
Abstract
Background
Although levodopa is considered the most effective pharmacotherapy for motor symptoms of Parkinson’s disease (PD), chronic use is associated with motor complications, including fluctuating response and unpredictable, involuntary movements called dyskinesia. ADS-5102 (amantadine) extended-release (ER) capsules (GOCOVRITM) is a recent US FDA-approved treatment for dyskinesia in PD patients. ADS-5102 is a high-dose, ER formulation of amantadine, administered orally once daily at bedtime, that achieves high plasma drug concentrations throughout the day.
Objective
In this study, we present pooled results from two randomized, double-blind, placebo-controlled, phase III ADS-5102 trials.
Patients and Methods
The two studies in PD patients with dyskinesia shared design and eligibility criteria, differing only in treatment duration. Results from common assessment time points were pooled.
Results
At 12 weeks, the least squares (LS) mean change in total score on the Unified Dyskinesia Rating Scale among 100 patients randomized to ADS-5102 and 96 patients randomized to placebo was − 17.7 (standard error [SE] 1.3) vs. − 7.6 (1.3) points, respectively (− 10.1 points, 95% confidence interval [CI] − 13.8, − 6.5; p < 0.0001). The relative treatment difference between groups was 27.3% (p < 0.0001). At 12 weeks, the LS mean change in OFF time was − 0.59 (0.21) vs. +0.41 (0.20) h/day, a difference of − 1.00 h/day (95% CI − 1.57, − 0.44; p = 0.0006). For both efficacy measures, a significant difference from placebo was attained by two weeks, the first post-baseline assessment, and was maintained throughout 12 weeks. In the pooled ADS-5102 group, the most common adverse events were hallucination, dizziness, dry mouth, peripheral edema, constipation, falls, and orthostatic hypotension.
Conclusions
These analyses provide further evidence supporting ADS-5102 as an adjunct to levodopa for treating both dyskinesia and OFF time in PD patients with dyskinesia.
Clinicaltrials.gov identifier: NCT02136914 and NCT02274766
Similar content being viewed by others
Avoid common mistakes on your manuscript.
ADS-5102 extended-release capsules are an oral formulation of amantadine administered once daily at bedtime in patients with Parkinson’s disease (PD) to treat dyskinesia associated with the use of levodopa. |
Pooled results from two randomized, double-blind, placebo-controlled, phase III trials confirm that treatment with ADS-5102 is associated with a significant improvement over placebo for dyskinesia in as early as two weeks and maintained over 12 weeks (relative treatment difference between groups = 27.3%; p < 0.0001). A significant improvement in the total daily duration of OFF time was also achieved. |
These results provide further evidence supporting ADS-5102 as an adjunct to levodopa for both dyskinesia and OFF time in PD patients with dyskinesia. |
1 Introduction
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons and reduced dopamine-mediated signaling from the substantia nigra [1]. To suppress the resulting motor symptoms, levodopa, a precursor of dopamine, is considered the most effective treatment [2, 3]. However, chronic levodopa use is associated with the eventual development of motor complications, consisting of fluctuations in therapeutic motor benefit, and dyskinesia [4,5,6]. During an ON period, PD motor symptoms are generally well-controlled, whereas during an OFF period (often referred to as ‘wearing off’, or OFF time) PD symptoms re-emerge, despite levodopa treatment [6,7,8]. Dyskinesia is another component of motor complications, and consists of unpredictable, involuntary movements that typically occur during the patient’s ON time [5, 6]. Motor fluctuations and dyskinesia are typically experienced by approximately 35–40% of patients with PD after approximately 4–6 years of dopaminergic treatment. After 10 years of treatment, approximately ≥ 90% of patients will be experiencing motor complications [5]. These problems may be severely detrimental to a patient’s daily activities, quality of life, and healthcare utilization [9,10,11,12]. Attempts to manage dyskinesia by reducing a patient’s antiparkinsonian regimen may succeed only at the risk of increasing OFF time [6, 13, 14]. Dyskinesia is suspected to result from dysregulated striatal excitation by glutamate, a neurotransmitter acting at N-methyl-D-aspartate (NMDA) glutamate receptors [15, 16].
Several small studies suggest that the immediate-release (IR) formulation of amantadine, a glutamate antagonist, ameliorates dyskinesia [17,18,19,20]; however, the IR drug has not been extensively studied in well-controlled trials [21] and the durability of its effect remains [22] disputed, particularly for those patients previously naive to amantadine treatment [22]. ADS-5102 (GOCOVRITM) is an orally administered, extended-release (ER) formulation of amantadine HCl [23]. Once-daily ADS-5102 treatment at bedtime is expected to achieve an amantadine pharmacokinetic profile unattainable with amantadine IR [24], including a slow rate of initial rise in plasma drug concentration during sleep to a high drug concentration throughout the day, when dyskinesia can be most troublesome. In a phase II/III dose-finding study of ADS-5102, mean amantadine plasma concentrations of approximately 1500 ng/mL were associated with reduction in dyskinesia [24]. This was followed by two randomized, double-blind, placebo-controlled, phase III clinical trials of ADS-5102 that confirmed reduction of dyskinesia, and in which an additional benefit of reduced daily OFF time was observed [25, 26].
