
Abstract
Growing evidence indicates that B cells play a key role in the pathogenesis of multiple sclerosis (MS). B cells occupy distinct central nervous system (CNS) compartments in MS, including the cerebrospinal fluid and white matter lesions. Also, it is now known that, in addition to entering the CNS, B cells can circulate into the periphery via a functional lymphatic system. Data suggest that the role of B cells in MS mainly involves their in situ activation in demyelinating lesions, leading to altered pro- and anti-inflammatory cytokine secretion, and a highly effective antigen-presenting cell function, resulting in activation of memory or naïve T cells. Clinically, B cell-depleting agents show significant efficacy in MS. In addition, many disease-modifying therapies (DMTs) traditionally understood to target T cells are now known to influence B cell number and function. One of the earliest DMTs to be developed, glatiramer acetate (GA), has been shown to reduce the total frequency of B cells, plasmablasts, and memory B cells. It also appears to promote a shift toward reduced inflammation by increasing anti-inflammatory cytokine release and/or reducing pro-inflammatory cytokine release by B cells. In the authors’ opinion, this may be mediated by cross-reactivity of B cell receptors for GA with antigen (possibly myelin basic protein) expressed in the MS lesion. More research is required to further characterize the role of B cells and their bidirectional trafficking in the pathogenesis of MS. This may uncover novel targets for MS treatments and facilitate the development of B cell biomarkers of drug response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
B cells play a key role in the pathogenesis of multiple sclerosis (MS); this likely involves their activation within lesions, leading to altered cytokine secretion and a predominant inflammatory environment. |
The therapeutic effect of glatiramer acetate (GA) appears to be mediated, in part, by activation of B cells, which results in a shift toward reduced inflammation. |
One possible explanation is that this involves cross-reactivity of B cell receptors for GA with antigen(s) expressed in MS lesions. |
1 Introduction
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system (CNS) that is characterized by demyelination and loss of axons and neurons, leading to acceleration of brain volume loss [1,2,3,4]. These pathophysiological changes result in neurological, physical, cognitive, and psychological impairments [5]. MS is an immune-mediated disease that predominantly involves the adaptive immune system, although cells of the innate immune system (e.g., natural killer cells, dendritic cells, and astrocytes) are also implicated [6, 7].
The key cells of the adaptive immune system are T lymphocytes (T cells) and B lymphocytes (B cells). Historically, T cells have been considered the main drivers in the pathogenesis of MS. This was supported by observations including the higher number of T cells than B cells in MS lesions [8, 9] and the transfer of experimental autoimmune encephalomyelitis (EAE)—the most widely used animal model of MS—to naïve recipient animals by T cells [10, 11]. However, it is now understood that B cells play a pivotal role throughout the course of MS [12]. Most recently, it has been shown that specific B cell-depleting agents have robust efficacy in patients with MS [13]. This, in turn, has prompted a re-examination of the mechanism of action of disease-modifying therapies (DMTs) traditionally thought to target T cells [14, 15].
In this review, we provide an overview of the evidence for the role of B cells in the pathogenesis of MS, and evaluate the preclinical and clinical data implicating B cells in the mechanism of action of one of the earliest DMTs to be developed for MS—glatiramer acetate (GA).
2 The Brain and the Immune System in Multiple Sclerosis (MS)
Historically, the brain was thought to be immunoprivileged, based on its isolation from the immune system by the blood–brain barrier (BBB), its perceived lack of lymphatic drainage, and the immunocompetence of microglia, an innate immune cell within the CNS [16]. In this context, MS was viewed as a peripherally driven disease, whereby peripherally activated immune cells gained access to the CNS via a compromised BBB [17]. B cells were thought to mature in the periphery before migrating to the CNS via one or more cerebrovascular pathways: via the choroid plexus into the cerebrospinal fluid (CSF) (across the blood–CSF barrier); via the parenchymal vessels into the perivascular space (across the BBB); or via the post-capillary venules into the subarachnoid and Virchow–Robin spaces (also across the BBB) [18]. The transmigration of B cells across the BBB, like that of T cells, is thought to be mediated by chemokines and adhesion molecules [18,19,20]. Once inside the CNS, B cells undergo clonal expansion and somatic hypermutation in ectopic lymphoid follicles, which are generally identified in the subarachnoid space near the meninges [21,22,23].
Several observations have since altered these fundamental concepts. In 2012, it was demonstrated for the first time that B cell trafficking between the periphery and CNS could be bidirectional [24]. Three years later, it was reported that the CNS does have a functional lymphatic system [25, 26]. This transports CSF and lymphocytes mainly into the deep cervical nodes [26, 27]. Immunohistochemical analyses have demonstrated that the meningeal lymphatic fluid contains both B and T cells, indicating that it provides a novel route for trafficking lymphocytes, including antigen-carrying B cells [28], out of the CNS [18, 26, 29]. B cell populations move back into the circulation via the thoracic duct, circulate to the brain, and then cross the BBB (e.g., via the post-capillary venules), leading to new inflammation that characterizes MS [18]. These B cells infiltrate the brain parenchyma, circulate in the CSF, populate ectopic lymphoid follicles in the subarachnoid space, and transit back to the peripheral lymphoid compartment [18]. It has also been suggested that effector B cells (and T cells) may be ‘imprinted’ with CNS-specific trafficking programs in the cervical lymph nodes to allow them to target CNS antigens [27].
Another concept that has evolved in recent years relates to the site of clonal expansion of B cells. It is now clear that this can take place in the periphery, CNS, or cervical lymph nodes [12]. In one study, which examined paired CNS and cervical lymph node autopsy tissue samples from five patients with MS, the founding clones and much of the subsequent maturation appeared to take place in the cervical lymph nodes [30]. It has been suggested that this could provide a mechanism for epitope spreading, a phenomenon whereby the antigenic target of the CNS inflammatory attack shifts over time as injury exposes additional epitopes [12].
