fluid (CSF) [7]. Considering the fact that cladribine is only given a few treatment days per year and the estimated terminal half-life of cladribine is only 1 day [8, 9] it remains unclear if the drug causes long-lasting changes in the absence of ongoing direct drug-related effects. Siponimod is a sphingosine-1-phosphate (S1P) receptor modulator with high affinity for S1PR1 and S1PR5, retaining lymphocytes within the lymph nodes, thus decreasing their entry into the CNS [10]. Because of the small size and lipophilic nature of siponimod, it can efficiently cross the BBB [11]. Since S1P receptors are expressed on CNS-resident cells, including astrocytes, microglia, and oligodendrocytes, an effect of siponimod on CNS-resident cells is possible [12].
To date the only approved drug for the treatment of PPMS is the CD20 B cell-depleting antibody ocrelizumab [13, 14]. CD20 is expressed on B cells across different stages of maturation, ranging from pre-B cells in the bone marrow to short-lived plasmablasts, while long-lived, antibody-producing plasma cells completely downregulate CD20 expression [15, 16]. Ocrelizumab significantly reduced the risk of disability and rate of brain atrophy as compared to the placebo group in a phase III study of PPMS patients [17]. However, the clinical benefit of ocrelizumab treatment in this trial was only moderate. As the results of the study revealed that the effect of ocrelizumab was higher in younger patients and patients with increased disease activity [17], a B cell-depleting therapy might be most efficient in disease stages characterized by acute inflammation. Since there is no current evidence to suggest that ocrelizumab can penetrate the CNS, it remains questionable if the antibody acts on CNS-established B cells.
Although RRMS and PMS have many similarities, the development of efficacious therapies for PMS has been hindered by a number of factors. In particular, the clinical definition and diagnosis of PMS has been a challenging concept, and only recently was defined as clinical worsening of disability independent of clinical relapses [1]. Based on this, the development of effective drugs for the treatment of PMS is challenging since specific surrogates and clinical outcome measurements are lacking [18]. Furthermore, the lack of known biological targets specific for PMS hinders the development of efficient drugs. The mechanisms leading to relapses are well investigated; however, understanding of the pathological mechanisms of PMS is still limited. The current understanding includes a broad range of processes including inflammation and neurodegeneration. Therefore, the understanding of the pathomechanisms of PMS, the identification of specific targets, and a more refined clinical trial design will likely result in the expansion of treatment options for progressive MS patients.
In this review, we summarize the current understanding of the pathomechanisms of PMS as well as review recent advances in the treatment of progressive MS with a focus on Bruton's tyrosine kinase (BTK) inhibition. Although inhibition of BTK is also considered a promising therapeutic target for RRMS, we focus on the potential of BTK inhibition for the treatment of PMS. Relevant studies were identified based on a PubMed search using the terms “multiple sclerosis,” “progressive multiple sclerosis,” “progression of multiple sclerosis,” “BTK,” “BTKi,” “evobrutinib,” “tolebrutinib,” “remibrutinib,” orelabrutinib,” “fenebrutinib,” “BIIB091,” and clinicaltrial.gov regarding MS trials.
2 Pathology and Pathomechanisms of Progressive Multiple Sclerosis
To therapeutically target PMS, it is important to understand key pathological processes driving the disease. Several mechanisms have been proposed to drive PMS, including processes that are dependent on inflammatory lesions and processes that are independent of lesions. These processes include inflammation and neurodegeneration. Inflammation in PMS is understood to occur behind an intact or repaired BBB with continuous involvement of CNS-established hematopoietic cells and the involvement of CNS-resident cells [19, 20]. Infiltrating hematopoietic cells includes B cells, T cells, and myeloid cells, which are found in the leptomeninges and blood vessels of the CNS [21]. The degree of hematopoietic infiltrates found within the meninges is directly correlated with the degree of axonal loss in the normal-appearing white matter (NAWM) [22, 23]. It is likely that demyelination and neurodegeneration may be driven by soluble factors produced in the meningeal inflammatory infiltrates. Indeed, subpial cortical lesions are frequently found near B cell follicles [24]. The presence of oligoclonal bands in the CSF of MS patients as well as B cell follicular structures in patients with SPMS, which correlate with a higher rate of disability and disease progression, support the notion that B cells may play a major role in PMS [24]. B cell functions that could be relevant in PMS include antibody production, cytokine secretion, antigen-presentation, and ectopic formation of follicle-like structures [25, 26]. Since the inflammation is supposed to be CNS compartmentalized, it may be possible that CNS-established B cells are protected from drugs that cannot efficiently cross the BBB. Importantly, in MS it was found that B cell clones present in the CSF are also represented in the blood in both early and later disease stages [27] and that IgG representing the oligoclonal bands are linked to circulating peripheral B cells [28]. These data demonstrate that B cells circulate between the CNS and peripheral compartments, and therefore treatments affecting B cells in the periphery might have consequences for those that populate the CNS.
Chronic active, also called smoldering, lesions are most common in patients with PMS and are typified by a rim of iron-rich activated microglia and macrophages, cells that seem to drive the expansion of established lesions [23]. Importantly, many studies do not differentiate between these cells and only recently microglia could be distinguished through the marker TMEM119 and were found to be predominantly present in slowly expanding lesions (SELs) [29, 30]. SELs are characterized by a hypomyelinated core and a rim containing iron-rich cells, which represent a subgroup of chronic active lesions that show gradual expansion over time [31].