In this study, we present the results of a pooled analysis of these two phase III trials using prespecified methodology.
2 Methods
EASE LID (ClinicalTrials.gov identifier: NCT02136914) was conducted at 44 North American sites between May 2014 and July 2015, and EASE LID 3 (NCT02274766) was conducted at 39 sites in the US and Western Europe between October 2014 and December 2015. Except for a longer treatment duration in EASE LID (up to 25 weeks) than in EASE LID 3 (up to 13 weeks), the studies had the same design and the same criteria for patient enrollment. The results from these studies have been published separately [25, 26]. For the pooled efficacy evaluations presented here, the analyses were restricted to the assessment time points common to both studies (baseline and the double-blind treatment weeks 2, 8, and 12). For the pooled safety evaluations presented here, descriptive statistics encompass the full duration of study-drug exposure.
2.1 Study Participants
Patients were required to be aged 30–85 years, to have a clinical diagnosis of PD by United Kingdom Parkinson Disease Society Brain Bank criteria [27], and to be receiving treatment with levodopa at least three times daily in an antiparkinsonian regimen that had been stable for at least 30 days. They had to be experiencing at least 1 h/day (two half-hour periods) of ON time with troublesome dyskinesia between 9:00 am and 4:00 pm, as documented on two consecutive days prior to treatment day 1 (baseline) by patient entries in a 24-h PD home diary [28]. The dyskinesia was required to cause at least mild functional impairment, as documented at screening and baseline by a score ≥ 2 on item 4.2 of the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS–UPDRS) [29]. On this scale, the scoring is 0 = no impact, 1 = slight, 2 = mild, 3 = moderate, and 4 = severe.
Patients were excluded for reasons including atypical parkinsonism, acute or major psychiatric disorder that would affect the subject’s ability to complete study assessments, neurosurgical intervention in PD, dyskinesia not caused by dopaminergic stimulation in PD, an estimated glomerular filtration rate (eGFR) < 50 mL/min/1.73 m2, and use of amantadine within the previous 30 days.
2.2 Study Design
By a centralized, interactive, web-based response system, enrolled patients in each study were randomized in a 1:1 ratio to take ADS-5102 or matching placebo once daily at bedtime. ADS-5102 was administered at 137 mg for the first week (administered as one capsule containing active drug, or one capsule containing placebo), and 274 mg (two 137-mg capsules) thereafter. For the two-capsule dose, the 274 mg amantadine content is equivalent to 340 mg of amantadine HCl. For the final week, the dose was reduced to 137 mg. Throughout the studies, each patient’s regimen of antiparkinsonian medications, including levodopa, remained unchanged.
2.3 Efficacy Measures
During screening, each patient was trained in PD home-diary completion and received diaries to be completed on the two days preceding the baseline visit. Patients also completed diaries on the two days preceding each subsequent visit. The diaries [28] required patients to categorize their predominant clinical status during each half-hour interval of the 24-h day as ‘asleep’, ‘OFF’, ‘ON with no dyskinesia’, ‘ON with nontroublesome dyskinesia’, or ‘ON with troublesome dyskinesia’.
At each visit, the Unified Dyskinesia Rating Scale (UDysRS) [30] and the MDS–UPDRS [29] were completed by a qualified rater during the patient’s ON time, at least 30 min after a routine levodopa dose, while the patient was experiencing typical dyskinesias. If possible, the same rater performed each patient’s ratings at approximately the same time of day. The Clinician’s Global Impression of Change (CGI–C) [31] was completed at each visit and required the investigator to rate how much the patient’s PD had improved or worsened after treatment relative to the baseline state. The UDysRS is a 26-item assessment of involuntary movements, with two primary sections: Historical (Part I and Part II, patient reported) and Objective (Part III and Part IV, physician assessed). Part I assesses the presence and impact of ON dyskinesia on patient’s experiences of daily living, Part II assesses the presence and impact of OFF dystonia on experiences of daily living, and Parts III and IV assess the intensity and disability of dyskinesia in seven body regions. The overall score can range from 0 to 104 points, with higher scores reflecting greater disease severity [30]. The MDS–UPDRS is a 4-part, multi-item rating scale intended to assess non-motor experiences of daily living (Part I), motor experiences of daily living (Part II), motor function (Part III), and motor complications (motor fluctuations and dyskinesia; Part IV) in patients with PD. Part IV, item 4.1, determines the percentage of time spent per waking day with dyskinesia, while item 4.2 measures the functional impact of the dyskinesia by determining the degree to which dyskinesias impact daily activities and social interactions. Part IV, item 4.3, measures the amount of time spent in the OFF state, while item 4.4 measures the functional impact of motor fluctuations (impact on activities and social interactions). Higher scores indicate greater disability [29].
