
Abstract
Background
Given the limited treatment options for younger children with attention-deficit/hyperactivity disorder (ADHD), a clinical study for SHP465 treatment was warranted.
Objectives
We aimed to evaluate the pharmacokinetics, safety, and tolerability of SHP465 mixed amphetamine salts (MAS) 6.25 mg after multiple once-daily doses in children aged 4–5 years with ADHD.
Methods
In this open-label multicenter study, SHP465 MAS 6.25 mg once daily was administered for 28 days to children aged 4–5 years with ADHD; baseline ADHD Rating Scale-5 total score ≥ 28 (boys) or ≥ 24 (girls) and Clinical Global Impression-Severity scale score ≥ 4. Blood samples were collected in the pharmacokinetic-rich group predose on day 1 week 1 and day 7 week 4 (predose, postdose at 2, 5, 8, 12, 16, 24, and 48 hours); and in the pharmacokinetic-sparse group predose on day 1 weeks 1, 2, and 3 and 24 hours postdose on day 7 week 4 . Key pharmacokinetic parameters included maximum plasma drug concentration (Cmax), plasma trough drug concentration, time to Cmax during a dosing interval (tmax), area under the concentration–time curve from time 0 to time of last collected sample, area under the concentration–time curve over the dosing interval (24 h) at steady state (AUCtau,ss), first-order rate constant associated with the terminal phase of elimination, terminal half-life (t1/2), total clearance of drug from plasma after oral administration, and apparent volume of distribution at steady state. Safety endpoints included treatment-emergent adverse events and vital signs.
Results
Mean ± standard deviation age and body mass index of 24 participants (66.7% male) were 4.8 ± 0.41 years and 17.2 ± 3.18 kg/m2, respectively. The most common ADHD was the combined presentation (91.7%); ratings were 50% markedly ill and 45.8% moderately ill on the Clinical Global Impression-Severity scale. Plasma d-amphetamine and l-amphetamine steady state was attained by predose on treatment day 8, consistent with the half-life. Peak steady-state plasma concentration (median tmax) for both d-amphetamine and l-amphetamine occurred at 7.92 h postdose on day 7 week 4 and thereafter declined monoexponentially, with a geometric mean t1/2 of 10.4 and 12.3 h for d-amphetamine and l-amphetamine, respectively. For both d-amphetamine and l-amphetamine, Cmax and AUCtau,ss were comparable between children aged 4 years (n = 3) and children aged 5 years (n = 8) regardless of sex. In total, 14 treatment-emergent adverse events were reported by 45.8% (11/24) of participants. Five treatment-emergent adverse events, reported for four (16.7%) participants, were considered treatment related; affect lability occurred in two (8.3%) participants, and insomnia, accidental overdose, and increased blood pressure each occurred in one (4.2%) participant.
Conclusions
In children aged 4–5 years with ADHD, following multiple once-daily administrations of SHP465 MAS 6.25 mg, the pharmacokinetic profile of plasma d-amphetamine and l-amphetamine was generally consistent among participants. Between-individual variability of plasma d-amphetamine and l-amphetamine steady-state exposure was low to moderate. SHP465 MAS was generally well tolerated in this study.
Trial Registration
ClinicalTrials.gov, NCT03327402 (31 October, 2017).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Once-daily administration of SHP465 mixed amphetamine salts 6.25 mg to children aged 4–5 years with attention-deficit/hyperactivity disorder yielded a pharmacokinetic profile of plasma d-amphetamine and l-amphetamine that was generally consistent among participants. |
Between-individual variability of plasma d-amphetamine and l-amphetamine steady-state exposure was low to moderate. |
In this study, SHP465 mixed amphetamine salts 6.25 mg was generally well tolerated, and no unexpected treatment-related adverse events were reported; observed adverse events of insomnia, blood pressure increased, and affect lability are consistent with known side effects of prescription amphetamines. |
1 Introduction
Global prevalence of attention-deficit/hyperactivity disorder (ADHD) in children is 7.2% [1]. A 2016 national survey indicated that 9.4% of children in the USA between the ages of 2 and 17 years have had an ADHD diagnosis, including 2.4% of children between the ages of 2 and 5 years [2]. The clinical presentation of ADHD is heterogeneous, characterized by a persistent pattern of inattention, hyperactivity-impulsivity, or a combination of inattention and hyperactivity [3]. In children and teenagers with ADHD, the symptoms as well as sleep problems are usually noticeable before the age of 6 years [4, 5]. The most common sleep problem in children with ADHD is “difficulty falling asleep” and often includes frequent night-time awakenings [4]. For children aged 4–5 years with ADHD, the American Academy of Pediatrics recommends evidence-based behavioral treatments as first-line therapy. Pharmacologic treatment may be considered for children with moderate-to-severe ADHD who have inadequate improvement in symptoms with nonpharmacologic therapy [6]. Amphetamine is the only stimulant approved by the US Food and Drug Administration (FDA) for ADHD in children younger than 6 years of age [6].