Microglia contribute to a chronic inflammatory milieu through multiple ways: Their activation results in increased production of cytokines and chemokines as well as reactive oxygen species (ROS) and reactive nitrogen species (RNS) [32]. Additionally, they can act as antigen-presenting cells and phagocytose myelin debris [33, 34]. These mechanisms not only contribute to the inflammatory component of the disease but also drive neurodegenerative processes. The release of ROS and RNS contributes to mitochondrial and axonal damage, and this finally leads to degeneration of neurons, axons, and oligodendrocytes [35]. The contention that microglia activation plays a major role in progressive disease forms was further strengthened in a study that investigated microglia activity by positron emission tomography (PET) of the mitochondrial translocator protein TSPO in RRMS and PMS patients [36]. This study showed that microglia activity correlates with disease disability and prognosis in PMS patients, but not in RRMS patients. However, it needs to be considered that TSPO is also detectable in astrocytes and macrophages [37, 38].
Additionally, PMS patients show diffuse white matter abnormalities in the NAWM, consisting of inflammatory infiltrates, mainly CD8+ T cells, diffuse axonal injury, reactive astrocytic scarring, and microglia activation [23]. Failure of regeneration and remyelination is supposed to be one reason behind the transition from RRMS to SPMS, since remyelinated shadow plaques are present in RRMS patients but are lacking in progressive MS patients [23]. One possibility why remyelination is mostly lacking in PMS may be impaired repair mechanisms. The mechanisms of myelin repair are not fully understood; however, reduced repair and impaired axonal regeneration are related to age and a lifelong oxidative stress environment and especially oligodendrocytes have been shown to be sensitive to oxidative stress, leading to an exhaustion in myelination capacity [39]. Reactive astrogliosis is present in the NAWM and white and grey matter in MS patients, leading to continuous production of pro-inflammatory factors, ROS, and RNS [40]. These events contribute to neuroaxonal and mitochondrial damage and can thereby increase neurodegeneration.
These multifactorial processes associated with PMS present several biological targets and pathways for potential therapeutic intervention. However, as these processes occur within a compartmentalized CNS, immunomodulatory agents must be capable of entering the CNS to counteract detrimental neuroinflammation. Many agents are currently tested in phase II and III trials that have more than one potential mechanism of action that could benefit PMS. One auspicious target is the inhibition of the enzyme Bruton’s Tyroisne Kinase (BTK). Since BTK inhibition is already approved for the treatment of B cell malignancies, it was first investigated as an improvement for the B cell-based therapy in MS [41]. As it was recognized that BTK also mediates the activity of macrophages and microglia [42], it gained importance for the treatment of PMS. Therefore, BTK inhibition provides dual approaches to disarm both the lymphocyte and microglia/macrophage cells that are dysregulated in MS.
3 Bruton's Tyrosine Kinase (BTK) Expression and Signaling Pathways
Bruton's Tyrosine Kinase (BTK) is part of the Tec (tyrosine kinase expressed in hepatocellular carcinoma) family of kinases, consisting of Tec, BTK, ITK (interleukin (IL) 2-inducible T-cell kinase), BMX (bone-marrow tyrosine kinase gene on the X chromosome), and RLK (resting lymphocyte kinase). Structurally very similar, members of the Tec family possess a pleckstrin homology (PH) domain at the N-terminus (except for RLK). Adjacent lies a short Tec homology (TH) domain that is formed by one or two proline-rich regions (PRR) in combination with the BTK homology (BH) domain. The C-terminus is formed by Src homology (SH) 2 as well as SH3 domains and a catalytic kinase domain [41, 43, 44].
Most cells of the immune system express at least one member of the Tec family of kinases. Importantly, the expression of BTK itself is not restricted to a single cell type but spreads across a number of cells of hematopoietic origin, and furthermore extends to other immune cells such as microglia [42, 45]. Within the hematopoietic lineage, the expression of BTK has been confirmed in B cells, dendritic cells, mast cells, neutrophils, macrophages, platelets, erythrocytes, and hematopoietic stem cells [43, 46,47,48].
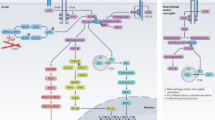
Similar to its expression in multiple cell types, BTK plays a role in the signaling cascades of a variety of immunologically relevant receptors, including the B cell receptor (BCR), toll-like receptors (TLRs), Fc receptors (FcRs), and chemokine receptors (Fig. 1). Most prominently, the engagement and aggregation of the BCR leads to conformational changes that activate a Lck/Yes novel tyrosine kinase (Lyn)-mediated phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs). This generates a signaling complex consisting of Syk, Lyn, the adaptor molecules B cell linker protein (BLNK) and Cbl-interacting protein of 85 kDa (CIN85), phospholipase C-gamma 2 (PLC-γ2), and BTK. PLC-γ2 then mediates the cleavage of the plasma membrane lipid phosphatidylinositol-4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3). IP3 initiates the release of calcium from the endoplasmic reticulum (ER), which in turn activates transcription factors, such as nuclear factor “kappa-light-chain-enhancer” of activated B cells (NF-κB) and nuclear factor of activated T cells (NFAT). NF-κB induces B cell proliferation and can induce immunoglobulin class switching. NFAT stimulates immune cell effector functions, for example by initiating cytokine production and release. Similar to IP3, DAG can act as a second messenger and activate protein kinase C (PKC) and, together with Syk, engage the mitogen-activated protein kinase (MAPK) pathway and increase survival and accelerate proliferation [47, 49, 50]. Its prominent place in the BCR signaling cascade, relaying incoming, extracellular signals into cellular responses, highlights the importance of the role of BTK in B cell function.