2.4 Efficacy Analyses
In each study, the primary efficacy measure was change from baseline to 12 weeks in each patient’s UDysRS total score. Secondary assessments included change at 12 weeks in each patient’s PD-diary-based clinical states: OFF time, and ON time without troublesome dyskinesia (the sum of ON time with no dyskinesia and ON time with nontroublesome dyskinesia), UDysRS historical (Parts I and II) and objective (Parts III and IV) scores, MDS–UPDRS, and CGI–C.
For each efficacy measure, mean change in each treatment group was tested for a statistically significant difference between ADS-5102 and placebo using a linear mixed model with repeated measures, with change from baseline as the dependent variable and baseline value as a covariate. The model included categorical effects for treatment group, study visit (weeks 2, 8, and 12), and interaction between treatment group and visit. The prespecified efficacy analysis population was the modified intent-to-treat (mITT) population, comprising all randomized patients who were exposed to the study drug and providing at least one post-baseline UDysRS assessment. All analyses were set at a two-sided, 5% significance level.
Secondary assessments were analyzed using a fixed-sequence hierarchical gatekeeping procedure to control the overall type 1 error rate. For the hierarchical analysis, the comparisons of ADS-5102 versus placebo for ON time without troublesome dyskinesia and ADS-5102 versus placebo for OFF time at week 12 were performed using linear mixed models with repeated measures methods using two-sided tests at the 5% level of significance. A specified comparison was considered confirmatory only if the primary efficacy analysis and all previously conducted secondary analyses were statistically significant (p < 0.05).
As descriptive analyses, UDysRS data from patients who provided such data at both baseline and 12 weeks were utilized to calculate the placebo-corrected change in UDysRS total score in patient subgroups stratified by sex, age, median baseline UDysRS total score, median baseline MDS–UPDRS item 4.2 score, and median baseline OFF-time duration.
Diary data from all patients who provided such data at both baseline and 12 weeks were utilized to characterize the proportions of patients with specified magnitudes of improvement in each PD-diary-based clinical state, and the proportion of those who experienced complete resolution (i.e. reduction to 0.0 h/day) of OFF time and ON time with troublesome dyskinesia.
2.5 Safety Measures
The safety-analysis population included all patients exposed to the study drug. Safety parameters included adverse events (AEs), serious AEs (SAEs), and adverse drug reactions (ADRs), defined as those events that were expected to occur as a result of ADS-5102 treatment and reported by the investigators, throughout study-drug exposure.
2.6 Ethical Conduct
Each study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Before the start of each study, each site received approval from an Institutional Review Board, Research Ethics Board, or Independent Ethics Committee. Prior to any study procedures, written informed consent was obtained from each patient.
3 Results
3.1 Study Participants
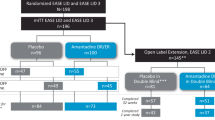
Among 189 patients screened in EASE LID, 126 were randomized. The study’s mITT population comprised 121 patients, of whom 63 received ADS-5102 and 58 received placebo. In these groups, 52 and 51 patients, respectively, completed 12 weeks. Among 114 patients screened in EASE LID 3, 77 were randomized. The study’s mITT population comprised 75 patients, of whom 37 received ADS-5102 and 38 received placebo. In these groups, 29 and 35 patients, respectively, completed 12 weeks. Across the two studies, the pooled mITT population comprised 100 patients randomized to ADS-5102 and 96 randomized to placebo. The pooled safety population included an additional two patients, both in EASE LID, who received placebo but did not provide a post-baseline UDysRS assessment (Fig. 1).

Pooled population used in this analysis arising from the EASE LID and EASE LID 3 trials. aThe pooled safety population included an additional two patients, both in EASE LID, who received placebo but did not provide a post-baseline UDysRS assessment. mITT modified intent-to-treat, UDysRS Unified Dyskinesia Rating Scale
Baseline characteristics of the pooled mITT population are summarized by study and treatment group in Table 1. On average, the patients were aged 64.7 years (range 34–82 years), with a 9.7-year duration of PD (range 1.0–26.8), a 7.7-year duration of levodopa treatment (range 0.1–14.0), a 3.8-year duration of dyskinesia (range 0.1–14.0), and a mean UDysRS total score of 40.1 points (range 8.0–76.0). They reported a mean 2.8 h/day of OFF time (range 0.0–9.5) and 4.9 h/day of ON time with troublesome dyskinesia (range 0.0–13.3). Except for levodopa dosage, which, on average, was higher in EASE LID than in EASE LID 3, and baseline use of dopamine agonists and monoamine oxidase-B (MAO-B) inhibitors, which was less common in EASE LID than in EASE LID 3, the ADS-5102 and placebo groups were well-balanced across the studies and between the groups in each study.