Mydayis® (mixed salts of a single-entity amphetamine product; MAS) extended-release capsules for oral use is FDA approved in the USA for treating ADHD in individuals aged 13−17 years at a maximum daily dose of 25 mg and in adults at a maximum daily dose of 50 mg [7]. Each MAS capsule contains equal amounts (by weight) of dextroamphetamine sulfate, amphetamine sulfate, dextroamphetamine saccharate, and amphetamine aspartate monohydrate, resulting in a 3:1 ratio of dextro- (d) amphetamine to levo- (l) amphetamine base equivalent [7]. Under the SHP465 investigational product name, SHP465 MAS produced significantly greater reductions in ADHD Rating Scale (ADHD-RS) scores than placebo in adults, adolescents, and children diagnosed with ADHD [8,9,10,11]. Based on data from two randomized, placebo-controlled, phase III clinical studies, SHP465 was well tolerated [11, 12] and in one study demonstrated superiority over placebo in reducing ADHD symptoms in children (aged 6–12 years) and adolescents [11]. Although doses between 12.5 and 25 mg once daily (QD) were superior to placebo, a lower dose of 6.25 mg QD failed to reduce ADHD symptoms in this population [11, 12].
Another mixed amphetamine salt-based stimulant, Adderall®, is FDA approved in the USA for treating ADHD in adults and children aged 3 years and older [13], while Adderall XR® is approved for individuals aged ≥ 6 years [14]. To date, there are no published phase I studies of a lower dose of SHP465 MAS in children aged 4–5 years with ADHD. In response to a pediatric written request from the FDA, additional SHP465 MAS studies (NCT03327402, NCT03325894, NCT03325881) were conducted to assess the effects of lower doses of SHP465 MAS in children aged 4–12 years with ADHD. A phase I study (NCT03327402) was designed to assess the pharmacokinetics (PK), safety, and tolerability of 6.25 mg SHP465 MAS QD in children aged 4–5 years diagnosed with ADHD.
2 Methods
2.1 Study Design
This phase I, open-label, multi-dose study (NCT03327402) was conducted at eight US clinical research centers (CRCs). Based on the PK sampling scheme, there were two cohorts (PK rich [nine samples/participant] and PK sparse [four samples/participant]). Enrollment was stratified by sex to ensure that two or more of the ten participants in the PK-rich cohort and three or more of the ten participants in the PK-sparse cohort were female. The study included a screening period of up to 32 days, a treatment period (approximately 30 days for the PK-rich cohort and approximately 29 days for the PK-sparse cohort), and a follow-up visit or phone call that occurred approximately 1 week after the last dose of the investigational product. Although this was a phase I PK study, participants received SHP465 MAS oral doses for approximately 30 days in order to obtain 1-month safety data. Following the screening visit, eligible participants returned to the CRC to reconfirm eligibility and enter the treatment period on day 1 of treatment week 1. Participants taking ADHD medication during the screening period were required to discontinue medication use ≥ 7 days before the first SHP465 MAS dose was administered. Participants taking medications, including cytochrome P450 2D6 (CYP2D6) inhibitors, were required to undergo a washout period of 30 days before the baseline visit; antihistamines, herbal and other over-the-counter preparations, including melatonin, had a 14-day washout period. Baseline was defined on day 1 of week 1 before the first dose of SHP465 MAS.
The study was conducted in accordance with guidelines of the International Council for Harmonisation of Good Clinical Practice, the principles of the Declaration of Helsinki, and applicable FDA regulations. Relevant documentation was approved by institutional review boards (see Declarations section). Before initiating any study-related procedures, written informed consent was obtained from the participant’s parent or legally authorized representative (LAR) and, if applicable, assent was obtained from the participant.
2.2 Study Participants
Boys or girls aged 4–5 years with a primary ADHD diagnosis based on Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria were enrolled in the study. Participants were required to have baseline ADHD-Rating Scale-5 (ADHD-RS-5) Child, Home Version scores of ≥ 28 for boys and ≥ 24 for girls, and ≥ 4 (moderately ill) on the Clinical Global Impression-Severity (CGI-S) scale. Participants were also required to have undergone an adequate course of nonpharmacologic treatment or to have had severe enough ADHD symptoms to be considered by the investigator for enrollment without prior nonpharmacologic treatment, and to have never used ADHD pharmacotherapy, or to have used ADHD pharmacotherapy with unacceptable efficacy and/or tolerability.