Signaling pathways involving Bruton’s tyrosine kinase (BTK). BTK is placed within the pathways of the B cell receptor (BCR), the Fc receptor (FcR), as well as in the Toll-like receptor (TLR) and chemokine receptor cascades. It has a central place in relaying extracellular signaling to downstream pathways such as the nuclear factor “kappa-light-chain-enhancer” of activated B cells (NFkB), nuclear factor of activated T cells (NFAT), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK) cascades. Solid arrows correspond to strong, direct involvement of BTK whereas dotted arrows indicate a weaker involvement. ITAM immunoreceptor tyrosine-based activation motif
Besides the well-studied relevance of BTK in the BCR signaling cascade, it is also involved in the signaling of Fc receptors, and therefore the recognition of specific antigens by immunoglobulins. Because BTK is placed within the signaling pathways of both activating and inhibiting FcRs, its involvement can amplify or attenuate the induced signals [46, 51].
Additionally, BTK plays a role in the recognition of damage-associated molecular patterns (DAMPs) and/or pathogen-associated molecular patterns (PAMPs), which is critical in the early detection of dangers such as infections or damage. These signals are identified by cells using TLRs. BTK has been described to play a role in TLR2, TLR3, TLR4, TLR7/8, and TLR9 signaling. BTK influences the TLR pathways through interactions with myeloid differentiation primary response 88 (MYD88), a central player of all TLR cascades (with the exception of TLR3). Additionally, interactions of BTK with Toll/IL-1 receptor domain containing adaptor protein (Tirap or MAL) as well as interleukin-1 receptor-associated kinase 1 (IRAK-1) can bridge BTK to TLR-induced signal transduction [52,53,54,55]. Conclusively, BTK plays a role in the recognition and activation of immune receptors through BCR, FcR, and TLR signals. However, BTK also regulates the recruitment of immune cells by facilitating the signals of chemokine receptors. For example, the stimulation by CXCL-12 induces the activation of BTK [41]. Additionally, the expression and function of CXCR4, a chemokine receptor that regulates key B cell functions such as trafficking into lymphoid structures, is regulated by a complex mechanism involving BTK [47, 56].
Taken together, because of the expression of BTK in multiple cells and its involvement in various signaling cascades, the effects of BTK inhibition depend on the distinct cellular environment and context. However, with the exception of inhibiting FcRs, BTK inhibition will block the relay of mainly pro-inflammatory signals, therefore an inhibition will primarily attenuate immune responses. To what extent this can be compensated over time and how continuous exposure to BTK inhibition changes the composition (maturation and differentiation) of immune cell subsets still needs to be addressed.
4 Experimental Data in Multiple Sclerosis (MS) Models
So far, six BTK inhibitors—evobrutinib, tolebrutinib, fenebrutinib, remibrutinib, orelabrutinib, and BIIB091—have been tested for the treatment of MS patients [57]. While all these inhibitors are small molecules, they differ in their distinct characteristics. According to their mode of action and based on their mode of binding to BTK, the inhibitors can be classified into two types: irreversible inhibitors (evobrutinib, tolebrutinib, remibrutinib, orelabrutinib), which form a covalent bond with the amino acid residue Cys481 in the ATP-binding site of BTK [50, 58,59,60], and reversible inhibitors (fenebrutinib and BIIB091), which bind to specific pockets in the SH3 domain by weak, reversible forces (hydrogen bonds or hydrophobic interactions), inducing an inactive conformation of the kinase [61,62,63].
The binding mode of the inhibitor defines the potency of the molecule [58]. Covalent binding increases the potency of the drug; however, non-covalent binding is a weaker binding and therefore decreases potency, but also toxicity and risks associated with chronic use [50]. Targeting of the Cys481 shows high selectivity, as only ten other kinases contain the same region of the receptor [58]. Binding to the SH3 pocket is even more selective since only three kinases were inhibited off-target by fenebrutinib [61], and BIIB091 only inhibited one off-target kinase [62]. Importantly, a mutation in the Cys481 region can lead to a reduction in the compound potency and might reduce the therapeutic efficacy in patients with these mutations, which was found in patients on BTK inhibitors experiencing cancer relapses [64].
Recently published and early results suggest that due to molecular size and properties, the inhibitors are able to cross the BBB [65,66,67], but preliminary evidence shows that the inhibitors differ in their capacity to efficiently cross the BBB and accumulate within the CNS [68]. A study presented at a scientific meeting compared the CNS exposure and potency of evobrutinib, tolebrutinib, and fenebrutinib. All three agents, administrated as a single oral dose of 10 mg/kg daily, achieved similar CSF concentration in cynomolgus monkeys. Tolebrutinib CSF concentrations exceeded the estimated IC90, while both evobrutinib and fenebrutinib failed to reach exposure levels approaching their IC90 value [68]. Specifically, their brain penetrance underlines the reasoning for their respective positioning as either sole peripheral drugs used for RMS patients or compounds that are also investigated for their potential in progressive disease forms [50].
