
Abstract
Anti-CD20 therapies have demonstrated considerable efficacy in the treatment of relapsing multiple sclerosis, constituting a high-efficacy treatment approach for reducing relapse risk and mitigating disability progression. These therapies have been shown to strongly deplete circulating B cells and small subsets of CD3+ CD4 and CD8 T cells that express low levels of CD20. While the clinical profiles of the various anti-CD20 monoclonal antibodies used in treating multiple sclerosis are well-described in the literature, greater understanding of the implications of their distinct molecular and pharmacological attributes is needed. In this review, we focus on four anti-CD20 monoclonal antibodies—rituximab, ocrelizumab, ofatumumab, and ublituximab—that are currently used, approved, or in late-stage clinical development for the treatment of multiple sclerosis. We provide clinical perspectives on the potential implications of differences in molecular structures, target epitopes, dosing regimens, mechanisms and impact on B-cell depletion and reconstitution, immunogenicity, administration-related reactions, and infection risks.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.14912757. |
The ongoing development and approval of anti-CD20 monoclonal antibody therapies, including rituximab, ocrelizumab, ofatumumab, and ublituximab, has represented a major advance in the care of patients with several autoimmune conditions, including multiple sclerosis (MS). |
These anti-CD20 molecules offer robust control of MS disease activity and generally excellent tolerability and safety. |
Differences in their molecular structures, target epitopes, dosing regimens/route of administration, and mechanisms of B-cell depletion may lead to varying clinical effects in terms of B-cell depletion and reconstitution patterns, immunogenicity, administration-related reactions, and infection risks. |
1 Introduction
The central role of B cells in the pathogenesis of multiple sclerosis (MS) is underscored by the potent clinical efficacy of B-cell-depleting monoclonal antibodies (mAbs) in the treatment of MS [1]. B cells are thought to contribute to MS through several mechanisms, including modulation of other immune cell responses (e.g., through antigen presentation and cytokine secretion) and autoantibody production [2,3,4]. B-cell mAbs directed against CD20, a surface marker expressed on pre-B cells, naïve B cells, and memory B cells, strongly deplete circulating B cells and small subsets of CD3+ CD4 and CD8 T cells that express low levels of CD20, while sparing antibody secreting plasma cells that do not express CD20 [5, 6]. These therapies represent a high-efficacy treatment approach with a favorable risk–benefit profile that substantially decreases MS relapses and mitigates disability progression in individuals with relapsing MS [7,8,9,10,11,12,13,14]. Current evidence suggests that the use of these therapies early in the course of disease, versus withholding until later, may result in improved clinical outcomes for people with MS [15,16,17].
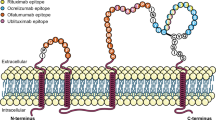
Despite their common target, the anti-CD20 mAbs have distinct molecular and pharmacological attributes. In this review, we provide clinical perspectives on the potential implications of differences in molecular structures (Table 1), target epitopes (Table 1 and Fig. 1), dosing regimens (Table 2), mechanisms of B-cell depletion, kinetics of B-cell depletion and reconstitution, and immune-mediated responses. Although the focus of this article is to review clinical data from MS studies of four anti-CD20 mAbs [rituximab (RTX), ocrelizumab (OCR), ofatumumab (OMB), and ublituximab (UTX)], we draw from the broader experience with these mAbs in oncology and rheumatology to provide additional potential clinical insights.

(adapted from Fox et al. [14])
Target epitopes of anti-CD20 monoclonal antibodies of interest
2 Molecular and Pharmacological Attributes of Anti-CD20 Monoclonal Antibodies
RTX is a murine-human chimeric mAb [18] that is approved for a number of indications in oncology and rheumatology (Table 3) [19, 20]. It was the first anti-CD20 mAb to be tested in MS and has been used as an off-label MS therapy [5]. RTX has been studied in relapsing-remitting MS (RRMS) and primary progressive MS (PPMS) [7, 9, 21], with the same dosing regimen approved for rheumatoid arthritis (RA) [19] comprising two 1000 mg intravenous infusions administered 2 weeks apart (at weeks 0 and 2), followed by subsequent 1000 mg infusions every 6 months [7, 21]. A subcutaneous formulation of RTX is currently approved for cancer treatment [20], however this formulation has not been investigated in MS. At least one full dose of RTX via intravenous infusion is required prior to subcutaneous injection administered by a healthcare professional. Additionally, premedication is required before each dose of RTX [20].
OCR, a humanized mAb that targets an almost identical epitope to that of RTX [18], was the first anti-CD20 mAb approved for MS, and is indicated for the treatment of relapsing forms of MS (RMS) and PPMS [22] based on the results of the phase III trials OPERA I/II [23] and ORATORIO [8, 24], respectively. OCR was approved for intravenous administration at a dose of 300 mg over ≥ 2.5 h for the first two doses, with subsequent doses of 600 mg delivered over ≥ 3.5 h every 6 months, with premedication recommended prior to each infusion to mitigate systemic reactions [22]. A shorter OCR infusion time of 2 h was recently approved for the 600 mg doses [25]. OCR was evaluated in RA in phase III clinical trials at doses of 200 or 500 mg on background methotrexate treatment, however development in RA was terminated due to the lack of additional benefit over existing therapies, including RTX [26]. The safety and tolerability of subcutaneously administered OCR is currently being investigated in a phase Ib open-label trial in RMS and PPMS (NCT03972306).
OMB, a fully human mAb that targets a unique epitope that includes the small extracellular loop on the exposed outcrop of CD20 (Fig. 1) [18], was recently approved in the US for the treatment of RMS [27] following completion of the phase III ASCLEPIOS I and II trials [13]; it represents the first subcutaneously administered B-cell-depleting therapy for MS. OMB is approved as a subcutaneous self-administered injection of 20 mg once monthly (three initial doses administered weekly starting at week 0, followed by once-monthly dosing starting at week 4) and does not require premedication [27]. Alternatively, intravenously administered OMB is approved for the treatment of chronic lymphocytic leukemia (CLL) at an initial dose of 300 mg, followed by subsequent doses of 1000 or 2000 mg at different intervals depending on indication, with recommended premedication before each infusion [28]. Intravenous OMB has been studied in a phase II study in RRMS [12]. It was also undergoing phase II and III clinical trials in RA but studies were terminated to refocus clinical development on the subcutaneous delivery route (NCT00611455, NCT00603525, NCT00655824) [29].