Participants were excluded if they had a controlled or uncontrolled Axis I or II disorder or any disorder that contraindicated SHP465 MAS use, concurrent illness, disability, or a condition that might confound safety assessments, increase participant risk, or prohibit the participant from completing the study. Participants were also excluded if they were considered a suicide risk, previously made a suicide attempt, or had a history of/currently demonstrating active suicidal ideation, a family history of sudden cardiac death or ventricular arrhythmia, a history of symptomatic cardiovascular disease or other serious cardiac conditions, a clinically significant electrocardiogram, or blood pressure ≥ 95th percentile. Additional exclusion criteria included height or weight ≤ 5th percentile for age and sex at screening; having a documented allergy, hypersensitivity, or intolerance to amphetamine or to any excipients in SHP465 MAS; having used ADHD medication with acceptable efficacy and/or tolerability, ADHD medication within 7 days before SHP465 MAS administration, or any medication within 30 days (or five half-lives) before SHP465 MAS administration.
2.3 Treatment
Eligible participants returned to the CRC on day 1 of treatment week 1 to initiate 28 days of treatment with 6.25 mg SHP465 QD. On day 1 (8 a.m. ± 1 h) of weeks 1, 2, 3, and 4, treatment was administered at the CRC, and SHP465 MAS capsules were dispensed to the parent/LAR for use for the rest of the week (days 2−7). Participants were instructed to either swallow the SHP465 MAS capsule whole or to sprinkle the contents of the capsule onto 1 tablespoon of applesauce and consume it in its entirety immediately without chewing.
2.4 Study Endpoints
Key primary PK parameters included maximum plasma drug concentration (Cmax) and plasma trough drug concentration, time to Cmax during a dosing interval (tmax), area under the concentration–time curve (AUC) from time 0 to last collected sample (AUC0–tlast) and over the 24-h dosing interval at steady state (AUCtau,ss), terminal half-life, the first-order rate constant associated with the terminal phase of elimination, total body clearance for extravascular administration, and apparent volume of distribution at steady state. The key secondary PK endpoints were observed plasma analyte concentration at 12, 16, and 24 h after dose administration (C12, C16, and C24, respectively), AUC from 5 to 12 h postdose (AUC5–12), AUC from 12 to 16 h postdose (AUC12–16), and AUC from 16 to 24 h postdose (AUC16–24).
For the rich PK sample population, blood samples were collected predose on day 1 of week 1 and on day 7 of week 4 and postdose on day 7 of week 4 at 2, 5, 8, 12, 16, 24, and 48 h. For the sparse PK sample population, blood samples were collected predose on day 1 of weeks 1, 2, and 3 and postdose on day 7 of week 4 at 24 h. Plasma (K2EDTA) d-amphetamine and l-amphetamine concentrations were determined using a validated liquid chromatography with tandem mass spectrometry method at QPS, LLC (Newark, DE, USA). The assay was linear over a range of 0.1–100 ng/mL (lower limit of quantitation, 0.1 ng/mL).
Safety and tolerability endpoints included treatment-emergent adverse events (TEAEs; coded using the Medical Dictionary for Regulatory Activities, version 20.1); changes in vital signs (blood pressure and heart rate), electrocardiogram results, weight, and body mass index. In addition, other safety endpoints were TEAEs of particular interest (insomnia, weight decreased, and decreased appetite) and pre-defined TEAEs of special interest [psychiatric events including psychosis/mania, suicide ideation, aggression, and other miscellaneous psychiatric events (see Appendix 1)]. These were defined as TEAEs of particular and special interest because they are of concern with the stimulant class of medication and, therefore, the sponsor carefully monitored these TEAEs during the course of the study. The Columbia-Suicide Severity Rating Scale (C-SSRS) [15] was used to assess suicide risk. Sleep problems were assessed based on the Children’s Sleep Habits Questionnaire (CSHQ), while overall quality of sleep was assessed by Pediatric Sleep Questionnaire (PSQ) [16, 17]. The occurrence of TEAEs was assessed from the time of informed consent through every study visit. Vital signs were assessed at screening, predose on day 1 of each treatment week, 5 and 48 h postdose on day 7 of week 4 (PK-rich sampling only) or 24 h postdose on day 7 of week 4 (PK-sparse sampling only). Electrocardiograms were assessed at screening, predose on day 1 of each treatment week (only weeks 1–3 in the PK-sparse sampling), and then 5 and 48 h postdose on day 7 of week 4 (PK-rich sampling only) or 24 h postdose on day 7 of week 4 (PK-sparse sampling only). Body weight was assessed at screening, predose on day 1 of each treatment week, and then 24 h postdose on day 7 of week 4 (PK-sparse sampling only) or 48 h postdose on day 7 of week 4 (PK-rich sampling only).