The inhibitors were analyzed in various models and cell types: The inhibitor evobrutinib demonstrated beneficial immunological effects in the EAE (experimental autoimmune encephalomyelitis) mouse model in vivo [69]. Evobrutinib inhibited antigen-triggered activation and maturation of B cells and the release of pro-inflammatory cytokines. Furthermore, evobrutinib impaired the capacity of B cells to act as antigen-presenting cells for the development of encephalitogenic T cells. This ultimately reduced CNS infiltration and ameliorated clinical and histological EAE [69]. Since BTK is also expressed in myeloid cells, evobrutinib was further investigated in human-derived macrophages in vitro [70]. Inhibition with evobrutinib hindered pro-inflammatory macrophage differentiation and promoted differentiation towards an anti-inflammatory macrophage phenotype [70]. Another study showed that treatment of evobrutinib favored remyelination in both ex vivo mouse cerebellar slices and in vivo transgenic Xenopus tadpoles [48]. Importantly, both models of demyelination are independent of adaptive immunity, and immunohistochemistry revealed that most cells expressing BTK are microglia, in which the expression of BTK increased upon demyelination [48].
BIIB091 was shown to block human and mouse B cell activation, proliferation, and differentiation in vitro [71]. Furthermore, it was shown that BII091 blocks myeloid cell function in vitro. In depth, pre-treatment with BII091 blocked ROS production in human neutrophils, human basophil degranulation, and TNF-α production of human monocytes [71]. Furthermore, a number of preliminary data presented at scientific meetings indicate the mode of action of BTK inhibition [66, 72,73,74,75]. The effect of evobrutinib was investigated in vitro in primary murine microglia upon cytokine and TLR stimulation, and revealed a reduced microglial activation. Furthermore, in vivo in a passive EAE model evobrutinib was given as pre-treatment and induced a downregulation of disease-associated molecules involved in activation and antigen-presentation on microglial cells [72] (Fig. 2).

Proposed central nervous system (CNS) effects of Bruton's tyrosine kinase (BTK) inhibitors. The inhibition of BTK is thought to (a) block the activation of microglial cells, (b) promote the remyelination of axons by affecting oligodendrocytes and their precursors, and (c) prevent the extravasation of peripheral immune cells into the CNS. Green arrows indicate a promoting effect of BTK inhibitors whereas red symbols correspond to inhibitory effects
Tolebrutinib reduced EAE severity and blocked BCR-mediated activation of B cells as well as Fc-receptor activation of macrophages and microglia cells in vitro [66]. Tolebrutinib was further investigated in the in vivo cuprizone model, a model of toxic demyelinating, and inhibition of BTK revealed a benefit by decreased myelin loss [73]. Fenebrutinib, given as pretreatment in vivo in the EAE model, was shown to reduce mean clinical signs, which was associated with reduced microglial activation [76]. As the inhibitors were often given as a preventive treatment, it remains to be evaluated if the effect is associated with inhibition of disease development or to what extent BTK inhibitors exert a direct effect on microglia. The described studies indicate that BTK inhibitors potentially have a dual mode of action targeting both the innate and the adaptive immune response.
5 Clinical Trials of BTK Inhibitors in MS
Two of the aforementioned BTK inhibitors have so far completed testing in RMS patients, evobrutinib and tolebrutinib. Evobrutinib, which binds BTK in a covalent manner, demonstrated significantly fewer enhancing lesions through 24 weeks of observation in a phase II trial testing against dimethyl fumarate (DMF) and placebo (NCT02975349) [77]. There was no significant effect of evobrutinib on the annualized relapse rate (ARR) or disability progression [78]. However, during the open-label extension of this study, an improvement of the ARR in evobrutinib-treated patients was maintained through week 108. Additionally, analysis of the probability and time to first qualified relapse (QR) favored patients initiated on a high dose of evobrutinib in comparison to lower doses or placebo [79]. In the planned phase III clinical trials, EVOLUTION RMS 1/2 (NCT04338022/NCT04338061), recruiting 930 patients, the efficacy and safety of evobrutinib will be compared to teriflunomide through a 96-week treatment period. In addition to ARR as the primary endpoint, safety, various MRI parameters, as well as the time to first occurrence of 12- and 24-week confirmed disability progression, will be analyzed [80, 81].
The inhibitor tolebrutinib has demonstrated a dose-dependent reduction of gadolinium-enhancing lesions in a 16-week phase IIb clinical trial involving 125 participants with RMS (NCT03996291). The investigated doses ranged from 5 to 60 mg, the highest dose being the most efficacious. All tolebrutinib doses were well tolerated [82]. Tolerability was confirmed in the open-label extension (OLE) up to 48 weeks of observation [83, 84]. Additionally, tolerability of tolebrutinib was confirmed in healthy volunteers. In this study, up to 120 mg single exposure or 90 mg repeated dosing of tolebrutinib was well tolerated and showed a high level of BTK coverage in the periphery [68]. In the upcoming phase III GEMINI 1/2 trials in RMS (NCT04410978/NCT04410991), tolebrutinib will be tested against teriflunomide with regard to efficacy to reduce the relapse rate (primary objective) as well as disability progression, MRI lesions, cognitive performance, and quality of life (secondary objectives) [85, 86]. In two upcoming trials, tolebrutinib will be tested separately in SPMS (1,290 participants, NCT04411641) and PPMS (990 participants, NCT04458051). Both trials—HERCULES in SPMS and PERSEUS in PPMS—will primarily analyze tolebrutinib´s effects on delaying disability progression. The secondary objectives are MRI lesions, cognitive performance, physical function, and quality of life [87, 88]. Lastly, safety, tolerability, and pharmacodynamics will also be analyzed in all trials. Recently, the FDA placed a partial hold on tolebrutinib phase III clinical trials over concerns of drug-induced liver injuries. Importantly, most affected subjects had a historical predisposition for liver injury and laboratory values for tracking liver injuries were found to be reversible upon tolebrutinib cessation. Currently, subject enrolment is progressing with updated trial protocols and improved safety monitoring [89, 90].