UTX, an intravenously administered murine-human chimeric mAb that targets a distinct epitope from other anti-CD20 mAbs (Fig. 1) [14, 30], is currently in late-stage clinical development for the treatment of MS. It has been studied in a phase II trial in RMS [14, 30] and was also evaluated in RMS in the recently completed ULTIMATE I and II phase III trials [31]. UTX was evaluated in the phase III trials as a 150 mg dose over 4 h, followed by a 1 h 450 mg infusion 2 weeks later and every 24 weeks thereafter up to week 96 [31, 32]. Premedication was required before each infusion in the phase II study [14]. Phase II and III studies of UTX for various oncology indications are currently underway (Table 3).
3 Clinical Perspectives Related to the Molecular and Pharmacological Attributes of Anti-CD20 Monoclonal Antibodies
3.1 Differences in the Mechanism of B-Cell Depletion
In vitro mechanisms by which anti-CD20 mAbs can deplete CD20-expressing cells include cross-linking-induced apoptosis, antibody-dependent cellular cytotoxicity (ADCC), and complement-dependent cytotoxicity (CDC), with the latter two thought to be the main mechanisms acting in vivo [18, 33, 34]. Variations in molecular structure and target epitopes, as summarized in Table 1 and Fig. 1, are thought to contribute to differences in the predominant mechanisms of B-cell depletion across the agents. The mechanisms of action of anti-CD20 mAbs are also depicted in the video below. These differences in mechanisms of B-cell depletion and relative potencies of depletion are thought to have dosing implications [35, 36].
Mechanisms of action of anti-CD20 monoclonal antibodies (MP4 219470 kb)
RTX depletes B cells through a combination of CDC and ADCC [18], with higher levels of CDC than ADCC [34]. OMB, which also mediates higher CDC levels than ADCC [34], has been shown in in vitro studies to have twofold greater ADCC compared with RTX, and tenfold higher CDC activity in RTX-sensitive tumor cell lines [37, 38]. As shown in Fig. 1, OMB binds to the small extracellular loop on the exposed outcrop of CD20, which allows it to attach closer to the cell membrane than RTX, OCR, or UTX, possibly accounting for its high degree of CDC activity [39]. When compared with OCR, OMB induced greater complement-dependent B-cell lysis in vitro (77.1% vs. 7.1%) after a 2-h exposure, and maintained effectiveness in inducing CDC when complement addition was delayed for 6 h [36]. This enhanced CDC potency is thought to account for OMB’s demonstrated efficacy at a low dose compared with the higher doses required for RTX, OCR, and UTX [36].
In contrast to RTX and OMB, OCR and UTX exhibit higher levels of ADCC than CDC [34, 40]. Compared with RTX, OCR exhibits two- to fivefold greater ADCC activity with enhanced binding to low-affinity variants of Fcγ receptor IIIa (FCγRIIIa), and three- to fivefold lower CDC activity [35]. UTX, which also has enhanced affinity for FCγRIIIa receptors through its glycoengineered Fc segment, has more pronounced ADCC effects than RTX, OCR, and OMB [14, 41]. This ADCC difference likely enables lower intravenous dosing and shorter infusion regimens versus those used with RTX and OCR [42].
3.2 B-Cell Depletion and Reconstitution Patterns with Anti-CD20 Monoclonal Antibodies
Gauging the actual depth and breadth of B-cell depletion with anti-CD20 therapies is challenging since measurements in the blood do not necessarily reflect the extent of B-cell depletion within the tissues. The available evidence indicates that the anti-CD20 regimens used in MS result in rapid and near-complete depletion of circulating B cells, with varying rates of B-cell reconstitution (with the latter potentially providing indirect insights into the extent of preceding tissue depletion).
In RTX phase I and II clinical trials in RRMS [7, 9] and the phase II/III trial in PPMS [21], two 1000 mg intravenous infusions of RTX administered 2 weeks apart (at weeks 0 and 2) resulted in rapid and near complete (> 95%) depletion of circulating B cells by week 2, which was sustained through week 24 [7, 9, 21]. In the RRMS trials, B cells were reconstituted to a mean of 30.7–34.5% of baseline by week 48 following the last RTX dose [7, 9]. In the PPMS trial, 35% of the RTX-treated patients had recovered peripheral B-cell counts above the lower limit of normal (LLN; 80 cells/μL) at 50 weeks after their last dose [21]. Similarly, 40% of patients who discontinued treatment early had recovered peripheral B-cell counts above the LLN (80 cells/μL) within 48 weeks of their last RTX dose [21]. Prolonged peripheral B-cell depletion lasting more than 3 years after a single course of RTX treatment (on background methotrexate) was found in a small proportion of RA patients (approximately 4%) [19].
In the phase III OPERA I and II trials of OCR in RMS, CD19+ cell counts (a surrogate marker for B-cell counts in those treated with anti-CD20 mAbs) were almost completely depleted by week 2 after the first OCR dose of 600 mg, and remained extensively depleted through week 96, with infusions every 24 weeks [23]. Similar findings were reported in the phase III ORATORIO trial in PPMS, which utilized the same dosing regimen as the OPERA trials, with almost complete CD19+ cell depletion from week 2 through to week 120 [8]. B-cell levels increased to either baseline or the LLN in 90% of patients within 2.5 years after the last OCR infusion, with a median time to repletion of 72 weeks (range 27–175 weeks) [22]. Studies of OCR in RA suggested that a greater number of infusions was not associated with longer time to B-cell repletion (defined as return of CD19+ levels to baseline or ≥80 cells/mL, whichever was lower), but that repletion may be more prolonged in patient populations with more severe, long-term disease [26].