3.2 Efficacy: Unified Dyskinesia Rating Scale
The time course of change from baseline in UDysRS total score is presented by pooled treatment group in Fig. 2a. At each assessment time point, the least squares (LS) mean change was significantly greater in the ADS-5102 group than in the placebo group (p < 0.0001 at each time point). At 12 weeks, the LS mean (standard error [SE]) change was − 17.7 (1.3) vs. − 7.6 (1.3) points in the placebo group, an LS mean difference of − 10.1 (1.8) points (95% confidence interval [CI] − 13.8, − 6.5; p < 0.0001). This represents a relative LS mean change from baseline to week 12 of 41.1% for the ADS-5102 group and 13.9% for the placebo group, a percentage treatment difference of 27.3% (p < 0.0001) [Table 2]. For the historical, patient-reported portions of the UDysRS (Parts 1 and 2), the LS mean change was − 10.7 (0.9) vs. − 5.1 (0.8) points, an LS mean difference of − 5.6 (1.2) points (95% CI − 8.0, − 3.3; p < 0.0001). For the physician-rated portions (Parts 3 and 4), the LS mean change was − 7.0 (0.8) vs. − 2.5 (0.8), an LS mean difference of − 4.5 (1.1) points (95% CI − 6.6, − 2.4; p < 0.0001).

Time course of change from baseline in a UDysRS total score, b OFF time, c ON time with troublesome dyskinesia, and d ON time without troublesome dyskinesia, by pooled treatment group. LS least squares, SE standard error, UDysRS Unified Dyskinesia Rating Scale
The cumulative distribution of change in UDysRS total score at 12 weeks is presented by pooled treatment group in Fig. 3. Approximately half of the ADS-5102 group had a score reduction (improvement) of 17 points or more, while approximately half of the placebo group had a reduction of 8 points or more. Every degree of score reduction was more frequent for ADS-5102 than for placebo, producing a leftward shift (signifying improvement) of the entire ADS-5102 curve relative to the placebo curve.

Cumulative distribution of change in UDysRS total score at 12 weeks by pooled treatment group. UDysRS Unified Dyskinesia Rating Scale
Subgroup analyses of 12-week change in UDysRS total score are presented in Fig. 4a. The treatment effect of ADS-5102 was consistent across the following predefined subgroups: sex, age, and baseline dyskinesia severity (baseline UDysRS total score and baseline MDS–UPDRS, Part IV, item 4.2 score). Among 46 ADS-5102 recipients and 40 placebo recipients with UDysRS baseline scores below the median (< 40), the LS mean difference in UDysRS total score change at 12 weeks was − 6.6 points (95% CI − 11.7, − 1.6), favoring ADS-5102. Among 54 ADS-5102 recipients and 56 placebo recipients with baseline scores equaling or exceeding the median (≥ 40), the LS mean difference in score change was − 12.4 points (95% CI − 17.7, − 7.2), also favoring ADS-5102. For patient subgroups stratified by baseline MDS–UPDRS item 4.2 score, a similar pattern was seen. Among 49 ADS-5102 recipients and 48 placebo recipients with a baseline score of 2, the LS mean difference in UDysRS total score change at 12 weeks was − 8.4 points (95% CI − 12.9, − 3.9), while among 51 ADS-5102 recipients and 48 placebo recipients with a baseline score of 3 to 4, the difference was − 12.1 points (95% CI − 17.8, − 6.3).

Subgroup analyses of 12-week change in a UDysRS total score and b OFF time (2.5 h is the median baseline OFF time). a n = 100 for ADS-5102 and 96 for placebo. b n = 46 for ADS-5102 and 41 for placebo. c n = 54 for ADS-5102 and 55 for placebo. d n = 48 for ADS-5102 and 41 for placebo. e n = 54 for ADS-5102 and 48 for placebo. fMedian value at baseline in the mITT population. g n = 46 for ADS-5102 and 40 for placebo. h n = 54 for ADS-5102 and 56 for placebo. i n = 49 for ADS-5102 and 48 for placebo. j n = 51 for ADS-5102 and 48 for placebo. k n = 45 for ADS-5102 and 49 for placebo. CI confidence interval, LS least squares, MDS–UPDRS Movement Disorder Society–Unified Parkinson’s Disease Rating Scale, UDysRS Unified Dyskinesia Rating Scale
3.3 Efficacy: Movement Disorder Society–Unified Parkinson’s Disease Rating Scale
At week 12, changes in MDS–UPDRS showed a statistically significant treatment effect for ADS-5102 for the Part IV total score (p < 0.0001), as well as for the item 4.1 (p = 0.0015), item 4.2 (p < 0.0001), item 4.3 (p < 0.0001), and item 4.4 scores (p = 0.0166) [Table 3]. The treatment difference from baseline to 12 weeks was not statistically significant for the MDS–UPDRS Parts I, II, and III combined (− 3.2; p = 0.1275), nor for Parts II and III combined (− 2.2; p = 0.2282).