2.5 Study Questionnaires
The C-SSRS is a 19-item semi-structured interview with questions used to assess suicide risk [15]. It is designed to capture the occurrence, severity, and frequency of suicide-related thoughts and behaviors during the assessment period with most items rated on a dichotomous scale (yes or no) or 3-point or 5-point Likert scales [18, 19]. Although the C-SSRS is not validated in children, it was included in the protocol at the request of the FDA and it was completed by a clinician who was trained and experienced in the evaluation of preschool children with ADHD. The C-SSRS (pediatric/cognitively impaired version) “baseline” version was completed at the screening visit, and the “since last visit” version was completed at all post-screening visits.
The CSHQ is a 33-item questionnaire designed to screen for the most common sleep problems in children. Items are rated on a 3-point scale (“usually” [5–7 times/week], “sometimes” [2–4 times/week], and “rarely” [0–1 time/week]; some items are scored in reverse) and grouped into eight subscales (bedtime resistance, sleep-onset delay, sleep duration, sleep anxiety, night awakenings, parasomnias, sleep-disordered breathing, and daytime sleepiness). The total sleep disturbance score was calculated as the sum of subscale scores, excluding scores for the “needs parent in room to sleep” and “afraid of sleeping alone” items. Higher scores are indicative of greater sleep disturbance. The CSHQ was conducted at each visit to the site starting with the baseline visit (predose on day 1 of week 1) and was completed by the participant’s parent/LAR.
The PSQ is a seven-item questionnaire designed to assess overall quality of sleep (average time to sleep, sleep latency, frequency of interrupted sleep, duration of interrupted sleep, total sleep time, and sleep quality over the last week) with pharmacologic treatment. The PSQ was completed by the parent/LAR with the participant and the responses were reviewed by the clinician during the study visit at each visit to the site starting with the baseline visit (predose on day 1 of week 1).
2.6 Analysis Sets
The PK analysis was based on the PK analysis set (participants from the safety set having one or more PK blood sample collected). Pharmacokinetic parameters for d-amphetamine and l-amphetamine were calculated using noncompartmental analysis (Phoenix® WinNonlin®; Certara, Princeton, NJ, USA) from the PK-rich sampling population only and are reported using descriptive statistics. Safety and tolerability data are reported in all enrolled participants who took one or more doses of the investigational product (safety set) and were summarized with descriptive statistics.
3 Results
3.1 Participant Demographics and Clinical Characteristics
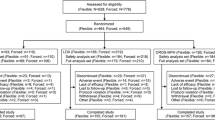
Of 33 screened participants, 24 were enrolled and included in both the safety set and PK set, and 22 (91.7%) completed the study (Fig. 1). One participant was withdrawn from the study by the parent/LAR, and one participant discontinued for another reason (visit out of window). No participant discontinued treatment or the study because of a TEAE. The mean ± standard deviation (SD) duration of exposure to SHP465 MAS was 3.9 ± 0.64 weeks, and the average daily dose was 6.2 ± 0.26 mg.

Patient disposition. LAR legally authorized representative
Most participants were male (66.7%), with an equal proportion white (50%) and black (50%); mean ± SD age was 4.8 ± 0.41 years (Table 1). A combined ADHD presentation was the most common among study participants (91.7%), and the majority were rated as moderately or markedly ill (45.8% and 50.0%, respectively) on the CGI-S scale. No patient received a stimulant as a prior medication (i.e. 30 days before study treatment), although one patient had received prior treatment with a stimulant (i.e. more than 30 days before study entry).