Time to B-cell repletion with OMB treatment is faster than with the other anti-CD20 mAbs [34]. The phase III ASCLEPIOS trials [13] and phase II APLIOS bioequivalence study [43], which utilized the same dosing regimen of subcutaneous OMB 20 mg (initial doses administered at weeks 0, 1, and 2, followed by monthly maintenance doses starting at week 4) in RMS patients, reported rapid and near-complete B-cell depletion (≤10 cells/μL) in approximately 82–85% of patients by week 2. B cells recovered over the LLN in at least 50% of patients at 24–36 weeks post-treatment discontinuation [27]. B-cell levels in RRMS patients receiving two intravenous infusions of OMB 100, 300, and 700 mg 2 weeks apart (significantly higher doses compared with the subcutaneous OMB dose) were repleted to greater than or equal to LLN (100 cells/mm3) by week 48 in a few patients in each OMB dose group; repletion was observed in all patients during the individualized follow-up period of up to 104 weeks [12]. The faster B-cell repletion with subcutaneous OMB suggests differences in the depth and breadth of depletion in the tissues and could potentially have important safety implications, particularly in terms of infection risks [34].
The phase II study of UTX in RMS showed significant reductions in mean B-cell counts from 7.3% at baseline to 0.2% at 24 h after the first UTX dose of 150 mg [14]. All patients treated with UTX had ≥ 95% peripheral CD19+ B-cell depletion from baseline (> 99% median depletion) within 2 weeks after their second UTX infusion (by week 4 of active treatment). B-cell depletion persisted predose at weeks 24 and 48 (24 weeks after patients received their third UTX dose), with no significant recovery of B cells observed [14, 30]. Further pharmacodynamic analyses from the recently completed phase III trials should provide additional data on the kinetics of B-cell depletion and reconstitution with UTX.
As described above, current MS dose regimens for RTX, OCR, OMB, and UTX result in near-complete depletion of circulating B cells; however, it remains unclear whether such extent of depletion is necessary in MS to elicit a robust treatment effect. The MIRROR phase IIb trial [10] studying four dosing regimens of subcutaneously administered OMB in patients with RRMS found that OMB had a robust clinical effect in reducing the rate of cumulative new gadolinium-enhancing magnetic resonance imaging (MRI) lesions, even with the dose regimens that resulted in partial B-cell depletion to approximately 5–25% of baseline. No additional cumulative benefits on measures of relapsing disease were demonstrated with the higher dose that resulted in greater B-cell depletion to < 2% of baseline levels [10].
As noted, the dose-dependent differences in the kinetics of B-cell reconstitution suggest that near-complete peripheral B-cell depletion is associated with varying depths of depletion in the tissues. Time to onset of B-cell repopulation was dose-dependent in the MIRROR phase IIb trial [10], with repletion starting at approximately week 30 for the highest subcutaneous OMB dose of 60 mg administered every 4 weeks (after receiving the last OMB dose at week 20), versus approximately week 16–18 for the lower doses of 3, 30, and 60 mg every 12 weeks (after receiving the last OMB dose at week 12) [10, 44]. Rates of B-cell repopulation were similar in all dose groups, with repletion to the LLN achieved by the two more highly depleted higher-dose groups by approximately 14 months versus approximately 11 months for the two lower-dose groups [10]. Dose-dependent reconstitution kinetics were also found in a phase II RRMS trial studying three doses of intravenously administered OMB [12]. After treatment with two infusions of OMB 100, 300, and 700 mg 2 weeks apart, B-cell levels started to increase approximately 12–16 weeks, approximately 20 weeks, and approximately 24 weeks after the respective OMB intravenous dose, with faster repletion rates observed with the lower doses [12]. Similar findings of dose-dependent B-cell reconstitution were also observed for intravenously administered OCR in a 72-week phase I/II dose-ranging study in RA evaluating a combination of OCR versus placebo and methotrexate [45]. Based on an analysis of B-cell subsets in the peripheral blood of RA patients treated with RTX, immature B cells were found to reconstitute first, with a concomitant increase in circulating plasma cells, followed by an increase in the number of mature naïve B cells [46]. Repopulation of memory B cells was slow and delayed, with levels remaining significantly reduced (<50% of baseline) for more than 2 years [46]. Similar findings were observed in a recent study of B-cell reconstitution patterns following RTX treatment in RRMS patients [47].
Of interest, the presence of wearing-off symptoms such as fatigue and coordination/motor problems has been reported prior to the next OCR infusion (every 6 months or more, in the case of extended dosing schedules), which may be due to B-cell repletion in some patients; however, further study is warranted to research this phenomenon and an observational study is currently underway (NCT04478591).
3.3 Immune Responses to Anti-CD20 Monoclonal Antibodies
3.3.1 Immunogenicity
Immune responses generated against therapeutic proteins such as anti-CD20 mAbs may have important efficacy and safety implications [48,49,50,51]. Murine-human chimeric mAbs, including RTX and UTX, are considered more likely to elicit immunogenic responses, including antidrug antibodies (ADAs), than humanized (OCR) and fully human (OMB) anti-CD20 mAbs [18, 52]. Route of administration also likely affects immunogenicity. However, it should be noted that since the detection of ADAs is highly dependent on the specificity and sensitivity of the assay as well as the study design and patient population, comparisons across studies may be misleading [53]. ADA data are currently unavailable for UTX.
In the phase II HERMES trial of intravenous RTX in RRMS, human anti-chimeric antibodies (HACA) developed by week 48 in 24.1% (14/58) of patients who completed study treatment [7]. During the treatment and follow-up phases of the phase II/III OLYMPUS trial of intravenous RTX in PPMS, 7.0% (20/286) and 6.3% (9/143) of patients receiving RTX and placebo, respectively, had HACA [21]. HACA positivity was not found to have an effect on efficacy response or the types of AEs observed [7, 21]. A large, pooled safety analysis of the RTX RA clinical trial program, which detected ADAs in 11% (273/2578) of intravenous RTX-treated patients receiving background methotrexate, also indicated that ADA positivity was not associated with increased infusion-related reactions (IRRs) or other adverse events [19, 54].