3 The Role of B Cells in MS
Historically, MS has been viewed as an autoimmune condition regulated predominantly by T cells activated in the periphery, most likely via molecular mimicry or bystander activation [28]. Thus, the pathophysiology of MS was thought to involve suppression of regulatory (anti-inflammatory) T cells and subsequent expansion of the autoreactive (pro-inflammatory) T helper cell (Th) types Th1 and Th17 [31]. However, evidence for a key role for B cells has been accumulating over recent years, from both animal and human studies. This includes the localization of B cells in specific CNS compartments in MS, including the CSF, and in post-mortem brain and lesion samples (Table 1).
The most recent evidence for B cell involvement has been generated by clinical trials of B cell-depleting therapies (rituximab, ocrelizumab, and ofatumumab), which have shown significant reductions in inflammatory lesions and clinical endpoints (relapses and MS-related disability in relapsing-remitting MS [RRMS] and disability progression in primary-progressive MS [PPMS]) [13, 17]. Although it has been shown that a subset of T cells express CD20, and that anti-CD20 therapies deplete this subset [45,46,47], such T cells constitute only 3–5% of circulating T cells in humans [48] and any therapeutic effects of their depletion has yet to be determined [49].
As a consequence of the results of studies of B cell-depleting therapies, the mechanism of action of more established DMTs thought to target T cells has been re-evaluated in light of the emerging importance of B cells in the pathogenesis of MS [14, 15, 17]. Indeed, a number of DMTs, including alemtuzumab [50], daclizumab [51], fingolimod [52, 53], dimethyl fumarate [54], cladribine [55], and GA (see Sect. 4), have now been shown to exert an effect on B cells.
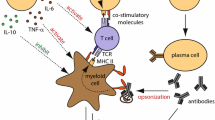
B cells have a number of functions within the immune system that could contribute to the tissue damage that characterizes MS. These include antibody production, antigen presentation, cytokine release, and release of factors that contribute to the formation and/or maintenance of persisting immune cell aggregates in the meninges (Fig. 1) [12, 17, 56].

Roles of effector and regulatory B cells in MS. B cells can modulate inflammation by a presenting antigen to effector T cells (which produce pro-inflammatory Th cells) or anti-inflammatory Tregs; b producing antibodies, which stimulate complement activation when in contact with antigen; and c producing pro-inflammatory and anti-inflammatory cytokines. In MS, there is a shift in the balance, with pro-inflammatory effects in the CNS predominating, leading to demyelination. There is also evidence of ongoing activation and clonal expansion of B cells in ectopic lymphoid follicles (d) in the CNS in MS. The mechanism of action of GA is thought to involve a shift toward reduced inflammation by increasing anti-inflammatory cytokine release and/or reducing pro-inflammatory cytokine release from B cells (1). In the authors’ opinion, the most likely sequence of events is that GA activates B cells in the periphery (2) and following entry to the CNS, these GA-specific B cells are reactivated, resulting in secretion of anti-inflammatory cytokines, which results in bystander suppression of disease activity. For further information and supporting references, please see the text. CNS central nervous system, GA glatiramer acetate, IFN interferon, IL interleukin, MS multiple sclerosis, TGF transforming growth factor, Th T helper cell, TNF tumor necrosis factor, Treg regulatory T cell
3.1 Antibody Production
In autoimmune conditions, autoreactive B cells can be activated to produce autoantibody-secreting plasmablasts via both T cell-dependent and T cell-independent pathways [57]. For example, T cell-independent activation of B cells can be mediated by Toll-like receptors on B cells [58], which enhances antibody production and other B cell functions, including antigen presentation and cytokine production [59]. T cell-independent activation of B cells can also be mediated by B cell-activating factor (BAFF) [60], which is produced by astrocytes and serum levels of which are elevated in a number of immune disorders, including MS [58]. Activation of receptors for BAFF results in the survival of plasmablasts generated from memory B cells by preventing apoptosis of immunoglobulin (Ig)-secreting cells, leading to a cycle of continuous antibody-mediated inflammation and tissue destruction [58, 61, 62].
A role for autoreactive, pathological antibodies in the pathophysiology of MS has been widely investigated. Central to this line of research is the presence of oligoclonal (Ig) bands in the CSF, a hallmark diagnostic finding in MS [56]. However, no single specific antigen has been identified as being recognized by these intrathecal antibodies [63] and it remains unclear whether they actively contribute to the pathogenesis of MS [56]. In addition, histopathological analysis of demyelinating lesions from patients with MS show that only a subset contain Ig and activated complement at sites of active myelin destruction [38, 64]. Furthermore, B cell-depleting agents do not decrease class-switched antibody-producing plasma cells and, thus, total IgA or IgG plasma concentrations are not reduced [28, 36, 41,42,43, 65]. This is not surprising as the target of B cell-depleting agents—CD20—is not present on plasma cells [36, 66]. Finally, rituximab has been reported to have no consistent effect on IgG concentrations or oligoclonal band numbers in the CSF of patients with MS [66]. Taken together, these data indicate that it is unlikely that antibodies secreted from B cells play a major role in the pathogenesis of MS.