The other compounds have so far not been tested in MS patients but completed clinical trials for another indication or are currently recruiting or will soon enter clinical testing in MS. Fenebrutinib, a non-covalent BTK inhibitor, recently entered phase III clinical trials in MS patients. So far, in phase II studies in rheumatoid arthritis (NCT02833350) [89] and systemic lupus erythematosus (NCT02908100) [90], fenebrutinib has demonstrated a very high potency and a safety profile consistent with alternative immunomodulatory treatments [91, 92]. Importantly, there was no evidence of an increased risk of infections [75, 76, 91, 92]. In RMS patients, the FENhance study (NCT04586023/NCT04586010) will now directly compare fenebrutinib, teriflunomide, and placebo, and evaluate the efficacy and safety of fenebrutinib on disability progression as well as relapse rate in 736 participants [93]. Furthermore, the FENopta trial (NCT05119569) will include 102 RMS patients to assess the effect of fenebrutinib on brain MRI [new gadolinium (Gd)-enhancing lesions] as well as safety and pharmacokinetics [94].
Because BTK inhibitors possess the ability to enter and accumulate within the CNS, even when the BBB remains closed, this is a highly promising approach in progressive disease forms. Therefore, several of the developed BTK inhibitors will also enter clinical testing for their efficacy in progressive MS forms. Fenebrutinib will be tested in 946 PPMS patients for safety as well as efficacy on disease progression [95]. In this trial, FENtrepid (NCT04544449), fenebrutinib will be compared to ocrelizumab, which has so far demonstrated a mild clinical benefit when administered to PMS patients [17, 96].
While remibrutinib has not completed clinical trials in MS, it was well tolerated in healthy subjects and asymptomatic atopic diathesis patients [97]. This highly selective BTK inhibitor is currently recruiting 800 RMS patients for a phase III clinical trial (NCT05147220/NCT05156281). Compared against teriflunomide, this study will focus on the ARR (primary outcome) as well as confirmed disability progression, neurofilament light chain levels, new Gd-enhancing lesions and physical function [98].
Two more BTK inhibitors have been developed and will be tested for their efficacy in RMS. A study using orelabrutinib is currently recruiting 160 RRMS patients and will analyze the formation of new Gd-enhancing lesions as well as the ARR (NCT04711148) [50, 99]. Lastly, BIIB091 is a reversible BTK inhibitor developed for the treatment of MS that has so far demonstrated the ability to block B cell-activation in healthy subjects (NCT03943056) [62, 71, 100].
Taken together, the high number of inhibitors targeting BTK that are currently tested or being developed for the treatment of MS patients highlight the potential that is seen in this therapeutic approach. Furthermore, the ability of BTK inhibitors to enter and accumulate within the CNS unravels novel treatment options, especially in progressive disease forms. A detailed overview on the completed as well as planned/ongoing clinical trials can be found in Table 1.
6 Conclusion
MS progression is assumed to be driven largely by a CNS-intrinsic inflammatory interplay of chronically activated CNS-resident cells, such as microglia and CNS-trapped hematopoietic immune cells. Other factors such as mitochondria injury, oxidative stress, or iron neurotoxicity likely contribute to chronic progression in MS [101]. This pathophysiological cascade is substantially different from MS relapse biology, and accordingly, all agents designed to control de novo CNS infiltration of immune cells basically failed to control MS progression independent of CNS lesion formation and acute relapses. Within the next decade, new targets within the CNS driving MS progression have to be discovered and harnessed therapeutically. BTK may be one such target, as it is centrally involved in the activation of immune cells such as B cells, but also in the chronic activation of microglia. The various currently developed BTK inhibitors are all relatively small molecules with a respective ability to cross the BBB. Experimental data suggest that BTK inhibition of cells within the CNS may downregulate chronic inflammation and thereby ameliorate processes associated with chronic progression. Based on these data, several BTK inhibitors are in clinical development both for relapsing forms of MS as well as for PMS. Results from these trials are expected with high anticipation and will be communicated soon.
In perspective, these trials will doubtlessly reveal whether and to what extent BTK inhibition may be a valuable approach to therapeutically counteract MS progression. In view of the urgent therapeutic need in PMS, some of these trials will likely generate an overall positive result. In that case, it would of course be of great interest which of the investigated molecules exerts the best clinical benefit at the lowest risk profile. Ideally, a direct comparative study testing BTK inhibitors head-to head in progressive MS would generate these results. Further, since MS relapse biology and continuous progression of MS are nowadays recognized to involve differential immunological and molecular cascades in different compartments, a clear assessment of BTK inhibition to control MS progression in the absence of focal CNS inflammation may pave the way into a new era of combination therapy in MS [5], one drug preventing relapse development and the other agent controlling MS progression.
Change history
23 November 2022
Missing Open Access funding information has been added in the Funding Note.
References
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Stuve O, Kita M, Pelletier D, Fox RJ, Stone J, Goodkin DE, et al. Mitoxantrone as a potential therapy for primary progressive multiple sclerosis. Mult Scler. 2004;10(Suppl 1):S58-61.
Jain KK. Evaluation of mitoxantrone for the treatment of multiple sclerosis. Expert Opin Investig Drugs. 2000;9(5):1139–49.