Compared with RTX, OCR (a humanized mAb) is less likely to elicit ADAs [52]. In RMS patients who received OCR 600 mg across both the OPERA I and II trials, binding ADAs were detected in 0.4% (3/825) of patients and neutralizing antibodies were detected in 0.1% (1/825) of patients [23]. The ORATORIO trial reported a 1.9% (9/486) ADA incidence among OCR-treated PPMS patients over 120 weeks, with a 0.2% (1/486) incidence of neutralizing antibodies [8]. Although there is a low incidence of ADAs and neutralizing antibodies for OCR in MS trials, the clinical implications on efficacy and safety are uncertain. However, pooled data of over 2700 patients with RA from four phase III OCR clinical trials suggest no apparent association between human anti-human antibody (HAHA) positivity and corresponding CD19 counts or disease activity levels in any of the treatment groups [26].
As a fully human mAb, OMB is expected to be the least immunogenic anti-CD20 mAb [34]. The ASCLEPIOS trials of subcutaneous OMB 20 mg injection had a 0.2% (2/946) ADA incidence, and had no reports of neutralizing antibodies [13]. In a phase III trial of a 700 mg dose of intravenous OMB in biologic-naïve RA patients stable on methotrexate, none of the 260 OMB-treated patients developed ADAs during the 24-week, double-blind, placebo-controlled period [29]. In more than 926 patients with CLL, anti-OMB antibodies were observed in <1% of patients after treatment with intravenously administered OMB at an initial dose of 300 mg followed by subsequent doses of 1000 or 2000 mg at different intervals [28]. Overall, the incidence of ADAs and neutralizing antibodies for OMB is very low across doses, routes of administration, and indications.
Alongside the data above, studies in oncology of the intravenous and subcutaneous formulations of RTX provide some insights into the impact of other factors on immune response to mAbs, including route of administration, dosing regimen, the patients’ immunologic status, and concomitant therapies [48,49,50]. In patients with low-grade lymphoma, particularly follicular lymphoma (FL), receiving single-agent intravenous RTX, ADAs were detected in 1.1% (4/356) of patients [19]. This relatively low immunogenicity level following intravenous RTX treatment in patients with FL, compared with those with autoimmune disease such as MS and RA described above, may be due to the immune status of cancer patients [55]. Moreover, ADA levels were similar between the intravenous and subcutaneous formulations of RTX in previously untreated patients with FL (1.9% with 375 mg/m2 intravenous RTX vs. 2.0% with 1400 mg subcutaneous RTX) and in CLL patients (15% with 500 mg/m2 intravenous RTX and 12% with 1600 mg subcutaneous RTX) on combination therapy with chemotherapy agents [20]. Differences in doses and timing of administration, and use of different concomitant chemotherapy agents, may account for the variations in ADA levels observed between the FL and CLL patient populations [55].
3.3.2 Infusion/Injection-Related Reactions
Administration-related reactions are the most common adverse events in patients treated with mAbs [34, 56]. Infusion- and injection-related reactions are generally mild-to-moderate with all four anti-CD20 mAbs, with most reactions occurring with administration of the first dose and diminishing with subsequent doses [7, 8, 13, 14, 21, 23].
In the RTX studies in MS, the percentage of patients who experienced IRRs ranged from 67.1 to 78.3% following the initial infusion, with subsequent decrease to 20.3% to 22.6% after the second infusion at week 2 [7, 21] and 4.9% after the eighth infusion at week 74 [21]. Grade 3 infusion-associated adverse events were observed in 7.4% of RTX-treated patients [7]; no grade 4 infusion-associated adverse events were reported in either trial [7, 21]. In the phase III OPERA I/II and ORATORIO trials of OCR in MS, 34.3–39.9% of patients receiving OCR reported one or more IRRs [8, 23]. Serious infusion reactions were experienced by 0.3% of OCR-treated MS patients, some of whom required hospitalization [22]. The safety and tolerability of switching RMS patients from RTX to OCR has been studied in a phase III trial (NCT02980042) [57]. The rates of any IRR were similar between patients who switched to OCR after two or more courses of RTX and those who remained on RTX (14% on day 1 of infusion), and no Grade 3 or higher IRRs were observed in either group. It was suggested that B-cell levels could be partially related to IRR frequency, with 26.2% of those with CD19 and/or CD20 > 1% experiencing IRRs, versus 5.4% if ≤ 1% [57]. In the phase II study of UTX in MS, IRRs were experienced by 50% of UTX-treated patients; all were severity grade 1 or 2, and 77% of a total of 141 UTX infusions did not result in an IRR [14]. Due to the prevalence of IRRs in their clinical trials, intravenous administration of RTX, OCR, and UTX include premedication prior to administration to lessen these adverse events (Table 2).
The subcutaneous route of administration has been suggested to be associated with reduced instances of administration-related reactions compared with intravenous infusions [56]. The incidence of injection-related systemic reactions (IRSRs) in MS patients receiving a subcutaneous injection of OMB in the phase III ASCLEPIOS trials was 20.2% versus 15.5% in patients receiving placebo injections in the oral teriflunomide group [13]; 99.8% of symptoms observed were mild to moderate in severity [27], and most IRSRs occurred after the first injection. Injection-site reactions were experienced by 10.9% of OMB-treated patients versus 5.6% of those receiving placebo injections in the teriflunomide arm [13]. Subcutaneously administered OMB does not require premedication (Table 2). Although in a different patient population with different dosing, the proportion of CLL patients receiving intravenous OMB in combination with chemotherapy agents who experienced IRRs ranged from 25 to 56% after the first infusion and decreased with subsequent infusions; premedication is required prior to each infusion of intravenous OMB [28].
More frequent administration-related reactions have been suggested to occur with higher doses and shorter administration times. The phase II MIRROR study of subcutaneous OMB in RMS demonstrated a dose-dependent effect on IRSRs; they were more frequently reported with the 60 mg regimens than the 30 mg regimens or with placebo [10]. A study of RRMS patients that compared two dosing regimens for OCR (600 mg infused over 3.5 h vs. 2 h) reported a similar rate of IRRs (26.5% vs. 28.8%) following the first treatment, but IRRs following the second and third doses occurred more frequently with the shorter infusion regimen [58]. IRRs after the first infusion for UTX-treated patients in the phase II study were more common for patients receiving faster infusions of the 150 mg priming dose; IRR frequency did not increase with faster infusion rates or higher doses for subsequent infusions [14].