3.2 Antigen Presentation
B cells act as highly efficient antigen-presenting cells (APCs), provided that antigen is first specifically recognized, internalized, and processed before being presented to T cells via major histocompatibility complex (MHC) [67]. This leads to activation of T cells [56] and it is plausible that this contributes to the pathogenesis of MS. Indeed, it has been shown that memory B cells from some patients with RRMS elicit CD4+ T cell proliferation in response to myelin antigens [36], suggesting that memory B cells may be involved in the pathogenesis of MS. However, disease activity does not appear to be related to CD4 counts [68]. In addition, anti-thymocyte globulin (an antibody against human T cells) and therapies targeting CD3 or CD4 have not proven effective in MS [69,70,71,72,73,74,75]. This suggests that the antigen-presenting activity of peripheral B cells may not be central to their pathogenic role in MS; instead, they may proliferate in situ when they come into contact with their target antigen, as demonstrated in MS plaques [12]. Alternatively, their pathogenicity may not depend on an interaction with pre-existing memory T cells in the circulation, as memory B cells are capable of inducing naïve T cells to mature into memory T cells in the absence of other competent APCs [76]. In addition, under certain conditions, memory B cells can function independently of T cells, as discussed in Sect. 3.1.
3.3 Cytokine Production
The most likely mechanisms by which B cells play a role in the pathogenesis of MS are the production of pro-inflammatory cytokines and the support of an inflammatory environment, which includes activated astrocytes that can produce pro-inflammatory cytokines, including tumor necrosis factor (TNF), and induce nitric oxide synthase activity, leading to increased nitric oxide production [77]. Activated B cells can produce both pro-inflammatory cytokines (e.g., interferon [IFN]-γ, TNF-α, and interleukin [IL]-6) and anti-inflammatory cytokines (e.g., IL-10 and transforming growth factor-β) [17, 56]. It has been shown that B cells from patients with MS show aberrant pro-inflammatory cytokine responses to stimuli, including increased release of IFN-γ and TNF-α [17]. It has been suggested that this contributes, at least in part, to new relapsing MS disease via bystander activation of disease-relevant pro-inflammatory T cells, although it is acknowledged that pro-inflammatory cytokines have multiple functions in different pathological contexts [78]. Secretion of TNF-α by B cells can also stimulate astrocytes to produce BAFF, which, in turn, amplifies B cell-dependent autoimmunity [77, 79]. These data suggest that there may be a reciprocal interaction between B cells and astrocytes in the pathogenesis of MS. This is an interesting observation given that astrogliosis is one of the pathological hallmarks of MS [6].
The role of regulatory B cells—which foster an anti-inflammatory environment via their interaction with regulatory T cells and APCs—in the pathogenesis of MS is currently unclear [17]. Some, but not all, studies have shown that B cells from patients with MS have a reduced capacity to secrete IL-10 and that patients with MS have a lower proportion of IL-10-producing regulatory B cells than healthy individuals without MS [17].
3.4 Meningeal Infiltrates
B cell-rich infiltrates in the meninges may form ectopic lymphoid follicles, such that B cell maturation can be sustained locally within the CNS, thus forming an immunocompetent niche [39]. These B cell follicles, which have been found in patients with secondary-progressive MS (SPMS), are associated with meningeal inflammation that correlates with gray matter cortical demyelination and an accelerated clinical course [22, 39, 80]. As B cell follicles have been found adjacent to subpial cortical lesions, it has been suggested that soluble factors diffusing from the follicles play a pathogenic role in SPMS [39]. Further evidence for the production of toxic factors by B cells in patients with MS comes from the observation that, unlike B cells from control subjects, those from patients with MS can (under ex vivo conditions) induce the death of neurons and oligodendrocytes via secreted factors [81]. The cytotoxicity was independent of Ig and multiple cytokines and was not complement mediated, but involved apoptosis. The identity of the secreted factors has yet to be determined, but they are primarily > 300 kDa in size, and potential candidates are proteins, lipids, and/or microRNAs present in extracellular vesicles and exosomes [81].
Unlike SPMS, B cell follicles do not appear to be present in PPMS [39, 82], although there are significant sampling biases due to the rarity of tissue samples from patients with early MS and PPMS. There are significant clusters of T and B cells in the meninges of patients with PPMS and diffuse meningeal inflammation is thought to play a role in the pathogenesis of cortical gray matter lesions, as well as in an increased rate of clinical progression in this MS phenotype [82]. Meningeal inflammation has also been detected in patients with RRMS; although this is to a lesser extent than observed in patients with progressive disease, it nevertheless indicates that such inflammation is not exclusively a late phenomenon [83]. Furthermore, the presence of inflammation was associated with patient age and disease severity [84], suggesting that it may not differ between phenotypes when these factors, along with disease duration, are taken into account. Interestingly, in a recent study using ultra-high-field magnetic resonance imaging (MRI) in 29 patients with MS (21 of whom had RRMS), 26 (90%) had at least one focus of leptomeningeal enhancement [85], implying a greater prevalence of inflammation than detected previously with less sensitive MRI techniques and histological studies.
4 Glatiramer Acetate (GA): B Cell-Mediated Mechanisms
GA has traditionally been viewed as a T cell-targeting agent. Its beneficial effects were thought to reflect induction of GA-specific T cells, resulting in a shift of the T cell balance from a dominant pro-inflammatory phenotype (Th1/Th17) to an anti-inflammatory phenotype (Th2/T regulatory cells) [86,87,88]. However, emerging data indicate that GA also targets B cells, as summarized in Table 2 and discussed in more detail in Sects. 4.1–4.6.