3.4 Efficacy: Parkinson’s Disease-Diary Findings
The time courses of change from baseline in OFF time, in ON time with troublesome dyskinesia, and in ON time without troublesome dyskinesia are presented by pooled treatment group in Figs. 2b–d, respectively. For OFF time, the LS mean treatment difference for ADS-5102 was a reduction of 1.00 h/day (95% CI − 1.57, − 0.44; p = 0.0006) [Table 3]. For ON time with troublesome dyskinesia, the treatment effect of ADS-5102 was a reduction of 1.46 h/day (95% CI − 2.25, − 0.67; p = 0.0003), and for ON time without troublesome dyskinesia, there was an improvement of 2.42 h/day (95% CI +1.48, +3.37; p < 0.0001) for the ADS-5102 group over placebo.
For each state, the distribution of 12-week change is presented by pooled treatment group in Figs. 5a–c. By week 12, 25% of the ADS-5102 group had a complete resolution of OFF time, compared with 14% in the placebo group; 68% of the ADS-5102 group had an increase > 2.0 h/day in ON time without troublesome dyskinesia, compared with 40% in the placebo group; and 52% of the ADS-5102 group had a complete resolution of ON time with troublesome dyskinesia, compared with 23% in the placebo group. Across the distributions, increase in ON time without troublesome dyskinesia reflected decreases both in ON time with troublesome dyskinesia and OFF time.

Distribution of 12-week change in a OFF time, b ON time without troublesome dyskinesia, and c ON time with troublesome dyskinesia, by pooled treatment group (12-week completers). hrs/d hours/day
In the pooled mITT population, the median amount of OFF time at baseline was 2.5 h/day. Among 45 ADS-5102 recipients and 49 placebo recipients with baseline OFF time below the median, the LS mean difference in OFF-time decrease at 12 weeks was − 0.8 h/day (95% CI − 1.49, − 0.06), favoring ADS-5102 (Fig. 4b). Among 55 ADS-5102 recipients and 47 placebo recipients with baseline OFF time equaling or exceeding the median, the LS mean difference was − 1.2 h/day (95% CI − 2.08, − 0.32), also favoring ADS-5102.
3.5 Efficacy: Clinician’s Global Impression of Change
After 12 weeks of treatment, 36 subjects (37.5%) in the placebo group and 76 subjects (76.0%) in the ADS-5102 group showed improvement in PD symptoms relative to baseline. Fifteen subjects (15.6%) in the placebo group and 57 subjects (57.0%) in the ADS-5102 group showed moderate to marked improvement. The difference between the treatment groups was statistically significant (p < 0.0001 from the Cochran–Mantel–Haenszel mean score test [using equally spaced scores] stratified by study) [Fig. 6].

Change from baseline for CGI–C at week 12 by pooled treatment group. CGI–C Clinician’s Global Impression of Change
3.6 Safety
ADRs reported by the investigators throughout study-drug exposure (up to 25 weeks in EASE LID and up to 13 weeks in EASE LID 3) are summarized by study and treatment group, as well as for the pooled treatment groups, in Table 4. Most ADRs were of mild to moderate intensity, and transient. In the pooled ADS-5102 group, the most common ADRs (reported in > 10% of pooled patient data) were hallucination (visual or auditory), dizziness, dry mouth, peripheral edema, constipation, falls, and orthostatic hypotension.
In the pooled ADS-5102 group, ADRs led to premature discontinuation of treatment by 20 patients (20.0%) compared with 8 patients (8.2%) in the placebo-treated group. ADRs that led to treatment discontinuation in at least 2% of patients were hallucinations (8 vs. 0% placebo), dry mouth (3 vs. 0% placebo), peripheral edema (3 vs. 0% placebo), blurred vision (3 vs. 0% placebo), postural dizziness and syncope (2 vs. 0% placebo), abnormal dreams (2 vs. 1% placebo), dysphagia (2 vs. 0% placebo), and gait disturbance (2 vs. 0% placebo).
In the pooled ADS-5102 group, SAEs were reported in 11 patients (11.0%). The only SAEs considered to be related to the study drug were constipation and urinary retention, in the same patient (1.0%). One patient died of advanced PD during hospice care in the ADS-5102 group (not related).