Humanized (OCR) and fully human (OMB) anti-CD20 mAbs are theorized to have lower IRR incidence and severity compared with the murine/human chimeric RTX due to decreased immunogenicity [56]. The relatively lower CDC potency of OCR and UTX compared with RTX and OMB has also been theorized to lower IRR incidence [34]. However, as there are no head-to-head trials comparing the incidence of administration-related reactions of the four mAbs, the clinical impact of these factors, including relative rates of hypersensitivity reactions, is yet to be determined.
3.3.3 Hypogammaglobulinemia and Infection Risk
A decline in serum immunoglobulin (Ig) levels is suggested to occur over time with anti-CD20 therapies. The effects of B-cell-depleting therapies on Ig levels, infection risk, long-term immunity, and response to vaccines are important considerations in routine MS disease management; these concerns have been heightened during the ongoing coronavirus disease 2019 (COVID-19) pandemic.
Across MS trials of RTX, OCR, and OMB, IgM levels were reduced more than IgG and IgA levels, while treatment with these mAbs was not associated with significantly increased infection risk in the short-term (up to 2.5 years) [7, 8, 12, 21, 23, 59]. Longer-term data regarding infection risk with OCR and OMB in MS patients are limited and additional data are expected in upcoming years. The longest-term safety data for anti-CD20 therapy comes from an analysis of 3194 patients with RA receiving RTX treatment (on background methotrexate) for up to 9.5 years. This study found an increased rate of serious infections among patients who developed low IgG levels. There were no differences in the serious infection rates for patients with low IgM or IgA levels [54]. In patients with MS treated with OCR, a 5.5-year assessment of serum Ig levels in the phase III (OPERA I, II and ORATORIO) and open-label extension studies also found an association between reductions in Ig levels occurring with more prolonged treatment and an increased rate of serious infections—this association was more pronounced with IgG than with IgM or IgA [60]. Mean levels of IgG and IgM were not observed to decrease below the LLN with monthly subcutaneous OMB treatment over up to 2.5 years compared with baseline in the phase III ASCLEPIOS trials; in individual patients with IgG or IgM levels below the LLN, no apparent association was observed between decreased Ig levels and infection risk [59]. Yet to be seen is whether the faster B-cell repletion of OMB compared with the other anti-CD20 mAbs (possibly reflecting differences in the extent of depletion in the tissues), along with sparing of splenic B cells with subcutaneous therapies compared with intravenous therapies, may lead to lower infection rates [34].
Rare cases of progressive multifocal leukoencephalopathy (PML), an opportunistic viral brain infection caused by the John Cunningham (JC) virus that can lead to death or severe disability, have occurred following treatment with RTX (in patients with hematological malignancies or autoimmune diseases receiving concurrent therapies) [19, 20], OCR [61], or intravenous OMB (in patients with CLL only, no confirmed cases in MS patients using subcutaneous dosing) [28]. In MS patients treated with RTX or OCR who developed PML, the cases were related to prior use of other DMTs such as natalizumab with a known association with PML [62, 63]. However, the first case of PML occurring with OCR monotherapy in a patient with MS without prior immunomodulatory DMT use was recently reported [61]. The occurrence, in a 78-year-old man with progressive MS following 2-year OCR treatment may have been due to both the immunomodulatory nature of OCR and age-related immunosenescence with a degree of lymphopenia [61]. This highlights the need to further evaluate the risk:benefit balance of anti-CD20 mAbs in patients with higher infection risks such as the growing elderly MS population [61, 63].
Whether the higher infection risks associated with anti-CD20 therapies due to their immunomodulatory nature increases the risk of severe COVID-19 has been of particular interest during the ongoing pandemic. Clinical evidence to date on the impact of RTX or OCR on COVID-19 severity has been mixed, with some studies suggesting either no association [64,65,66,67] or a slightly higher risk of hospitalization (but not of death) from COVID-19 [68,69,70]. Vaccine response has also been of particular concern during the pandemic. Potential differences in depth and breadth of B-cell depletion, and differential rates of reconstitution of B-cell subsets, may have important safety implications for humoral responses. Current evidence on the effect of B-cell-depleting therapies on humoral responses to vaccines is limited. The VELOCE phase IIIb study of OCR-treated patients with RMS showed that patients were able to mount a humoral response to non-live vaccines, albeit attenuated in comparison with levels seen in untreated patients or those receiving interferon-β1a [71]. A similar lack of humoral immune response was recently reported in a case study of a 44-year-old man with no evidence of disease activity for 3.5 years with OCR treatment, who received the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Pfizer vaccine [72]. The patient received his first vaccine dose 5 months after an OCR infusion, followed by the second vaccine dose 21 days later, then another OCR infusion 9 days after the second vaccine dose; seroconversion did not occur 27 days after the second vaccine dose [72]. The effects of OCR and OMB on vaccine response in patients with MS are being investigated in ongoing studies (NCT02545868, NCT04667117, NCT03650114). Recently issued COVID-19 messenger RNA (mRNA) vaccine guidance from the National Multiple Sclerosis Society encourages the vaccination of MS patients and suggests that the timing of vaccination could be coordinated with that of anti-CD20 treatment [73, 74].
4 Conclusion
Anti-CD20 therapies have been established as a highly effective and generally well-tolerated treatment for patients with MS. These therapies have broadened our understanding of the pathogenesis and progression of MS and, as a result of their efficacy and favorable safety profiles, have become a mainstay of treatment. Early initiation of anti-CD20 therapies has been shown to improve outcomes for MS patients, including reduced risk of relapses and slower disability accumulation [15,16,17]. Furthermore, these therapies may offer greater protection against relapse-associated worsening and disability progression independent of relapse [75], the latter of which is especially significant as recent findings suggest long-term disability worsening to be independent of relapse activity during the normal course of disease [76].