4.1 B Cell Antigen Presentation of GA
Jackson et al. [69] conducted a series of experiments using B cells from mice and from patients with MS, demonstrating that GA binds directly to murine and human B cell receptors (BCRs). When B cells from mice that were wild-type, MHC I-deficient, MHC II-deficient, or BCR-transgenic for hen-egg lysozyme were incubated with fluorochrome-conjugated GA, most of the observed GA binding was due to a specific interaction with the BCR. After binding to BCRs in cells from mice, GA was shown to be internalized and then presented by MHC II, with subsequent activation of purified murine CD4+ T cells. These data indicate that B cells act as APCs for GA [69]. Furthermore, the efficacy of GA in EAE is abrogated in mice with transgenic BCRs, indicating that the interaction of GA with BCRs is central to its mechanism of action [69]. It has also been shown that GA may bind directly to MHC II, without internalization and processing [110, 111]; as GA is composed of a mixture of peptides of varying lengths, this is more likely to occur for smaller peptides. However, a role for such non-specific binding is not supported by data showing that GA is ineffective or has inconsistent effects in other autoimmune conditions [112,113,114] (Joel Kaye, unpublished data).
The presentation of GA (by memory and naïve B cells) to ex vivo T cells from patients with RRMS has been shown to induce inflammatory cytokine secretion in only a small number of samples [36]. Instead, it has been suggested that memory cells contribute to the expansion of regulatory T cells in patients being treated with GA [36].
While the present review focuses on the effects of GA on B cells, there is also evidence to suggest that GA has an effect on myeloid-derived APCs; for further information see Prod’homme and Zamvil [115].
4.2 Effect of GA on the Amount of Circulating B Cells
GA alters the profile of circulating B cells in patients with MS. It has been shown to reduce the total frequency of B cells (as a percentage of all lymphocytes), plasmablasts, and memory cells (as a percentage of all CD19+ B cells) in patients with RRMS [96]. GA treatment has also been shown to induce a specific and significant decrease in circulating CD19+ B cells, although the investigators acknowledged that the specific B cell subsets affected (pro- or anti-inflammatory) remain to be established [94]. GA increased the number of naïve B cells in patients with RRMS [96], while B1 cells [99] and serum IgG concentrations seemed to be unaffected [17].
It is also interesting to note that GA has been shown to downregulate the B cell survival factors BAFF and a proliferation-inducing ligand (APRIL), potentially leading to apoptosis of autoreactive B cells [90]. Expression of these cytokines is associated with EAE and MS progression, and the investigators suggested that their downregulation contributes to the therapeutic benefits of GA [90].
4.3 GA and Antibody Production
Almost all patients treated with GA generate non-neutralizing anti-GA (IgG) antibodies [86, 92, 93, 98, 101, 103]. The predominant IgG isotype varies between studies and may change over time (see Table 2), but several studies have shown a persistent increase in IgG4 antibodies [86, 92, 101], which do not activate complement or bind to the Fc receptor on effector cells [86]. These findings are consistent with a shift to Th2 responses, as anti-inflammatory cytokines regulate isotype switching to IgG4 in plasma cells [92]. It is notable, however, that anti-GA antibody concentrations do not appear to correlate with clinical outcomes [92, 101].
4.4 Effect of GA on Cytokine Production by B Cells in Humans
Several studies have shown that GA alters cytokine production by B cells. More specifically, GA appears to promote a shift toward reduced inflammation by increasing anti-inflammatory cytokine release and/or reducing the release of pro-inflammatory cytokines.
In the clinical setting, Jackson et al. [69] evaluated the effect of GA on the secretion of a range of cytokines from purified B cells from treatment-naïve patients with MS. They demonstrated reduced concentrations of the pro-inflammatory cytokines IL-6 and TNF-α in 50% of the samples, with no consistent alteration in other cytokines [69]. In another study evaluating cytokine production by B cells from GA-treated patients with MS, GA was found to restore the ability of B cells to produce IL-10 (anti-inflammatory) and to reduce the secretion of lymphotoxin-α [96]. Taken together with the results for antigen presentation described in Sect. 4.1, these data suggest that the mechanism of action of GA on B cells includes the presentation of GA to peripheral T cells in the context of an anti-inflammatory cytokine setting [69]. More specifically, it is the authors’ opinion that GA-specific B cells enter the brain during the acute inflammation that characterizes MS; these B cells bind to myelin basic protein (MBP) or another CNS antigen, promoting bystander suppression of disease activity by activating B cells to produce anti-inflammatory cytokines that inhibit the cells driving MS. In this context, it is interesting to note that GA is not effective in animal models of lupus [113], has shown inconsistent outcomes in animal models of rheumatoid arthritis [114] (Joel Kaye, unpublished data), and was not effective in humans with neuromyelitis optica [112], suggesting its mechanism of action may be specific to MS pathophysiology.
4.5 The Role of B Cells in the Effect of GA in Experimental Autoimmune Encephalomyelitis in Mice
Studies have shown that when purified B cells are transferred from GA-treated mice to recipient mice with EAE, EAE is suppressed [69, 91, 97]. In mice that received GA-treated B cells, the onset and extent of disease presentation were significantly reduced compared with controls [91]; these mice also had substantially fewer neurological deficits [97].
Furthermore, the beneficial effects of GA were attenuated in B cell-deficient animals [97], suggesting that B cells are important for the protective effects of GA in EAE. The beneficial effects of GA in this context are thought to reflect a bias toward an anti-inflammatory environment [90, 97]. In agreement with the data in humans, EAE mice treated with GA demonstrated increased production of anti-inflammatory cytokines (IL-4, IL-10, and IL-13) by B cells and reduced production of pro-inflammatory cytokines (IL-6, IL-12, and TNF-α) [90].
4.6 Other Effects of GA on B Cells
Ireland et al. [96] evaluated whether B cells from patients with MS receiving GA were functionally distinct from those of treatment-naïve patients by exposing peripheral B cells from both sets of patients to stimulatory conditions that would induce the major pathways of B cell activation. They observed a lack of proliferative responses to high-dose CD40 ligand combined with other activation signals in B cells from patients receiving GA therapy [96]. The investigators suggested that GA may alter pathways that are important for integrating these signals with other functional pathways modulated by GA.