3.2 Pharmacokinetics
After multiple doses of treatment with SHP465 MAS 6.25 mg QD, the mean plasma d-amphetamine and l-amphetamine concentration–time profiles were characterized by a sustained absorption phase (Fig. 2). Table 2 presents PK parameters based on timepoints collected from the rich sample population. Mean ± SD peak steady-state plasma drug concentrations for d-amphetamine (32.8 ± 10.37 ng/mL) and l-amphetamine (10.4 ± 3.44 ng/mL) were observed at a median tmax of 7.9 h postdose. Thereafter, d-amphetamine and l-amphetamine concentrations decreased, with a geometric mean half-life of 10.4 and 12.3 h, respectively. Mean plasma trough d-amphetamine and l-amphetamine steady-state concentrations were attained by predose on day 8 of treatment. Because of sparse sampling, an accurate estimate of steady-state attainment cannot be provided. Between-individual variability, as measured by the geometric mean coefficient of variation, in steady-state exposure to d-amphetamine and l-amphetamine was low to moderate and comparable for both d-amphetamine and l-amphetamine. Peak and overall steady-state exposure (Cmax and AUCtau,ss) were comparable between 4-year-old (n = 3) and 5-year-old (n = 8) children, regardless of sex, for both d-amphetamine and l-amphetamine.

Steady-state plasma d-amphetamine and l-amphetamine concentration–time profiles (mean ± standard deviation) for the PK-rich cohort (PK set*), plotted on a linear concentration scale (a) or a logarithmic concentration scale (b) on day 7 of treatment week 4. PK pharmacokinetic, SD standard deviation. *Based on n = 11 for all timepoints except 48 h, for which n = 10
3.3 Safety and Tolerability
Overall, 45.8% of participants (11/24) reported a TEAE (Table 3). Most TEAEs were of mild severity (9/11), and no severe or serious TEAEs were reported. The only TEAE reported by more than one participant was affect lability, which occurred in two participants (8.3%). No TEAEs of special interest were reported. Of the TEAEs of particular interest, an event of insomnia and decreased appetite occurred in one (4.2%) participant each. Mean increases from baseline in systolic and diastolic blood pressure and decreases from baseline in pulse, body weight, and body mass index were observed at the final on-treatment assessment (FoTA; Table 3). Electrocardiogram parameters at FoTA, (normal, 83.3% vs abnormal, 16.7%) were similar to those observed at baseline (normal, 78.3% vs abnormal, 21.7%).
Suicidal behavior and active suicidal ideation were not reported on the C-SSRS at any visit during the study period, and there were no substantive changes in sleep as measured by the PSQ and CSHQ (Table 4). On the PSQ, time to fall asleep per night (mean ± SD) was shorter at FoTA compared with baseline (19.8 ± 11.75 vs 24.6 ± 12.50 min; Table 4), and length of time spent sleeping per night (mean ± SD) was slightly longer at FoTA (9.1 ± 1.41 vs 8.9 ± 1.41 h at baseline; Table 4). On the CSHQ, the sleep disturbance scores (mean ± SD) were 48.3 ± 8.24 at baseline and 45.4 ± 8.21 at FoTA (Table 4).
4 Discussion
The recommended starting dose of Mydayis® (MAS) is 12.5 mg QD and the maximum approved doses are 25 mg QD in adolescents aged 13–17 years, and 50 mg QD in adults. In phase III studies, SHP465 MAS has been shown to significantly improve ADHD symptoms in children, adolescents, and adults with ADHD, with a safety and tolerability profile consistent with amphetamine treatment [8,9,10,11]. In response to a pediatric written request from the FDA, additional SHP465 MAS studies (NCT03327402, NCT03325894, NCT03325881) were conducted to assess the effects of lower doses of SHP465 MAS in children aged 4–12 years. A dose of SHP465 MAS 6.25 mg QD was evaluated in children aged 6–12 years; although it was well tolerated, it failed to reduce ADHD symptoms [12].
In this phase I study, the pharmacokinetics, safety, and tolerability of SHP465 6.25 mg was assessed in children aged 4–5 years diagnosed with ADHD following once-daily administration for 28 days. Although this was a phase I PK study, study participants received SHP465 MAS doses for approximately 30 days in order to obtain 1-month safety data. Plasma d-amphetamine and l-amphetamine concentrations indicated that steady state was attained by predose on day 8 of treatment in all participants regardless of sex, consistent with the half-life and low-to-moderate between-individual PK variability. Because of sparse sampling, a more granular time calculation of the steady-state attainment could not be obtained. In general, peak and overall steady-state exposure, as measured by Cmax and AUCtau,ss, were comparable between 4-year-old and 5-year-old participants regardless of sex for both d-amphetamine and l-amphetamine. Mean steady-state (Cmax) for d-amphetamine and l-amphetamine was characterized by a sustained absorption phase and achieved peak steady-state concentration at a median tmax of 7.92 h postdose for both amphetamines. Thereafter, the plasma d-amphetamine and l-amphetamine concentrations declined monoexponentially, with geometric mean terminal half-life values of 10.4 h for d-amphetamine and 12.3 h for l-amphetamine. A dose of SHP465 MAS 6.25 mg was well tolerated in this study, with no serious or severe adverse events reported, similar to the study in children aged 6–12 years treated with the same dose [12]. Although the enzymes involved in amphetamine metabolism are poorly defined, in the liver, amphetamine is metabolized to benzoic acid and hippuric acid, and by CYP2D6 to the active metabolite 4-hydroxy-amphetamine [20, 21]. The CYP2D6 gene is polymorphic and many variants that affect function occur at different frequencies among populations [22]. Therefore, a risk of increased exposure to SHP465 MAS is possible with co-administration of CYP2D6 inhibitors [7].