The distinct molecular and pharmacological attributes of RTX, OCR, OMB, and UTX provide important insights into their varying clinical effects. Differences in their attributes have implications for B-cell-depleting mechanisms and potency, dosing, immunogenicity, administration-related reactions, and infection risks. Current MS dose regimens for RTX, OCR, OMB, and UTX result in near-complete depletion of circulating B cells. Dose-dependent differences in the kinetics of B-cell reconstitution suggest that near-complete peripheral B-cell depletion may be associated with varying depths of depletion in tissues, as evidenced by the greater time lag in onset of reconstitution following higher-dose versus lower-dose regimens [10, 12, 45]. It also remains unclear whether near-complete peripheral blood depletion is necessary in MS as a robust clinical effect may be elicited with regimens that only partially deplete circulating B cells [10].
Comparative analyses, postmarketing trials, registry data, and pediatric trials of anti-CD20 therapies in MS will be important in advancing our understanding of the potential impact of B-cell depletion and differential reconstitution patterns on long-term immunity, infection risk, and humoral responses. The available evidence indicates that RTX, OCR, OMB, and UTX are generally well-tolerated, however it will be important to gain further experience with these therapies. Longer-term studies are ongoing and will provide valuable information on both the safety and efficacy of these therapies in MS patients over time.
References
Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. 2018;83(1):13–26.
Comi G, Bar-Or A, Lassmann H, Uccelli A, Hartung HP, Montalban X, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13–23.
Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. 2018;19(7):696–707.
Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021;20(6):470–83.
Sabatino JJ Jr, Zamvil SS, Hauser SL. B-cell therapies in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(2):a032037.
Ancau M, Berthele A, Hemmer B. CD20 monoclonal antibodies for the treatment of multiple sclerosis: up-to-date. Expert Opin Biol Ther. 2019;19(8):829–43.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus Placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Bar-Or A, Calabresi PA, Arnold D, Markowitz C, Shafer S, Kasper LH, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. 2008;63(3):395–400.
Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: the MIRROR study. Neurology. 2018;90(20):e1805–14.
Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–87.
Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. 2014;82(7):573–81.
Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–57.
Fox E, Lovett-Racke AE, Gormley M, Liu Y, Petracca M, Cocozza S, et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult Scler. 2020;2020:1352458520918375.
Stankiewicz JM, Weiner HL. An argument for broad use of high efficacy treatments in early multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2020;7(1):e636.
Buron MD, Chalmer TA, Sellebjerg F, Barzinji I, Christensen JR, Christensen MK, et al. Initial high-efficacy disease-modifying therapy in multiple sclerosis. A nationwide cohort study. Neurology. 2020;95(8):e1041–51.
He A, Merkel B, Brown JWL, Zhovits Ryerson L, Kister I, Malpas CB, et al. Timing of high-efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol. 2020;19(4):307–16.
Klein C, Lammens A, Schafer W, Georges G, Schwaiger M, Mossner E, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22–33.
RITUXAN® (rituximab) [package insert]. 2020. https://www.gene.com/download/pdf/rituxan_prescribing.pdf. Accessed 3 Jun 2021.
RITUXAN HYCELA® (rituximab and hyaluronidase human) [package insert]. 2021. https://www.gene.com/download/pdf/rituxan_hycela_prescribing.pdf. Accessed 3 Jun 2021.
Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–71.
OCREVUS® (ocrelizumab) [package insert]. 2021. https://www.gene.com/download/pdf/ocrevus_prescribing.pdf. Accessed 3 Jun 2021.
Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 2017;376(3):221–34.
Wolinsky JS, Arnold DL, Brochet B, Hartung H-P, Montalban X, Naismith RT, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998–1009.
Roche. FDA approves Roche’s OCREVUS® (ocrelizumab) shorter 2-hour infusion for relapsing and primary progressive multiple sclerosis. 2020. https://www.roche.com/media/releases/med-cor-2020-12-14.htm. Accessed 1 Feb 2021.
Emery P, Rigby W, Tak PP, Dörner T, Olech E, Martin C, et al. Safety with ocrelizumab in rheumatoid arthritis: results from the ocrelizumab phase III program. PLoS One. 2014;9(2):e87379.
KESIMPTA® (ofatumumab) [package insert]. 2020. https://www.novartis.us/sites/www.novartis.us/files/kesimpta.pdf. Accessed 3 Jun 2021.
ARZERRA® (ofatumumab) [package insert]. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125326s062lbl.pdf. Accessed 3 Jun 2021.
Taylor PC, Quattrocchi E, Mallett S, Kurrasch R, Petersen J, Chang DJ. Ofatumumab, a fully human anti-CD20 monoclonal antibody, in biological-naive, rheumatoid arthritis patients with an inadequate response to methotrexate: a randomised, double-blind, placebo-controlled clinical trial. Ann Rheum Dis. 2011;70(12):2119–25.
Lovett-Racke AE, Gormley M, Liu Y, Yang Y, Graham C, Wray S, et al. B cell depletion with ublituximab reshapes the T cell profile in multiple sclerosis patients. J Neuroimmunol. 2019;332:187–97.
TG Therapeutics. TG Therapeutics Announces Positive Topline Results from the ULTIMATE I & II Phase 3 Studies Evaluating Ublituximab Monotherapy for the Treatment of Patients with Multiple Sclerosis. 2020. https://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-announces-positive-topline-results-ultimate-i-ii. Accessed 23 Feb 2021.
Steinman L, Fox E, Hartung HP, Alvarez E, Qian P, Wray S, et al. Study design and patient demographics of the ULTIMATE phase III trials evaluating ublituximab (UTX), a novel glycoengineered anti CD20 monoclonal antibody (mAb), in patients with relapsing multiple sclerosis (RMS). Mult Scler J. 2019;25(2 Suppl):357–580.
Sorensen PS, Blinkenberg M. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord. 2016;9(1):44–52.
Cotchett KR, Dittel BN, Obeidat AZ. Comparison of the efficacy and safety of anti-CD20 B cells depleting drugs in multiple sclerosis. Multiple Scleros Relat Disord. 2021;49:102787.
Morschhauser F, Marlton P, Vitolo U, Lindén O, Seymour JF, Crump M, et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann Oncol. 2010;21(9):1870–6.
Touil I, Perrot C, Elain G, Weckbecker G. Ofatumumab and ocrelizumab differentially induce human primary B cell lysis by complement dependent cytotoxicity. Mult Scler J. 2019;25(1 Suppl):157–65.