Visual hallucinations were reported by 18 patients in the ADS-5102 group compared with 3 patients in the placebo group. In 9 of the 18 patients reporting visual hallucination, the first occurrence was within the first 30 days of study-drug exposure. Of the 18 patients who reported visual hallucinations, 7 discontinued treatment, 3 had a dose interruption or reduction (to 137 mg), and 8 continued treatment at 274 mg (5 of these 8 patients experienced spontaneous resolution). Overall, for 9 of the 18 patients, the visual hallucinations resolved within two weeks of onset, and no patient required hospitalization or treatment with an antipsychotic medication. In the ADS-5102 group, the frequency of visual hallucination as a reported ADR showed no marked relation to use versus non-use of dopamine agonists or MAO-B inhibitors.
In the pooled ADS-5102 group, hallucination (visual or auditory) was reported in 16 of 52 (30.8%) patients aged ≥ 65 years, compared with 5 of 48 (10.4%) patients aged ≤ 65 years. ADS-5102 patients with a baseline eGFR of 50–89 mL/min/1.73 m2 had an increased rate of visual hallucinations (26.1%), compared with 6 of 54 patients (11.1%) with baseline eGFR ≥ 90 mL/min/1.73 m2.
Patients with major psychiatric disorders, including suicidal ideation, were excluded from the studies, although two patients receiving ADS-5102 experienced suicidal ideation—one was assessed as related to the study drug, and the other was assessed as not related to ADS-5102. The second patient, who attempted suicide, had stopped the study drug 4 days prior to the attempt. Both patients discontinued the study drug.
In general, vital signs and laboratory test results remained consistent with baseline values and were similar between treatment groups throughout the studies.
4 Discussion
ADS-5102 is an ER formulation of amantadine, and, when administered once-daily at bedtime, achieves high plasma drug concentrations (approximately 1500 ng/mL) throughout the day. These plasma concentrations cannot be obtained with amantadine IR when administered conventionally [24], and a phase II/III dose-finding study and two phase III clinical trials have shown that ADS-5102 reduces dyskinesia and also reduces daily OFF time [24,25,26]. As the phase III studies shared the same selection criteria and study design (except that EASE LID 3 had a longer treatment period) and the patient populations were comparable (Table 1), pooling of the patients allowed for further useful analysis.
This pooled analysis confirms that ADS-5102 showed a significant improvement over placebo for dyskinesia, as measured by the primary analysis of UDysRS total score, as early as two weeks (the first post-baseline assessment time point), and maintained this improvement over 12 weeks, with a mean treatment difference versus placebo of − 10.1 points at week 12 (Fig. 2a). More ADS-5102-treated subjects achieved any given level of UDysRS total score improvement, as seen by the leftward shift of the cumulative percentage curve (Fig. 3). For example, 50% of ADS-5102 patients achieved a score reduction of 17 points or more, versus approximately 20% of placebo patients. In addition, patient-reported and physician-rated UDysRS subscores (Parts I and II, and Parts III and IV, respectively) at 12 weeks showed comparable benefit versus placebo (Table 2).
No statistically significant change was observed in MDS–UPDRS Parts I, II, and III scores, showing that ADS-5102 has no significant effect of worsening the underlying PD, nor does it significantly impact the motor activities of daily living. The MDS–UPDRS Part IV score and the scores for time spent with dyskinesia (item 4.1) and functional impact of dyskinesia (item 4.2) both showed a significant beneficial treatment effect for ADS-5102, supporting the finding of the primary analysis. There was also significant improvement in the MDS–UPDRS Part IV items relating to motor fluctuations (items 4.3 and 4.4), which supports the observed decrease in OFF time due to ADS-5102 treatment.
For all the measured subgroups, the ADS-5102 group showed a greater benefit over placebo (Fig. 4). In addition, the benefit versus placebo was independent of baseline dyskinesia severity, as judged by the overlap in 95% CIs for the 12-week change in UDysRS total score in patient subgroups stratified by baseline UDysRS total score or baseline MDS–UPDRS item 4.2 score. However, the mean benefit was numerically greater in patients with more severe dyskinesia (baseline UDysRS total score ≥ 40, and MDS–UPDRS item 4.2 score 3–4).
As measured by PD home-diary entries, significant improvements in the total daily durations of PD clinical states were also achieved by two weeks, and were maintained through 12 weeks. At the end of 12 weeks, the placebo-corrected mean decrease in OFF time was approximately 1 h/day, irrespective of baseline OFF-time duration, and complete resolution of OFF time (and ON time with troublesome dyskinesia) was approximately twice as frequent in the ADS-5102 group as in the placebo group. A decrease in mean OFF time was associated with a decrease in mean ON time with troublesome dyskinesia, supporting the clinical relevance of the observed change in UDysRS total score. The subgroup analysis shows that the improvement occurs irrespective of the baseline OFF time, although the effect is slightly numerically greater for patients with longer (≥ 2.5 h) OFF times at baseline.