The most common adverse drug reactions in participants aged ≥ 13 years with ADHD treated with SHP465 MAS (incidence ≥ 5% and at a rate at least twice that of placebo) were decreased appetite, insomnia, nausea, decreased weight, and irritability [7]. Pediatric participants aged ≤ 12 years experienced higher plasma drug exposure and higher rates of adverse drug reactions, mainly insomnia and decreased appetite, than participants aged ≥ 13 years treated with the same dose [7]. In the current study of SHP465 6.25 mg QD in children aged 4–5 years, the overall TEAE frequency was 45.8% compared with 24.4% in children (aged 6–12 years) treated with the same dose of SHP465, 67.4% in adolescents and children treated with SHP465 MAS doses in a range of 12.5–25 mg, and 77.2–86.3% in adults treated with SHP465 MAS doses in a range of 25–75 mg [8, 11, 12]. In the present study, there were no discontinuations of treatment or the study because of a TEAE. Affect lability was the only TEAE that occurred in more than one (8.3%) participant. In contrast, in a study of atomoxetine in children (mean age 6.1 ± 0.58 years), mood lability was reported in 54.5% of participants [23], and a study of methylphenidate in children (mean age 4.8 ± 0.70 years) reported 64.3% of participants discontinued because of emotionality or irritability [24]. Changes in vital signs (with the exception of pulse rate) in the current study were minimal, consistent with a previous study in children (aged 6–12 years) at the same dose of SHP465 MAS 6.25 mg [12]. Historically, insomnia was the most frequently reported TEAE in studies conducted in adults and in children and adolescents [8,9,10,11]. In the current study, insomnia was reported for one participant (4.2%). Patients were not allowed to take melatonin during the study. In this study, children treated with SHP465 MAS 6.25-mg doses took less time to fall asleep and remained asleep at night for a longer period with fewer awakenings at FoTA compared with baseline, indicating improved sleep patterns with treatment. There were no substantive changes in other sleep-related parameters, including bedtime resistance, parasomnias, sleep anxiety, and length of time awake per night between baseline and FoTA as measured by the PSQ and CSHQ (no statistical comparisons for any measurements were performed), consistent with previous results with SHP465 6.25 mg in children (aged 6–12 years) and with the SHP465 MAS dose range of 12.5–25 mg in adults [8, 12].
Limitations of this study include that it was based on a small sample size owing to the young age of the participants. In addition, it was necessary to partially employ a sparse PK sampling method because of the age of the participants, which may have increased inter-individual variability. Because of very sparse sampling, a definitive estimate of steady-state attainment cannot be obtained. Further, the C-SSRS is not validated in children and adolescents; children aged 4–5 years do not understand the questions, thus the caregiver/parent answered in lieu of the participants.
5 Conclusions
In this study of children aged 4–5 years with ADHD, multiple doses of SHP465 MAS 6.25 mg QD were administered. Plasma d-amphetamine and l-amphetamine concentration–time profiles were characterized by a sustained absorption phase, achieving peak steady-state concentrations at 7.92 h postdose (median tmax) for both analytes; thereafter, plasma drug concentrations declined monoexponentially, with a geometric mean half-life of 10.4 and 12.3 h for d-amphetamine and l-amphetamine, respectively. Between individual-variability of plasma d-amphetamine and l-amphetamine steady-state exposures (Cmax and AUCtau,ss) was low to moderate with a geometric mean CV value range of 29.8−34.7%. A dose of SHP465 MAS 6.25 mg was generally well tolerated, with the general safety and tolerability profiles being consistent with previous observations for SHP465 MAS in children and adolescents [11, 12]. The data from this study show that 28-day administration of SHP465 MAS 6.25 mg was well tolerated by children aged 4–5 years and warrants further studies to explore its effectiveness in reducing ADHD symptoms in this age group.