Craigen JL, Mackus WJM, Engleberts P, Miller SR, Speller S, Chamberlain LC, et al. Ofatumumab, a human mab targeting a membrane-proximal small-loop epitope on CD20, induces potent NK Cell-mediated ADCC. Blood. 2009;114(22):1725.
Oflazoglu E, Audoly LP. Evolution of anti-CD20 monoclonal antibody therapeutics in oncology. mAbs. 2010;2(1):14–9.
Du J, Yang H, Guo Y, Ding J. Structure of the Fab fragment of therapeutic antibody Ofatumumab provides insights into the recognition mechanism with CD20. Mol Immunol. 2009;46(11–12):2419–23.
Bellon A, Sadoun A, Grivel K, Moulard M, Brune F, Prost J-F, et al. Comparison of Cell lysis mediated by LFB-R603 with that mediated by ofatumumab against cells expressing low levels of CD20. Blood. 2011;118(21):3913.
Le Garff-Tavernier M, Herbi L, de Romeuf C, Nguyen-Khac F, Davi F, Grelier A, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28(1):230–3.
Fox EJ, Mayer L, Aungst A, Mancione L, Rennie N, Roustan A, et al. Long-term safety, compliance, and effectiveness of ofatumumab in patients with relapsing multiple sclerosis: alithios phase 3b study. Mult Scler J. 2020;26:223–4.
Bar-Or A, Fox E, Goodyear A, Ludwig I, Bagger M, Häring D, et al. Onset of B-cell depletion with subcutaneous administration of ofatumumab in relapsing multiple sclerosis: results from the APLIOS bioequivalence study. In: Presented at the 5th Annual Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum; 2020; Phoenix, AZ. 2020.
ClinicalTrials.gov. Ofatumumab Subcutaneous Administration in Subjects With Relapsing-Remitting Multiple Sclerosis (MIRROR) Study Results. 2018. https://clinicaltrials.gov/ct2/show/results/ NCT01457924. Accessed 26 Feb 2021.
Genovese MC, Kaine JL, Lowenstein MB, Giudice JD, Baldassare A, Schechtman J, et al. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I/II randomized, blinded, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008;58(9):2652–61.
Roll P, Palanichamy A, Kneitz C, Dorner T, Tony H-P. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2377–86.
Nissimov N, Hajiyeva Z, Torke S, Grondey K, Brück W, Häusser-Kinzel S, et al. B cells reappear less mature and more activated after their anti-CD20-mediated depletion in multiple sclerosis. Proc Natl Acad Sci USA. 2020;117(41):25690–9.
US FDA. Guidance for industry: immunogenicity assessment for therapeutic protein products. In: US FDA. 2014.
European Medicines Agency. Guideline on Immunogenicity assessment of therapeutic proteins. European Medicines Agency.2017.
Hassanein M, Partridge MA, Shao W, Torri A. Assessment of clinically relevant immunogenicity for mAbs; are we over reporting ADA? Bioanalysis. 2020;12(18):1325–36.
Balsa A, Lula S, Marshall L, Szczypa P, Aikman L. The comparative immunogenicity of biologic therapy and its clinical relevance in psoriatic arthritis: a systematic review of the literature. Expert Opin Biol Ther. 2018;18(5):575–84.
Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. mAbs. 2010;2(3):256–65.
US FDA. Immunogenicity testing of therapeutic protein products—developing and validating assays for anti-drug antibody detection: guidance for industry. US FDA. 2019.
van Vollenhoven RF, Emery P, Bingham CO, 3rd, Keystone EC, Fleischmann RM, Furst DE, et al. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann Rheum Dis. 2013;72(9):1496–502.
Saffari F, Jafarzadeh A, Kalantari Khandani B, Saffari F, Soleimanyamoli S, Mohammadi M. Immunogenicity of rituximab, trastuzumab, and bevacizumab monoclonal antibodies in patients with malignant diseases. Int J Cancer Manag. 2018;11(11):e64983.
Du FH, Mills EA, Mao-Draayer Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights. 2017;8(1):12.
Alvarez E, Nair K, Shelton I, Selva S, Voge N, Zanganeh N, et al. Evaluating the tolerability and safety of switching from rituximab to ocrelizumab: infusion related reactions in relapsing forms of multiple sclerosis (P4.2-015). Neurology. 2019;92(15 Suppl):P4.2-015.
Hartung HP, Berger T, Bermel RA, Brochet B, Carroll WM, Holmøy T, et al. Shorter infusion time of ocrelizumab: results from the randomized, double-blind ENSEMBLE PLUS substudy in patients with relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2020;46:102492.
de Seze J, Bar-Or A, Correale J, Cross A, Kappos L, Selmaj K, et al. Effect of ofatumumab on serum immunoglobulin levels and infection risk in relapsing multiple sclerosis patients from the phase 3 ASCLEPIOS I and II trials. In: Presented at the Consortium of Multiple Sclerosis Centers (CMSC) Virtual Annual Meeting. 2020.
Derfuss T, Weber MS, Hughes R, Wang Q, Sauter A, Koendgen H, et al. ECTRIMS 2019—oral presentations: serum immunoglobulin levels and risk of serious infections in the pivotal Phase III trials of ocrelizumab in multiple sclerosis and their open-label extensions. Mult Scler J. 2019;25(2 Suppl):3–130.
Patel A, Sul J, Gordon ML, Steinklein J, Sanguinetti S, Pramanik B, et al. Progressive multifocal leukoencephalopathy in a patient with progressive multiple sclerosis treated with ocrelizumab monotherapy. JAMA Neurol. 2021;78(6):736–40.
Luna G, Alping P, Burman J, Fink K, Fogdell-Hahn A, Gunnarsson M, et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 2020;77(2):184–91.
Sul J, Patel A, Gordon ML, Steinklein J, Sanguinetti S, Pramanik B, et al. Progressive multifocal leukoencephalopathy in a patient on ocrelizumab monotherapy (4875). Neurology. 2020;94(15 Suppl):4875.
Chaudhry F, Bulka H, Rathnam AS, Said OM, Lin J, Lorigan H, et al. COVID-19 in multiple sclerosis patients and risk factors for severe infection. J Neurol Sci. 2020;418:117147.