The mechanism of action by which ADS-5102 reduces OFF time is unknown, although it could be related to the NMDA receptor antagonism by amantadine, which may block overactive glutamate transmission in the pathway that favors the OFF state in patients with motor complications, resulting in a reduction of OFF time [32].
Changes in the CGI–C also supported the primary analysis findings by showing a statistically significant treatment effect for ADS-5102 at week 12 compared with placebo (p < 0.0001), with 57.0% of subjects in the ADS-5102 group showing moderate to marked improvement in PD symptoms versus 15.6% of subjects in the placebo group.
In general, ADS-5102 was safe and well tolerated. Although visual hallucinations were a reported ADR primarily among patients receiving ADS-5102, hallucinations are a frequent occurrence in PD. Factors such as age, PD progression, cognitive impairment, sleep disorders, comorbidities, and concomitant medications are known to influence the risk of such events [33]. In this study, there was a higher number of ADRs of hallucinations in patients ≥ 65 years, as well as in those with eGFR 50–89 mL/min/1.73 m2.
In the pooled ADS-5102 group, the frequency of visual hallucination showed no marked relation to the use of dopamine agonists or MAO-B inhibitors. However, across the individual studies, the frequency of visual hallucination among ADS-5102 recipients was higher in EASE LID than in EASE LID 3 (23.8% vs. 8.1%, respectively). Although concomitant medication use was less frequent in EASE LID than in EASE LID 3 (46.0% vs. 56.8% for dopamine agonists, and 41.3% vs. 45.9% for MAO-B inhibitors, respectively), mean levodopa dosage was higher in EASE LID (905.6 vs. 671.9 mg/day). Use of anticholinergics was low in both studies, and all other baseline characteristics had similar values across the studies [25, 26]. Patients receiving ADS-5102 should be monitored for hallucinations, especially at the inception of treatment and after dose increases. Lastly, the ADRs of dry mouth, constipation, and nausea appear to be predicted by the anticholinergic activity of ADS-5102.
5 Conclusions
The unique pharmacokinetic characteristics of ADS-5102 may be important for its efficacy in reducing OFF time and dyskinesia, and its favorable tolerability [24]. There is a need for a pharmacotherapy that ameliorates dyskinesia and OFF time, potentially enabling optimization of levodopa dosing. The present analyses further support ADS-5102, administered once daily at bedtime, as an adjunct to levodopa for treating both dyskinesia and OFF time in PD patients.
References
Lindholm D, Makela J, Di Liberto V, Mudo G, Belluardo N, Eriksson O, et al. Current disease modifying approaches to treat Parkinson’s disease. Cell Mol Life Sci. 2016;73(7):1365–79.
Hornykiewicz O. A brief history of levodopa. J Neurol. 2010;257(Suppl 2):S249–52.
LeWitt PA, Fahn S. Levodopa therapy for Parkinson disease: a look backward and forward. Neurology. 2016;86(14 Suppl 1):S3–12.
Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. 2014;311(16):1670–83.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448–58.
Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72(21 Suppl 4):S1–136.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11–6.
Lees AJ. The on-off phenomenon. J Neurol Neurosurg Psychiatry. 1989;52(Suppl):29–37.
Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005;20(2):224–30.
Hechtner MC, Vogt T, Zollner Y, Schroder S, Sauer JB, Binder H, et al. Quality of life in Parkinson’s disease patients with motor fluctuations and dyskinesias in five European countries. Parkinsonism Relat Disord. 2014;20(9):969–74.
Rascol O, Perez-Lloret S, Damier P, Delval A, Derkinderen P, Destee A, et al. Falls in ambulatory non-demented patients with Parkinson’s disease. J Neural Transm (Vienna). 2015;122(10):1447–55.
Suh DC, Pahwa R, Mallya U. Treatment patterns and associated costs with Parkinson’s disease levodopa induced dyskinesia. J Neurol Sci. 2012;319(1–2):24–31.
Muller T, Woitalla D, Russ H, Hock K, Haeger DA. Prevalence and treatment strategies of dyskinesia in patients with Parkinson’s disease. J Neural Transm (Vienna). 2007;114(8):1023–6.
Schaeffer E, Pilotto A, Berg D. Pharmacological strategies for the management of levodopa-induced dyskinesia in patients with Parkinson’s disease. CNS Drugs. 2014;28(12):1155–84.
Chase TN, Oh JD. Striatal mechanisms and pathogenesis of parkinsonian signs and motor complications. Ann Neurol. 2000;47(4 Suppl 1):S122–9 (discussion S129–S130).