References
Thomas R, Sanders S, Doust J, et al. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994–1001.
Danielson ML, Bitsko RH, Ghandour RM, et al. Prevalence of parent-reported ADHD diagnosis and associated treatment among U.S. children and adolescents, 2016. J Clin Child Adolesc Psychol. 2018;47:199–212.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5TM, 5th edn. Arlington: American Psychiatric Publishing, Inc.; 2013.
Sung V, Hiscock H, Sciberras E, et al. Sleep problems in children with attention-deficit/hyperactivity disorder: prevalence and the effect on the child and family. Arch Pediatr Adolesc Med. 2008;162:336–42.
Bundgaard AF, Asmussen J, Pedersen NS, et al. Disturbed sleep and activity in toddlers with early signs of attention deficit hyperactivity disorder (ADHD). J Sleep Res. 2018;27: e12686.
Wolraich ML, Hagan JF Jr, Allan C, et al. Clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2019;144: e20192528.
Mydayis® (mixed salts of a single-entity amphetamine product). Full prescribing information. Lexington: Shire US Inc., 2019. https://www.shirecontent.com/PI/PDFs/Mydayis_USA_ENG.pdf
Frick G, Yan B, Adler LA. Triple-bead mixed amphetamine salts (SHP465) in adults with ADHD: results of a phase 3, double-blind, randomized, forced-dose trial. J Atten Disord. 2020;24:402–13.
Spencer TJ, Adler LA, Weisler RH, et al. Triple-bead mixed amphetamine salts (SPD465), a novel, enhanced extended-release amphetamine formulation for the treatment of adults with ADHD: a randomized, double-blind, multicenter, placebo-controlled study. J Clin Psychiatry. 2008;69:1437–48.
Weisler RH, Greenbaum M, Arnold V, et al. Efficacy and safety of SHP465 mixed amphetamine salts in the treatment of attention-deficit/hyperactivity disorder in adults: results of a randomized, double-blind, placebo-controlled, forced-dose clinical study. CNS Drugs. 2017;31:685–97.
Brams M, Childress AC, Greenbaum M, et al. SHP465 mixed amphetamine salts in the treatment of attention-deficit/hyperactivity disorder in children and adolescents: results of a randomized, double-blind placebo-controlled study. J Child Adolesc Psychopharmacol. 2018;28:19–28.
Mattingly G, Arnold V, Yan B, et al. A phase 3, randomized, double-blind study of the efficacy and safety of low-dose SHP465 mixed amphetamine salts extended-release in children with attention deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2020;30:549–57.
Adderall® (dextroamphetamine saccharate, amphetamine aspartate monohydrate, dextroamphetamine sulfate and amphetamine sulfate tablet). Horsham: Teva Select Brands, 2017. https://medlibrary.org/lib/rx/meds/adderall-4/
Adderall XR® (mixed salts of a single-entity amphetamine product). Full prescribing information. Lexington (MA): Shire US Inc., 2017. https://www.shirecontent.com/PI/PDFs/AdderallXR_USA_ENG.PDF
Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–77.
Chervin RD, Hedger K, Dillon JE, et al. Pediatric Sleep Questionnaire (PSQ): validity and reliability of scales for sleep-disordered breathing, snoring, sleepiness, and behavioral problems. Sleep Med. 2000;1:21–32.
Owens JA, Spirito A, McGuinn M. The Children’s Sleep Habits Questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep. 2000;23:1043–51.
Posner K. Suicidality issues in clinical trials: Columbia suicidal adverse event identification in FDA safety analyses. Food and Drug Administration, Division of Metabolism and Endocrinology Products Advisory Committee Meeting, June 13 2007. https://slideplayer.com/slide/5797943/. Accessed Sep 2021.
Cooper D. A study of the validity of the C-SSRS in assessing risk of suicidality in children and adolescents. ProQuest LLC, number 28090146. https://www.proquest.com/openview/923fcb325653d38be89829b2ae16d9ca/1?pq-origsite=gscholar&cbl=51922&diss=y. Accessed Sep 2021.
Asatoor AM, Galman BR, Johnson JR, et al. The excretion of dexamphetamine and its derivatives. Br J Pharmacol Chemother. 1965;24:293–300.
Markowitz JS, Patrick KS. Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet. 2001;40:753–72.
Taylor C, Crosby I, Yip V, et al. A review of the important role of CYP2D6 in pharmacogenomics. Genes (Basel). 2020;11:1295.