MS is associated with increased concentrations of certain cell-bound adhesion molecules on immune cells, including B cells, facilitating their extravasation into the CNS [102]. GA has been shown to reduce concentrations of these molecules on immune cells, including intercellular adhesion molecule-3 on B cells [102]. However, Sellner et al. [102] highlighted that, although these alterations may contribute to the efficacy of GA, the time course of the changes (which were most pronounced after 6 months) and their non-selective nature suggest that they are secondary to other immunoregulatory phenomena.
5 Summary and Future Perspectives
Evidence has been accumulating in recent years to support a central role for B cells in the pathogenesis of MS. Indeed, current data on the role of B cells fulfill eight of nine standard criteria for establishing causality [116]. Of course, the immunopathogenesis of MS is highly complex, with reciprocal interactions between immune cell populations and potentially glia (e.g., astrocytes) within the CNS; it is therefore difficult to dissect the roles of specific subsets. Nevertheless, the demonstration of a robust effect of B cell-depleting therapies in patients with MS provides evidence of the central role of B cells. The extent to which this is mediated by autoreactive antibodies is unclear, as no single antigen has been identified; studies of cell culture supernatants from B cell polyclonal stimulation cultures for anti-CNS antibody arrays have shown a large range of antibody responses (Stefanie Kuerten, unpublished data). Furthermore, evidence of antibody deposition in MS plaques only occurs in a subset of patients with MS [38, 64] and the B cell-depleting therapy rituximab, which is effective for treating MS, does not reduce antibody levels in the CSF [66]. It is more likely that B cells are involved in the development of inflammatory MS lesions via in situ activation in the MS plaque leading to alterations in pro- and anti-inflammatory cytokine secretion. It remains to be established whether specific B cell subsets play a specific role, or if the role of B cells differs between patients, changes over the course of MS, or varies according to the MS phenotype. Recently, the role of memory B cells as a target for DMTs has been highlighted [14, 117] and Baker et al. [14] have proposed that memory B cell depletion may represent a common, unifying mechanism of action for DMTs [14]. Interestingly, it has been suggested that the secretion of functionally diverse cytokines by B cells may play a role in determining MS disease phenotypes [118]. In addition, it has been shown that a subgroup of patients with MS display brain-reactive B cell responses that may be associated with clinical relapse [33]; the clinical relevance of this observation is not yet clear.
GA is a widely used DMT, the mechanism of action of which has been investigated mainly in the context of T cells. However, as summarized in the current article, it has now been shown to have a number of effects on B cells, which may underlie its efficacy in terms of reducing inflammatory lesions and relapse rates [119]. We speculate that these may include activation of GA-specific BCRs by cross-reactivity with antigen (possibly MBP) expressed in the acute MS lesion, leading to bystander suppression and possibly activation of other inhibitory T cell or B cell pathways. Thus, GA combines a mechanism of action that involves B cells, with a well-established and favorable safety profile [120]. Interestingly, patients with RRMS treated with GA can be subdivided into B cell responders and non-responders, based on the presence or absence of brain-specific B cells in the blood [100]. The presence of these cells has been correlated with responsiveness to GA [100], suggesting that, in the future, they may act as a marker by which to identify treatment responders. Another interesting area of research is the use of GA following rituximab induction therapy, on the basis that it may induce new B cell-dependent regulatory circuits as the B cell population recovers from induction. Results from a small study in patients with relapsing MS suggest that this approach may provide superior efficacy compared with GA monotherapy [121]. Perhaps the most intriguing area for further research is development of the potential effect of GA on regulatory B cells, which are stimulated in the periphery before entering the CNS to be activated within MS plaques. This raises the possibility of developing similar CNS antigens (possibly combined with an appropriate adjuvant) with a similar mechanism of action, which enhance the clinical effects but maintain the safety profile of GA.
6 Conclusion
In summary, the recent identification of lymphatic connections between the subarachnoid space and the deep cervical lymph nodes, and evidence of clonally related B cells in both the CNS and these lymph nodes suggest the presence of a specific brain to lymph node circuit for B cells in MS. This will require further investigation and may represent another target for future MS therapeutics. Other important advances include the demonstration that most approved DMTs for MS, including GA, have effects on B cells that may explain at least part of their efficacy. The converging lines of evidence suggest that B cells should be a key target of future research into both the causes and treatment of MS.
References
Carassiti D, Altmann DR, Petrova N, et al. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol Appl Neurobiol. 2018;44(4):377–90.
De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:93–9.
Kamm CP, Uitdehaag BM, Polman CH. Multiple sclerosis: current knowledge and future outlook. Eur Neurol. 2014;72:132–41.
Paolillo A, Pozzilli C, Gasperini C, et al. Brain atrophy in relapsing-remitting multiple sclerosis: relationship with ‘black holes’, disease duration and clinical disability. J Neurol Sci. 2000;174:85–91.
Ghasemi N, Razavi S, Nikzad E. Multiple sclerosis: pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017;19:1–10.
Domingues HS, Portugal CC, Socodato R, et al. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71.
Ludwin SK, Rao V, Moore CS, et al. Astrocytes in multiple sclerosis. Mult Scler. 2016;22:1114–24.
Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404.
Hauser SL, Bhan AK, Gilles F, et al. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–87.
Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–9.
Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1952–60.
Michel L, Touil H, Pikor NB, et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol. 2015;6:636.
Moreno Torres I, Garcia-Merino A. Anti-CD20 monoclonal antibodies in multiple sclerosis. Expert Rev Neurother. 2017;17:359–71.
Baker D, Marta M, Pryce G, et al. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine. 2017;16:41–50.