Danysz W, Parsons CG, Kornhuber J, Schmidt WJ, Quack G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents: preclinical studies. Neurosci Biobehav Rev. 1997;21(4):455–68.
Ory-Magne F, Corvol JC, Azulay JP, Bonnet AM, Brefel-Courbon C, Damier P, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology. 2014;82(4):300–7.
Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50(5):1323–6.
Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56(11):1383–6.
Wolf E, Seppi K, Katzenschlager R, Hochschorner G, Ransmayr G, Schwingenschuh P, et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov Disord. 2010;25(10):1357–63.
Crosby NJ, Deane KH, Clarke CE. Amantadine for dyskinesia in Parkinson’s disease. Cochrane Database Syst Rev. 2003;(2):CD003467.
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):141–3.
GOCOVRI (amantadine) extended release capsules for oral use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208944lbl.pdf.
Pahwa R, Tanner CM, Hauser RA, Sethi K, Isaacson S, Truong D, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study). Mov Disord. 2015;30(6):788–95.
Pahwa R, Tanner CM, Hauser RA, Isaacson SH, Nausieda PA, Truong DD, et al. ADS-5102 (Amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–9.
Oertel W, Eggert K, Pahwa R, Tanner CM, Hauser RA, Trenkwalder C, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord. 2017;32(12):1701–9.
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–4.
Hauser RA, Friedlander J, Zesiewicz TA, Adler CH, Seeberger LC, O’Brien CF, et al. A home diary to assess functional status in patients with Parkinson’s disease with motor fluctuations and dyskinesia. Clin Neuropharmacol. 2000;23(2):75–81.
Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23(15):2129–70.
Goetz CG, Nutt JG, Stebbins GT. The Unified Dyskinesia Rating Scale: presentation and clinimetric profile. Mov Disord. 2008;23(16):2398–403.
National Institute of Mental Health, Early clinical drug, evaluation unit (ECDEU). Clinical Global impressions. In: Guy W, editor. ECDEU assessment manual for psychopharmacology. National Institute of Mental Health: Rockville; 1976. p. 216–22.
Calon F, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T. Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol Dis. 2003;14(3):404–16.
Diederich NJ, Fenelon G, Stebbins G, Goetz CG. Hallucinations in Parkinson disease. Nat Rev Neurol. 2009;5(6):331–42.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Adamas Pharmaceuticals, Inc. funded these studies and analyses. Editorial assistance to the authors was provided by The Curry Rockefeller Group, LLC, and this assistance was funded by Adamas. Open access was funded by Adamas.
Conflicts of Interest
Lawrence W. Elmer has received honoraria for speaking engagements from Lundbeck, Novartis, UCB Pharma, and Teva Neuroscience; has served as a paid consultant for Teva Neuroscience and UCB Pharma; has received honoraria as a member of advisory boards for Lundbeck, Teva Neuroscience, and UCB Pharma; and has received unrestricted educational grant support from Teva Neuroscience. Jorge L. Juncos reports research grants from the National Institute of Child Health and Human Development (NICHD), the Michael J. Fox Foundation, Adamas Pharmaceuticals, US WorldMeds, Psydon, and Neurocrine. Carlos Singer has received honoraria from Neurocrine, Revance, US WorldMeds, TEVA, and Acorda, and has received grants from Allergan, Adams, Pfizer, Synovia, the Parkinson’s Foundation, National Institutes of Health (NIH), and the Huntington’s Disease Society of America. Daniel D. Truong has received research grants from Ispen, Merz, Auspex, Daiichi Sankyo Pharma, AbbVie, National Institute of Neurological Disorders and Stroke, Kyowa, and Neurocrine. Susan R. Criswell has no relevant disclosures or conflicts of interest. Sotirios Parashos has no relevant disclosures or conflicts of interest. Unrelated to the subject matter of this study, Sotirios Parashos has received consultancies from Dong-A, research support from the Park Nicollet Foundation, National Parkinson Foundation, NINDS, Patient-Centered Outcomes Research Institute, Acorda Therapeutics, Astellas, Pharma2B, and Sunovion Pharmaceuticals. Larissa Felt, Reed Johnson, and Rajiv Patni are employees of and own stock in Adamas Pharmaceuticals.
Additional information
The original version of this article was revised: Table 2, column heads ‘Placebo’ and ‘ADS-5102’ under the ‘Pooled’ section were incorrectly transposed. The column heads are now corrected.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License http://creativecommons.org/licenses/by-nc/4.0/, which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Elmer, L.W., Juncos, J.L., Singer, C. et al. Pooled Analyses of Phase III Studies of ADS-5102 (Amantadine) Extended-Release Capsules for Dyskinesia in Parkinson’s Disease. CNS Drugs 32, 387–398 (2018). https://doi.org/10.1007/s40263-018-0498-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-018-0498-4