Kratochvil CJ, Vaughan BS, Mayfield-Jorgensen ML, et al. A pilot study of atomoxetine in young children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2007;17:175–85.
Greenhill L, Kollins S, Abikoff H, et al. Efficacy and safety of immediate-release methylphenidate treatment for preschoolers with ADHD. J Am Acad Child Adolesc Psychiatry. 2006;45:1284–93.
Acknowledgements
We thank Yi Wang, PhD (Takeda Pharmaceuticals USA, Lexington, MA, USA) for contributing to the development of the study synopsis. Under the direction of the authors, Sonia Mohinta, PhD (ICON, North Wales, PA, USA) provided writing assistance for this manuscript. Funding to ICON for support in writing this manuscript was provided by Takeda Pharmaceuticals USA, Inc., Lexington, MA, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study and open-access publication fee were funded by Shire Development, LLC, a member of the Takeda group of companies, Lexington, MA, USA. Takeda Pharmaceuticals USA, Cambridge, MA, USA provided funding to ICON (North Wales, PA, USA) for support in writing and editing this manuscript.
Conflicts of Interest/Competing Interests
KI and BY are employees of Takeda Pharmaceuticals USA, and own stock in Takeda. AK is a consultant for Shire, a member of the Takeda group of companies, and owns stocks in Takeda. NM has served as a consultant for Shire, a member of the Takeda group of companies.
Ethics Approval
The study protocol, final approved informed consent document, and all supporting information were submitted to and approved by Schulman Associates Institutional Review Board, Cincinnati, OH, USA (part of Advarra Institutional Review Board since November 2017) and University Hospitals Cleveland Medical Center Institutional Review Board, Cleveland, OH, USA, and by the US Food and Drug Administration, as appropriate, before study initiation. The study was conducted in accordance with guidelines of the International Council for Harmonisation of Good Clinical Practice, the principles of the Declaration of Helsinki, and applicable FDA regulations.
Consent to Participate
Written informed consent was obtained from the participant’s parent or legally authorized representative and, if applicable, assent was obtained from the participant.
Consent for Publication
Not applicable.
Availability of Data and Material
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant’s data supporting the results reported in this article, will be made available within 3 months from an initial request to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
Code Availability
Not applicable.
Authors’ Contributions
KI designed the study; NM and KI performed the research; and KI, BY, and AK analyzed and interpreted the data. KI critically revised the draft manuscript. All authors critically reviewed the manuscript, agree to be fully accountable for ensuring the integrity and accuracy of the work, and read and approved the final manuscript.
Appendices
Appendix 1
Adverse events of special interest
Category | Preferred term/verbatim term |
|---|---|
Signs and/or symptoms of psychosis/mania | Hallucination (any type, including visual, auditory, tactile, mixed) |
Delusion (any type including somatic, persecutory, grandeur, reference) | |
Schizophrenia (any type) | |
Psychotic disorder | |
Transient psychosis | |
Acute psychosis | |
Paranoia | |
Childhood psychosis | |
Schizophreniform disorder | |
Schizoaffective disorder | |
Catatonia | |
Mania | |
Hypomania | |
Suicidal ideation and behavior | Depression suicidal |
Gunshot wound | |
Intentional self-injury | |
Non-accidental overdose | |
Overdose | |
Self-injurious behavior | |
Self-injurious ideation | |
Self-mutilation | |
Suicidal ideation | |
Suicidal attempt | |
Completed suicide | |
Aggression and violent behavior | Aggression |
Anger | |
Hostility | |
Homicidal ideation | |
Sexual offense | |
Murder | |
Imprisonment | |
Miscellaneous psychiatric events (include events with serious outcome only) | Abnormal behavior |
Agitation | |
Amnesia | |
Confusional state | |
Depressed mood | |
Depression | |
Disorientation | |
Emotional disorder | |
Emotional distress | |
Feeling abnormal | |
Memory impairment | |
Mood altered | |
Mood swings | |
Personality change | |
Thinking abnormal | |
Anxiety | |
Fearfulness | |
Phobia | |
Panic attack | |
Sleep disturbance | |
Tics | |
Obsessive or compulsive behavior | |
Trichotillomania |
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ilic, K., Kugler, A.R., Yan, B. et al. Pharmacokinetics, Safety, and Tolerability of SHP465 Mixed Amphetamine Salts After Administration of Multiple Daily Doses in Children Aged 4–5 Years with Attention-Deficit/Hyperactivity Disorder. CNS Drugs 36, 71–81 (2022). https://doi.org/10.1007/s40263-021-00870-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00870-5