Dooley J, Pauwels I, Franckaert D, et al. Immunologic profiles of multiple sclerosis treatments reveal shared early B cell alterations. Neurol Neuroimmunol Neuroinflamm. 2016;3:e240.
Carson MJ, Doose JM, Melchior B, et al. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65.
Staun-Ram E, Miller A. Effector and regulatory B cells in multiple sclerosis. Clin Immunol. 2017;184:11–25.
Blauth K, Owens GP, Bennett JL. The ins and outs of B cells in multiple sclerosis. Front Immunol. 2015;6:565.
Holman DW, Klein RS, Ransohoff RM. The blood–brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812:220–30.
Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129:200–11.
Franciotta D, Salvetti M, Lolli F, et al. B cells and multiple sclerosis. Lancet Neurol. 2008;7:852–8.
Serafini B, Rosicarelli B, Magliozzi R, et al. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–74.
Zuckerman NS, Hazanov H, Barak M, et al. Somatic hypermutation and antigen-driven selection of B cells are altered in autoimmune diseases. J Autoimmun. 2010;35:325–35.
von Budingen HC, Kuo TC, Sirota M, et al. B cell exchange across the blood–brain barrier in multiple sclerosis. J Clin Investig. 2012;122:4533–43.
Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–9.
Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41.
Engelhardt B, Carare RO, Bechmann I, et al. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016;132:317–38.
Claes N, Fraussen J, Stinissen P, et al. B cells are multifunctional players in multiple sclerosis pathogenesis: Insights from therapeutic interventions. Front Immunol. 2015;6:642.
Raper D, Louveau A, Kipnis J. How do meningeal lymphatic vessels drain the CNS? Trends Neurosci. 2016;39:581–6.
Stern JN, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. 2014;6:248ra107.
Korn T. Pathophysiology of multiple sclerosis. J Neurol. 2008;255(Suppl 6):2–6.
Villar LM, Masjuan J, Gonzalez-Porque P, et al. Intrathecal IgM synthesis is a prognostic factor in multiple sclerosis. Ann Neurol. 2003;53:222–6.
Hohmann C, Milles B, Schinke M, et al. Categorization of multiple sclerosis relapse subtypes by B cell profiling in the blood. Acta Neuropathol Commun. 2014;2:138.
Kuerten S, Pommerschein G, Barth SK, et al. Identification of a B cell-dependent subpopulation of multiple sclerosis by measurements of brain-reactive B cells in the blood. Clin Immunol. 2014;152:20–4.
Feldman A, Gurevich M, Huna-Baron R, et al. The role of B cells in the early onset of the first demyelinating event of acute optic neuritis. Investig Ophthalmol Vis Sci. 2015;56:1349–56.
Harp CT, Ireland S, Davis LS, et al. Memory B cells from a subset of treatment-naive relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-gamma production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol. 2010;40:2942–56.
Cornaby C, Gibbons L, Mayhew V, et al. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett. 2015;163:56–68.
Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17.
Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–104.
Magliozzi R, Howell OW, Reeves C, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–93.
Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. 2008;63:395–400.
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221–34.
Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88.
Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209–20.
Holley JE, Bremer E, Kendall AC, et al. CD20+ inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Mult Scler Relat Disord. 2014;3:650–8.
Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol. 2014;193:580–6.
Wilk E, Witte T, Marquardt N, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum. 2009;60:3563–71.
Schuh E, Berer K, Mulazzani M, et al. Features of human CD3+ CD20+ T cells. J Immunol. 2016;197:1111–7.
Forsthuber TG, Cimbora DM, Ratchford JN, et al. B cell-based therapies in CNS autoimmunity: differentiating CD19 and CD20 as therapeutic targets. Ther Adv Neurol Disord. 2018;11:1756286418761697.
Thompson SA, Jones JL, Cox AL, et al. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol. 2010;30:99–105.
Lin YC, Winokur P, Blake A, et al. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann Clin Transl Neurol. 2015;2:445–55.
Bail K, Notz Q, Rovituso DM, et al. Differential effects of FTY720 on the B cell compartment in a mouse model of multiple sclerosis. J Neuroinflammation. 2017;14:148.
Blumenfeld S, Staun-Ram E, Miller A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL-10 and TGFβ in patients with multiple sclerosis. J Autoimmun. 2016;70:40–51.
Li R, Rezk A, Ghadiri M, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol. 2017;198:691–8.
Mitosek-Szewczyk K, Tabarkiewicz J, Wilczynska B, et al. Impact of cladribine therapy on changes in circulating dendritic cell subsets, T cells and B cells in patients with multiple sclerosis. J Neurol Sci. 2013;332:35–40.
Lehmann-Horn K, Kronsbein HC, Weber MS. Targeting B cells in the treatment of multiple sclerosis: recent advances and remaining challenges. Ther Adv Neurol Disord. 2013;6:161–73.
Rawlings DJ, Metzler G, Wray-Dutra M, et al. Altered B cell signalling in autoimmunity. Nat Rev Immunol. 2017;17:421–36.
Mackay F, Groom JR, Tangye SG. An important role for B-cell activation factor and B cells in the pathogenesis of Sjogren’s syndrome. Curr Opin Rheumatol. 2007;19:406–13.
Buchta CM, Bishop GA. Toll-like receptors and B cells: functions and mechanisms. Immunol Res. 2014;59:12–22.
Krumbholz M, Theil D, Derfuss T, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med. 2005;201:195–200.
Avery DT, Kalled SL, Ellyard JI, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–97.
Dorner T. Crossroads of B cell activation in autoimmunity: rationale of targeting B cells. J Rheumatol Suppl. 2006;77:3–11.
Fraussen J, Claes N, de Bock L, et al. Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun Rev. 2014;13:1126–37.
Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7:115–21.
Ireland SJ, Blazek M, Harp CT, et al. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity. 2012;45:400–14.
Cross AH, Stark JL, Lauber J, et al. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180:63–70.
Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314:537–9.
Kousin-Ezewu O, Azzopardi L, Parker RA, et al. Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology. 2014;82:2158–64.
Jackson LJ, Selva S, Niedzielko T, et al. B cell receptor recognition of glatiramer acetate is required for efficacy through antigen presentation and cytokine production. J Clin Cell Immunol. 2014;5:15.
Kastrukoff LK, McLean DR, McPherson TA. Multiple sclerosis treated with antithymocyte globulin – a five year follow-up. Can J Neurol Sci. 1978;5:175–8.
Lindsey JW, Hodgkinson S, Mehta R, et al. Phase 1 clinical trial of chimeric monoclonal anti-CD4 antibody in multiple sclerosis. Neurology. 1994;44:413–9.
Llewellyn-Smith N, Lai M, Miller DH, et al. Effects of anti-CD4 antibody treatment on lymphocyte subsets and stimulated tumor necrosis factor alpha production: a study of 29 multiple sclerosis patients entered into a clinical trial of cM-T412. Neurology. 1997;48:810–6.
Rep MH, van Oosten BW, Roos MT, et al. Treatment with depleting CD4 monoclonal antibody results in a preferential loss of circulating naive T cells but does not affect IFN-gamma secreting TH1 cells in humans. J Clin Invest. 1997;99:2225–31.
van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled. MR-monitored phase II trial. Neurology. 1997;49:351–7.
Weinshenker BG, Bass B, Karlik S, et al. An open trial of OKT3 in patients with multiple sclerosis. Neurology. 1991;41:1047–52.
Rodriguez-Pinto D, Moreno J. B cells can prime naive CD4+ T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur J Immunol. 2005;35:1097–105.
Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol. 2015;6:180.
Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67:452–61.
Dalakas MC. Invited article: inhibition of B cell functions: implications for neurology. Neurology. 2008;79:2252–60.
Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–71.
Lisak RP, Nedelkoska L, Benjamins JA, et al. B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. J Neuroimmunol. 2017;309:88–99.
Choi SR, Howell OW, Carassiti D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012;135:2925–37.
Absinta M, Vuolo L, Rao A, et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology. 2015;85:18–28.
Zurawski J, Lassmann H, Bakshi R. Use of magnetic resonance imaging to visualize leptomeningeal inflammation in patients with multiple sclerosis: a review. JAMA Neurol. 2017;74:100–9.
Harrison DM, Wang KY, Fiol J, et al. Leptomeningeal enhancement at 7T in multiple sclerosis: frequency, morphology, and relationship to cortical volume. J Neuroimaging. 2017;27:461–8.
Basile E, Gibbs E, Aziz T, et al. During 3 years treatment of primary progressive multiple sclerosis with glatiramer acetate, specific antibodies switch from IgG1 to IgG4. J Neuroimmunol. 2006;177:161–6.
Neuhaus O, Farina C, Yassouridis A, et al. Multiple sclerosis: comparison of copolymer-1-reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA. 2000;97:7452–7.
Weber MS, Hohlfeld R, Zamvil SS. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics. 2007;4:647–53.
Bakshi S, Chalifa-Caspi V, Plaschkes I, et al. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets. 2013;17:351–62.
Begum-Haque S, Sharma A, Christy M, et al. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;219:47–53.
Begum-Haque S, Christy M, Ochoa-Reparaz J, et al. Augmentation of regulatory B cell activity in experimental allergic encephalomyelitis by glatiramer acetate. J Neuroimmunol. 2011;232:136–44.
Bomprezzi R, Schafer R, Reese V, et al. Glatiramer acetate-specific antibody titres in patients with relapsing/remitting multiple sclerosis and in experimental autoimmune encephalomyelitis. Scand J Immunol. 2011;74:219–26.
Brenner T, Arnon R, Sela M, et al. Humoral and cellular immune responses to Copolymer 1 in multiple sclerosis patients treated with Copaxone®. J Neuroimmunol. 2001;115:152–60.
Carrieri PB, Carbone F, Perna F, et al. Longitudinal assessment of immuno-metabolic parameters in multiple sclerosis patients during treatment with glatiramer acetate. Metabolism. 2015;64:1112–21.
Farina C, Vargas V, Heydari N, et al. Treatment with glatiramer acetate induces specific IgG4 antibodies in multiple sclerosis patients. J Neuroimmunol. 2002;123:188–92.
Ireland SJ, Guzman AA, O’Brien DE, et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol. 2014;71:1421–8.
Kala M, Rhodes SN, Piao WH, et al. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp Neurol. 2010;221:136–45.
Karussis D, Teitelbaum D, Sicsic C, et al. Long-term treatment of multiple sclerosis with glatiramer acetate: natural history of the subtypes of anti-glatiramer acetate antibodies and their correlation with clinical efficacy. J Neuroimmunol. 2010;220:125–30.
Rovituso D, Heller S, Schroeter M, et al. B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients. J Neuroimmunol. 2014;272:86–90.
Rovituso DM, Duffy CE, Schroeter M, et al. The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients. Sci Rep. 2015;5:14265.
Sellebjerg F, Hedegaard CJ, Krakauer M, et al. Glatiramer acetate antibodies, gene expression and disease activity in multiple sclerosis. Mult Scler. 2012;18:305–13.
Sellner J, Koczi W, Harrer A, et al. Glatiramer acetate attenuates the pro-migratory profile of adhesion molecules on various immune cell subsets in multiple sclerosis. Clin Exp Immunol. 2013;173:381–9.