Kingwell E, Koch M, Leung B, Isserow S, Geddes J, Rieckmann P, et al. Cardiotoxicity and other adverse events associated with mitoxantrone treatment for MS. Neurology. 2010;74(22):1822–6.
Costello F, Stuve O, Weber MS, Zamvil SS, Frohman E. Combination therapies for multiple sclerosis: scientific rationale, clinical trials, and clinical practice. Curr Opin Neurol. 2007;20(3):281–5.
Faissner S, Gold R. Oral therapies for multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(1):a032011.
Hermann R, Karlsson MO, Novakovic AM, Terranova N, Fluck M, Munafo A. The clinical pharmacology of cladribine tablets for the treatment of relapsing multiple sclerosis. Clin Pharmacokinet. 2019;58(3):283–97.
Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–26.
Giovannoni G, Soelberg Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler. 2018;24(12):1594–604.
Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–60.
Gentile A, Musella A, Bullitta S, Fresegna D, De Vito F, Fantozzi R, et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflammation. 2016;13(1):207.
Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328(1–2):9–18.
Frampton JE. Ocrelizumab: first global approval. Drugs. 2017;77(9):1035–41.
Frisch ES, Pretzsch R, Weber MS. A milestone in multiple sclerosis therapy: monoclonal antibodies against CD20-yet progress continues. Neurotherapeutics. 2021;18(3):1602–22.
Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180(1–2):63–70.
Klein C, Lammens A, Schafer W, Georges G, Schwaiger M, Mossner E, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22–33.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Tur C, Montalban X. Progressive MS trials: lessons learned. Mult Scler. 2017;23(12):1583–92.
Hochmeister S, Grundtner R, Bauer J, Engelhardt B, Lyck R, Gordon G, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol. 2006;65(9):855–65.
Abdelhak A, Weber MS, Tumani H. Primary progressive multiple sclerosis: putting together the puzzle. Front Neurol. 2017;8:234.
Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17(2):210–8.
Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol. 2010;68(4):465–76.
Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. 2018;9:3116.
Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089–104.
Lehmann-Horn K, Kinzel S, Weber MS. Deciphering the role of B cells in multiple sclerosis-towards specific targeting of pathogenic function. Int J Mol Sci. 2017;18(10):2048.
Kinzel S, Weber MS. B Cell-Directed Therapeutics in Multiple Sclerosis: Rationale and Clinical Evidence. CNS Drugs. 2016;30(12):1137–48.
Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. 2014;6(248): 248ra107.
Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol. 2014;75(2):266–76.
Jackle K, Zeis T, Schaeren-Wiemers N, Junker A, van der Meer F, Kramann N, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain. 2020;143(7):2073–88.
Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–46.
Beynon V, George IC, Elliott C, Arnold DL, Ke J, Chen H, et al. Chronic lesion activity and disability progression in secondary progressive multiple sclerosis. BMJ Neurol Open. 2022;4(1): e000240.
Simpson DSA, Oliver PL. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel). 2020;9(8):743.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–93.
Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18(12):905–22.
Sucksdorff M, Matilainen M, Tuisku J, Polvinen E, Vuorimaa A, Rokka J, et al. Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain. 2020;143(11):3318–30.
Lavisse S, Guillermier M, Herard AS, Petit F, Delahaye M, Van Camp N, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32(32):10809–18.
Zhang L, Hu K, Shao T, Hou L, Zhang S, Ye W, et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta Pharm Sin B. 2021;11(2):373–93.
Lassmann H, van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta. 2016;1862(3):506–10.
Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte Crosstalk in CNS Inflammation. Neuron. 2020;108(4):608–22.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–32.
Keaney J, Gasser J, Gillet G, Scholz D, Kadiu I. Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J Neuroimmune Pharmacol. 2019;14(3):448–61.
Torke S, Weber MS. Inhibition of Bruton s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Expert Opin Investig Drugs. 2020;29(10):1143–50.
Marcotte DJ, Liu YT, Arduini RM, Hession CA, Miatkowski K, Wildes CP, et al. Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010;19(3):429–39.
Geladaris A, Hausler D, Weber MS. Microglia: The Missing Link to Decipher and Therapeutically Control MS Progression? Int J Mol Sci. 2021;22(7):3461.
Whang JA, Chang BY. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov Today. 2014;19(8):1200–4.
Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57.
Martin E, Aigrot MS, Grenningloh R, Stankoff B, Lubetzki C, Boschert U, et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plast. 2020;5(2):123–33.
Corneth OBJ, Klein Wolterink RGJ, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105.
Garcia-Merino A. Bruton’s tyrosine kinase inhibitors: a new generation of promising agents for multiple sclerosis therapy. Cells. 2021;10(10):2560.
Ren L, Campbell A, Fang H, Gautam S, Elavazhagan S, Fatehchand K, et al. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcgamma receptor (FcgammaR) function. J Biol Chem. 2016;291(6):3043–52.
Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu D, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci U S A. 2012;109(15):5791–6.
Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278(28):26258–64.
Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454.
McDonald C, Xanthopoulos C, Kostareli E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology. 2021;164(4):722–36.
Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016;30(4):833–43.
Weber MS, Nicholas JA, Yeaman MR. Balancing potential benefits and risks of Bruton tyrosine kinase inhibitor therapies in multiple sclerosis during the COVID-19 pandemic. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1067.
Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62(17):7643–55.
Carnero Contentti E, Correale J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Expert Opin Emerg Drugs. 2020;25(4):377–81.
Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res. 2020;152: 104609.
Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227–45.
Hopkins BT, Bame E, Bajrami B, Black C, Bohnert T, Boiselle C, et al. Discovery and preclinical characterization of BIIB091, a reversible, selective BTK inhibitor for the treatment of multiple sclerosis. J Med Chem. 2022;65(2):1206–24.
Ma B, Bohnert T, Otipoby KL, Tien E, Arefayene M, Bai J, et al. Discovery of BIIB068: a selective, potent, reversible Bruton’s tyrosine kinase inhibitor as an orally efficacious agent for autoimmune diseases. J Med Chem. 2020;63(21):12526–41.
Brullo C, Villa C, Tasso B, Russo E, Spallarossa A. Btk inhibitors: a medicinal chemistry and drug delivery perspective. Int J Mol Sci. 2021;22(14):7641.
Boschert U, Crandall T, Pereira A, Higginbotham G, Wu Y, R. G, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by Evobrutinib (M2951) a novel Bruton’s tyrosine kinase inhibitor. ECTRIMS Online Library (2017), p P678. 2017(2017).
Francesco M, Wong M, LaStant J, Finkle D, Loewenstein N, Macsata R. PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. 2017;ECTRIMS Online Library (2017), p. P989.
Owens TD, Smith PF, Redfern A, Xing Y, Shu J, Karr DE, et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin Transl Sci. 2022;15(2):442–50.
Turner T, Brun P, Ofengeim D, Gruber R. Comparative CNS Pharmacology of Tolebrutinib Versus Other BTK Inhibitor Candidates for Treating MS. 2022;ACTRIMS 2022, P162.
Torke S, Pretzsch R, Hausler D, Haselmayer P, Grenningloh R, Boschert U, et al. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol. 2020;140(4):535–48.
Alankus Y-B, Grenningloh R, Haselmayer P, Bender A, Bruttger J. Inhibition of Bruton’s Tyrosine Kinase Prevents Inflammatory Macrophage Differentiation: A Potential Role in Multiple Sclerosis. 2019(2019 Annual Meeting of the Consortium of Multiple Sclerosis Centers).
Bame E, Tang H, Burns JC, Arefayene M, Michelsen K, Ma B, et al. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and Fc receptor signaling and downstream functions in B cells and myeloid cells. Clin Transl Immunol. 2021;10(6): e1295.
Geladaris A, Torke S, Weber M, Grenningloh R, Boschert U, Brück W. Targeting BTK in chronic CNS autoimmunity inhibits activation of microglia. 2021;ECTRIMS 2021(LB-ECTRIMS-2021-01631).
Gruber R, Dufault M, Chretien N, Proto J, Zhang M, Lamorte M, et al. Decoding bruton’s tyrosine kinase signalling in neuroinflammation. 2020;ECTRIMS 2020, P0311.
Weber M, Harp C, Bremer M, Goodyear A, Crawford J, Johnson A, et al. Fenebrutinib demonstrates the highest potency of Bruton tyrosine kinase inhibitors (BTKis) in phase 3 clinical development for multiple sclerosis (MS) (4437). Neurology. 2021;96(15 Supplement):4437.
Oh J, Cohen S, Isenberg D, Maurer M, Galanter J, Chu T, et al. The safety of fenebrutinib in a large population of patients with diverse autoimmune indications supports investigation in multiple sclerosis (MS) (4564). Neurology. 2021;96(15 Supplement):4564.
Weber M, Harp C, Goodyear A, Yuen T, Durk M, Kappos L. Fenebrutinib reduces disease activity in a mouse model of inflammatory multiple sclerosis, which is associated with reduced microglial activation. 2021;ECTRIMS 2021, P680.
ClinicalTrials. A Study of Efficacy and Safety of M2951 in Participants With Relapsing Multiple Sclerosis 2016, Access Date; https://clinicaltrials.gov/ct2/show/NCT02975349?term=evobrutinib&draw=3&rank=17
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380(25):2406–17.
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Martin EC, et al. Evobrutinib efficacy is maintained over two years in an open-label phase II study extension in patients with relapsing multiple sclerosis (4124). Neurology. 2021;96(15 Supplement):4124.
ClinicalTrials. Study of Evobrutinib in Participants With RMS (evolutionRMS 1). 2020. https://clinicaltrials.gov/ct2/show/NCT04338022?term=evobrutinib&draw=2&rank=10. Accessed 11 Apr 2022.
Montalban X, Arnold DL, Bar-Or A, Cross AH, Havrdova EK, Stuve O, et al. Rationale and design of two phase 3 randomized controlled trials (Evolution RMS 1&2) evaluating the Bruton’s tyrosine kinase inhibitor evobrutinib in patients with relapsing multiple sclerosis (4071). Neurology. 2020;94(15 Supplement):4071.
Reich DS, Arnold DL, Vermersch P, Bar-Or A, Fox RJ, Matta A, et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20(9):729–38.
Oh J, Syed S, Orogun L, Matta A, Dukovic D, Fox R. Safety and efficacy outcomes from the long-term extension study of tolebrutinib in patients with relapsing MS: Year 1 results. Mult Scler J. 2021;2021:571–2.
ClinicalTrials. Long Term Safety and Efficacy Study of Tolebrutinib (SAR442168) in Participants With Relapsing Multiple Sclerosis. 2019. https://clinicaltrials.gov/ct2/show/NCT03996291?term=tolebrutinib&draw=2&rank=5. Accessed 11 Apr 2022.