Louapre C, Collongues N, Stankoff B, Giannesini C, Papeix C, Bensa C, et al. Clinical characteristics and outcomes in patients with coronavirus disease 2019 and multiple sclerosis. JAMA Neurol. 2020;77(9):1079–88.
Montero-Escribano P, Matías-Guiu J, Gómez-Iglesias P, Porta-Etessam J, Pytel V, Matias-Guiu JA. Anti-CD20 and COVID-19 in multiple sclerosis and related disorders: a case series of 60 patients from Madrid, Spain. Multiple Scleros Relat Disord. 2020;42:102185.
Parrotta E, Kister I, Charvet L, Sammarco C, Saha V, Charlson RE, et al. COVID-19 outcomes in MS: observational study of early experience from NYU multiple sclerosis comprehensive care center. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e835.
Sormani MP, De Rossi N, Schiavetti I, Carmisciano L, Cordioli C, Moiola L, et al. Disease-modifying therapies and coronavirus disease 2019 severity in multiple sclerosis. Ann Neurol. 2021;89(4):780–9.
Sahraian MA, Azimi A, Navardi S, Ala S, Naser Moghadasi A. Evaluation of the rate of COVID-19 infection, hospitalization and death among Iranian patients with multiple sclerosis. Multiple Scleros Relat Disord. 2020;46:102472.
Simpson-Yap S, De Brouwer E, Kalincik T, Rijke N, Hillert J, Walton C, et al. SS02.04—First results of the COVID-19 in MS Global Data Sharing Initiative suggest anti-CD20 DMTs are associated with worse COVID-19 outcomes. In: 8th Joint ACTRIMS-ECTRIMS Virtual meeting. 2020.
Bar-Or A, Calkwood JC, Chognot C, Evershed J, Fox EJ, Herman A, et al. Effect of ocrelizumab on vaccine responses in patients with multiple sclerosis: the VELOCE study. Neurology. 2020;95(14):e1999–2008.
Khayat-Khoei M, Conway S, Rubinson DA, Jarolim P, Houtchens MK. Negative anti-SARS-CoV-2 S antibody response following Pfizer SARS-CoV-2 vaccination in a patient on ocrelizumab. J Neurol. 2021. https://doi.org/10.1007/s00415-021-10463-3.
National Multiple Sclerosis Society. COVID-19 vaccine guidance for people living with MS. 2020. https://www.nationalmssociety.org/coronavirus-covid-19-information/multiple-sclerosis-and-coronavirus/covid-19-vaccine-guidance. Accessed 18 Feb 2021.
National Multiple Sclerosis Society. Timing MS medications with COVID-19 mRNA vaccines. 2020. https://www.nationalmssociety.org/coronavirus-covid-19-information/multiple-sclerosis-and-coronavirus/covid-19-vaccine-guidance/Timing-MS-Medications-with-COVID-19-mRNA-Vaccines. Accessed 18 Feb 2021.
Kappos L, Wolinsky JS, Giovannoni G, Arnold DL, Wang Q, Bernasconi C, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132–40.
Cree BAC, Hollenbach JA, Bove R, Kirkish G, Sacco S, Caverzasi E, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–66.
Casan JML, Wong J, Northcott MJ, Opat S. Anti-CD20 monoclonal antibodies: reviewing a revolution. Hum Vaccin Immunother. 2018;14(12):2820–41.
Babiker HM, Glode AE, Cooke LS, Mahadevan D. Ublituximab for the treatment of CD20 positive B-cell malignancies. Expert Opin Investig Drugs. 2018;27(4):407–12.
Payandeh Z, Bahrami AA, Hoseinpoor R, Mortazavi Y, Rajabibazl M, Rahimpour A, et al. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomed Pharmacother. 2019;109:2415–26.
Acknowledgements
Medical writing support was provided by Grace Jeong, PhD and Akua Adu-Boahene, MD, MPH of Alphabet Health (New York, NY, USA), and was funded by Novartis Pharmaceuticals Corporation. This manuscript was developed in accordance with Good Publication Practice (GPP3) guidelines. Authors had full control of the content and made the final decision on all aspects of this publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors received no honoraria related to the development of this publication. Medical writing support and the open access fee was funded by Novartis Pharmaceuticals Corporation.
Conflict of interest
Amit Bar-Or has participated as a speaker in meetings sponsored by, and received consulting fees and/or grant support from, Accure, Atara Biotherapeutics, Biogen, BMS/Celgene/Receptos, GlaxoSmithKline, Gossamer, Janssen/Actelion, Medimmune, Merck/EMD Serono, Novartis, Roche/Genentech, and Sanofi-Genzyme. Susan O’Brien has received consulting fees from Amgen, Astellas Pharma, Celgene, GlaxoSmithKline, Janssen Oncology, Aptose Biosciences, Vaniam Group, AbbVie, and Alexion; grant support from Kite Pharma, Regeneron, and Acerta Pharma; and grant support and consulting fees from Gilead Sciences, Pharmacyclics, TG Therapeutics, Pfizer, and Sunesis Pharmaceuticals. Michael L. Sweeney has received speaking fees and served on advisory boards for Novartis and Genentech. Edward J. Fox has received compensation for research, consulting, speakers' bureau, and/or advisory work from AbbVie, Alexion, Biogen, Bristol-Myers Squibb, Chugai, EMD Serono, Genentech/Roche, MedDay, Novartis, Sanofi Genzyme, Teva, and TG Therapeutics. Jeffrey A. Cohen has received personal compensation for consulting from Adamas, Atara, Bristol-Myers Squibb, Convelo, MedDay, and Mylan; and for serving as an Editor of Multiple Sclerosis Journal.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
All authors made substantial contributions to the manuscript concept/design, critically reviewed and revised the manuscript drafts and drafts of the corresponding video, and provided final approval of the manuscript and video.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bar-Or, A., O’Brien, S.M., Sweeney, M.L. et al. Clinical Perspectives on the Molecular and Pharmacological Attributes of Anti-CD20 Therapies for Multiple Sclerosis. CNS Drugs 35, 985–997 (2021). https://doi.org/10.1007/s40263-021-00843-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00843-8