Teitelbaum D, Brenner T, Abramsky O, et al. Antibodies to glatiramer acetate do not interfere with its biological functions and therapeutic efficacy. Mult Scler. 2003;9:592–9.
Begum-Haque S, Christy M, Telesford K, et al. Glatiramer acetate treatment regulates CD44, receptor for osteopontin in EAE mice [abstract no. P1.214]. Neurology. 2014;82(10 Suppl):P1.214.
Begum-Haque S, Christy M, Wang Y, et al. Glatiramer acetate mediated expression of α4β1integrin (VLA-4) on T and B regulatory cells in EAE mice [abstract no. P02.109]. Neurology. 2012;78(1 Suppl.):P02.109.
Criscuolo C, Cianflone A, La Rocca C, et al. Identification of glatiramer acetate B lymphocytes targets in multiple sclerosis [abstract]. Mult Scler J. 2016;22:P409.
Begum-Haque S. Glatiramer acetate-conditioned regulatory B cells regulate osteopontin expression in experimental autoimmune encephalomyelitis (EAE) mice [abstract]. Mult Scler J. 2013;19:34474.
Hertzenberg D, Lehmann-Horn K, LaLive P, et al. Glatiramer acetate complements anti-CD20-mediated B-cell depletion in treatment of CNS autoimmune disease [abstract no. P04.124]. Neurology. 2012;78(P04):124.
Häusler D, Bernard CCA, Feldman L, et al. Glatiramer acetate immune modulates pathogenic B cell function and maintains clinical benefit after anti-CD20 induction therapy [abstract]. Mult Scler J. 2016;22:P666.
Fridkis-Hareli M, Teitelbaum D, Arnon R, et al. Synthetic copolymer 1 and myelin basic protein do not require processing prior to binding to class II major histocompatibility complex molecules on living antigen-presenting cells. Cell Immunol. 1995;163:229–36.
Fridkis-Hareli M, Teitelbaum D, Gurevich E, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells—specificity and promiscuity. Proc Natl Acad Sci U S A. 1994;91:4872–6.
Ayzenberg I, Schollhammer J, Hoepner R, et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol. 2016;263:575–82.
Borel P, Benkhoucha M, Weber MS, et al. Glatiramer acetate treatment does not modify the clinical course of (NZB × BXSB)F1 lupus murine model. Int Immunol. 2008;20:1313–9.
Zheng B, Switzer K, Marinova E, et al. Exacerbation of autoimmune arthritis by copolymer-I through promoting type 1 immune response and autoantibody production. Autoimmunity. 2008;41:363–71.
Prod’homme T, Zamvil SS. The evolving mechanisms of action of glatiramer acetate. Cold Spring Harb Perspect Med. 2018. https://doi.org/10.1101/cshperspect.a029249 (Epub 2018 Feb 12).
Disanto G, Morahan JM, Barnett MH, et al. The evidence for a role of B cells in multiple sclerosis. Neurology. 2012;78:823–32.
Bose T. Role of immunological memory cells as a therapeutic target in multiple sclerosis. Brain Sci. 2017;7:E148.
Piancone F, Saresella M, Marventano I, et al. B lymphocytes in multiple sclerosis: Bregs and BTLA/CD272 expressing-CD19+ lymphocytes modulate disease severity. Sci Rep. 2016;6:29699.
Caporro M, Disanto G, Gobbi C, et al. Two decades of subcutaneous glatiramer acetate injection: current role of the standard dose, and new high-dose low-frequency glatiramer acetate in relapsing-remitting multiple sclerosis treatment. Patient Prefer Adherence. 2014;8:1123–34.
Ziemssen T, Ashtamker N, Rubinchick S, et al. Long-term safety and tolerability of glatiramer acetate 20 mg/ml in the treatment of relapsing forms of multiple sclerosis. Expert Opin Drug Saf. 2017;16:247–55.
Honce JH, Nair KV, Stefan S, et al. Comparing rituximab induction therapy followed by glatiramer acetate therapy to glatiramer acetate monotherapy in MS patients on clinical and imaging [abstract]. Mult Scler J. 2016;22:P1187.
Acknowledgements
Nicky French of Anthemis Consulting Ltd provided editorial support, funded by Teva Pharmaceutical Industries, Frazer, PA, USA. The authors were not compensated and retained full editorial control over the content of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SK has received research funding and payment for lectures from Teva Pharmaceutical Industries. LJJ was an employee of Teva Pharmaceutical Industries at the time the manuscript was written and held equity in the company. JK is an employee of Teva Pharmaceutical Industries. TLV has received payment for activities such as advisory boards, lectures, and consultancy from Academic CME, Alcimed, Anthem Blue Cross, Genentech/Roche, Biogen IDEC, Novartis, CellGene, Serono, Epigene, Rocky Mountain MS Center, GLG Consulting, Ohio Health, TG Therapeutics, Topaz Therapeutics, Dleara Lawyers, and Teva Neuroscience; he has also received research support from Teva Neuroscience, NIH/NINDS (National Institutes of Health/National Institute of Neurological Disorders and Stroke), Rocky Mountain MS Center, Biogen, Actelion, Genentech/Roche, UT Southwestern, F. Hoffman–La Roche Ltd., and TG Therapeutics Ltd.
Funding
Editorial support and open-access fees were funded by Teva Pharmaceutical Industries, Frazer, PA, USA.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kuerten, S., Jackson, L.J., Kaye, J. et al. Impact of Glatiramer Acetate on B Cell-Mediated Pathogenesis of Multiple Sclerosis. CNS Drugs 32, 1039–1051 (2018). https://doi.org/10.1007/s40263-018-0567-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-018-0567-8