ClinicalTrials. Relapsing Forms of Multiple Sclerosis (RMS) Study of Bruton's Tyrosine Kinase (BTK) Inhibitor Tolebrutinib (SAR442168) (GEMINI 2) 2020, Access Date: 11.04.2022; Available from: https://clinicaltrials.gov/ct2/show/NCT04410991?term=tolebrutinib&draw=2&rank=8
ClinicalTrials. Relapsing Forms of Multiple Sclerosis (RMS) Study of Bruton's Tyrosine Kinase (BTK) Inhibitor Tolebrutinib (SAR442168) (GEMINI 1) 2020. https://clinicaltrials.gov/ct2/show/NCT04410978?term=tolebrutinib&draw=2&rank=9. Accessed 11 Apr 2022.
ClinicalTrials. Primary Progressive Multiple Sclerosis (PPMS) Study of Bruton's Tyrosine Kinase (BTK) Inhibitor Tolebrutinib (SAR442168) (PERSEUS). 2020, Access Date: 11.04.2022; Available from: https://clinicaltrials.gov/ct2/show/NCT04458051?term=tolebrutinib&draw=2&rank=7
ClinicalTrials. Nonrelapsing Secondary Progressive Multiple Sclerosis (NRSPMS) Study of Bruton's Tyrosine Kinase (BTK) Inhibitor Tolebrutinib (SAR442168) (HERCULES). 2020. https://clinicaltrials.gov/ct2/show/NCT04411641. Accessed 11 Apr 2022
Clinical Trials Arena. Sanofi Phase III tolebrutinib trials placed on partial hold by US FDA. 2022. https://www.clinicaltrialsarena.com/news/fda-hold-sanofi-tolebrutinib-trials/. Accessed 11 Aug 2022.
Sanofi. Media Update: Patient enrollment of phase III tolebrutinib trials paused in the U.S. 2022. https://www.sanofi.com/en/media-room/press-releases/2022/2022-06-30-05-30-00-2471767. Accessed 11 Aug 2022.
Cohen S, Tuckwell K, Katsumoto TR, Zhao R, Galanter J, Lee C, et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, phase II Trial (ANDES study). Arthritis Rheumatol. 2020;72(9):1435–46.
Isenberg D, Furie R, Jones NS, Guibord P, Galanter J, Lee C, et al. Efficacy, safety, and pharmacodynamic effects of the Bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2021;73(10):1835–46.
ClinicalTrials. A Study To Evaluate The Efficacy And Safety Of Fenebrutinib Compared With Teriflunomide In Relapsing Multiple Sclerosis (RMS) (FENhance) 2020. https://clinicaltrials.gov/ct2/show/NCT04586010?term=fenebrutinib&draw=2&rank=5. Accessed 11 Apr 2022.
ClinicalTrials. A Study to Investigate the Efficacy of Fenebrutinib in Relapsing Multiple Sclerosis (RMS) (FENopta) 2021. https://clinicaltrials.gov/ct2/show/NCT05119569?term=fenebrutinib&draw=2. Accessed 11 Apr 2022.
ClinicalTrials. A Study To Evaluate The Efficacy And Safety Of Fenebrutinib Compared With Ocrelizumab In Adult Participants With Primary Progressive Multiple Sclerosis (FENtrepid). 2020. https://clinicaltrials.gov/ct2/show/NCT04544449?term=fenebrutinib&draw=2&rank=6. Accessed 11 Apr 2022.
Wolinsky JS, Arnold DL, Brochet B, Hartung HP, Montalban X, Naismith RT, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998–1009.
Kaul M, End P, Cabanski M, Schuhler C, Jakab A, Kistowska M, et al. Remibrutinib (LOU064): a selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin Transl Sci. 2021;14(5):1756–68.
ClinicalTrials. Efficacy and Safety of Remibrutinib Compared to Teriflunomide in Participants With Relapsing Multiple Sclerosis. 2021. https://clinicaltrials.gov/ct2/show/NCT05147220?term=remibrutinib&draw=2&rank=1. Accessed 11 Apr 2022.
ClinicalTrials. A Phase 2 Study of Orelabrutinib in Patients With Relapsing-Remitting Multiple Sclerosis. 2021. https://clinicaltrials.gov/ct2/show/NCT04711148?term=orelabrutinib&draw=2. Accessed 11 Apr 2022.
ClinicalTrials. A Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study of BIIB091, a Bruton's Tyrosine Kinase (BTK) Inhibitor, in Healthy Adult Participants 2019. https://clinicaltrials.gov/ct2/show/NCT03943056?term=BIIB091&draw=2&rank=2. Accessed 11 Apr 2022.
Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol. 2022;18(1):40–55.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. This paper was not funded. We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Conflict of interest
A.G. has nothing to disclose. S.T. has received travel support from EMD Serono and research support from the Universitätsmedizin Göttingen (Startförderung). M.S.W. receives research support from the National Multiple Sclerosis Society (NMSS; PP 1660), the Deutsche Forschungsgemeinschaft (DFG; WE 3547/5-1), from Novartis, TEVA, Biogen-Idec, Roche, Merck and the ProFutura Programm of the Universitätsmedizin Göttingen.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
AG, ST and MSW participated in the concept, writing, and review of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Geladaris, A., Torke, S. & Weber, M.S. Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis: Pioneering the Path Towards Treatment of Progression?. CNS Drugs 36, 1019–1030 (2022). https://doi.org/10.1007/s40263-022-00951-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-022-00951-z